

Abstract
In visceral smooth muscles, both M2 and M3 muscarinic receptor subtypes are found, and produce two major metabolic effects: adenylyl cyclase inhibition and PLCβ activation. Thus, we studied their relevance for muscarinic cationic current (mICAT) generation, which underlies cholinergic excitation. Experiments were performed on single guinea-pig ileal cells using patch-clamp recording techniques under conditions of weakly buffered [Ca2+]i (either using 50 μM EGTA or 50–100 μM fluo-3 for confocal fluorescence imaging) or with [Ca2+]i ‘clamped' at 100 nM using 10 mM BAPTA/CaCl2 mixture.
Using a cAMP-elevating agent (1 μM isoproterenol) or a membrane-permeable cAMP analog (10 μM 8-Br-cAMP), we found no evidence for mICAT modulation through a cAMP/PKA pathway.
With low [Ca2+]i buffering, the PLC blocker U-73122 at 2.5 μM almost abolished mICAT, in some cases without any significant effect on [Ca2+]i. When [Ca2+]i was buffered at 100 nM, U-73122 reduced both carbachol- and GTPγS-induced mICAT maximal conductances (IC50=0.5–0.6 μM) and shifted their activation curves positively.
U-73343, a weak PLC blocker, had no effect on GTPγS-induced mICAT, but weakly inhibited carbachol-induced current, possibly by competitively inhibiting muscarinic receptors, since the inhibition could be prevented by increasing the carbachol concentration to 1 mM. Aristolochic acid and D-609, which inhibit PLA2 and phosphatidylcholine-specific PLC, respectively, had no or very small effects on mICAT, suggesting that these enzymes were not involved.
InsP3 (1 μM) in the pipette or OAG (20 μM) applied externally had no effect on mICAT or its inhibition by U-73122. Ca2+ store depletion (evoked by InsP3, or by combined cyclopiazonic acid, ryanodine and caffeine treatment) did not induce any significant current, and had no effect on mICAT in response to carbachol when [Ca2+]i was strongly buffered to 100 nM.
It is concluded that phosphatidylinositol-specific PLC modulates mICAT via Ca2+ release, but also does so independently of InsP3, DAG, Ca2+ store depletion or a rise of [Ca2+]i. Our present results explain the previously established ‘permissive' role of the M3 receptor subtype in mICAT generation, and provide a new insight into the molecular mechanisms underlying the shifts of the cationic conductance activation curve.
Keywords: Smooth muscle, muscarinic receptor, phospholipase C, InsP3, DAG, cAMP, carbachol, cationic current
Introduction
Mixed populations of muscarinic receptors (M2 and M3 subtypes) are co-expressed in gastrointestinal smooth muscles with the preponderance of the M2 subtype (75–80%). At a biochemical level, their activation produces two main effects: adenylyl cyclase (AC) inhibition and phospholipase C (PLC) activation (Caulfield, 1993; Eglen et al., 1996; Caulfield & Birdsall, 1998). A rise in intracellular free Ca2+ concentration ([Ca2+]i) for the contractile response occurs due to both Ca2+ store release (via the M3/PLC/D-myo-inositol 1,4,5,-trisphosphate (InsP3) system) and Ca2+ influx via L-type Ca2+ channels opened by membrane depolarization, which is evoked by the muscarinic receptor cationic current (mICAT). In recent years, several regulatory pathways that are important for mICAT generation and modulation have been revealed, including Ca2+/calmodulin/myosin light-chain kinase cascade (Kim et al., 1995; 1997), protein kinase C (Ahn et al., 1997; Kim et al., 1998b) and tyrosine kinases (Inoue et al., 1994). However, the relevance of the two major pathways, AC inhibition and PLC activation, has not been fully explored.
In visceral smooth muscles, muscarinic receptor-gated cationic channels are primarily opened due to M2 receptor/Go activation (or M2-Gi/Go link in airway myocytes), but M3 receptors also play an important role (Bolton & Zholos, 1997; Kang et al., 1997; Wang et al., 1997; Zholos & Bolton, 1997; Komori et al., 1998; Rhee et al., 2000; Yan et al., 2003). The most obvious mechanism involved is via Ca2+ release due to InsP3 formation, since these cationic channels, apart from being gated by activated G protein (likely the α-guanosine 5′-triphosphate (GTP) subunit of Go protein; Yan et al., 2003), are also highly sensitive to [Ca2+]i (Inoue & Isenberg, 1990b; Pacaud & Bolton, 1991); hence [Ca2+]i oscillations during InsP3-induced Ca2+ release (IICR) are accompanied by concurrent mICAT oscillations (Komori et al., 1993; Zholos et al., 1994; Gordienko et al., 1999). Indeed, after depletion of intracellular Ca2+ stores, methacholine failed to induce a [Ca2+]i increase or mICAT in airway myocytes (Wang et al., 1997). Also, in murine gastric myocytes, the current was inhibited by Gαq/Gα11 antibodies, as well as by the IICR blocker 2-APB (Lee et al., 2003).
However, Ca2+-dependent potentiation of mICAT cannot explain the permissive role of M3 receptor activation under conditions of strongly buffered [Ca2+]i (Bolton & Zholos, 1997; Zholos & Bolton, 1997). It is becoming increasingly evident that receptor-operated cationic currents in various smooth muscles are mediated by TRP channel proteins (Inoue et al., 2001; Walker et al., 2001; Jung et al., 2002; Lee et al., 2003). TRP functions and mechanisms of activation remain controversial and largely unknown, but one common property is the involvement of the PLC/phosphoinositide pathway in their activation or modulation either directly or via Ca2+ store depletion (Clapham et al., 2001).
Thus, in the present study, we investigated two possibilities, namely that some upstream events such as PLC activation per se may potentiate mICAT when [Ca2+]i is ‘clamped' or, alternatively, that Ca2+ store depletion may have an effect on mICAT generation. Our results are consistent with the former hypothesis, as we show that cationic channel opening is supported by PLC activation without the involvement of either InsP3 or 1,2-diacyl-sn-glycerol (DAG).
Methods
Cell preparation and current recording
Adult male guinea-pigs, weighing 300–400 g, were killed by dislocation of the neck followed by immediate exsanguinations, according to Schedule one of the Animals Scientific Procedures Act (1986). Single smooth muscle myocytes from the longitudinal muscle layer of the ileum were obtained after collagenase treatment, as previously described (Zholos & Bolton, 1994; 1997).
Whole-cell membrane current was recorded at room temperature using low-resistance borosilicate patch pipettes (1–3 MΩ) and an Axopatch 200B (Axon Instruments Inc., Foster City, CA, U.S.A.) voltage-clamp amplifier. mICAT was activated by applying carbachol to single cells externally or by intracellular application of guanosine 5′-O-(3-thiotriphosphate) (GTPγS) at 200 μM, by adding it to the pipette solution in order to activate G-proteins directly, bypassing muscarinic receptors. The holding potential was −50 mV, unless stated otherwise. The steady-state current–voltage (I–V) relationship was measured by applying slow-voltage ramps from a holding potential of −40 mV (6 s duration ramp pulses from 80 to −120 mV). This was preceded by a 0.6 s step to 80 mV to reach steady-state conditions before the ramp. Background current was measured before application of carbachol or immediately after break-through when patch pipettes were filled with GTPγS, and was digitally subtracted off-line.
Confocal imaging of fluo-3 fluorescence
Experimental chambers containing cells were placed on the stage of an Axiovert 100M inverted microscope attached to a LSM 510 laser-scanning unit (Zeiss, Oberkochen, Germany). Confocal x–y (at 2 Hz) or line-scan (at 200 Hz) imaging was performed using a Zeiss plan-Apochromat 63 × 1.4 NA oil-immersion objective. Fluo-3 fluorescence was excited by the 488 nm line of a 200 MW Argon ion laser (Laser-Fertigung, Hamburg, Germany) and the illumination intensity was set with an acousto-optical tuneable filter. The emitted fluorescence was captured with the confocal detector at wavelengths above 505 nm. The SCSi interface of the confocal microscope was hosted by a Pentium PC (32-bit Windows NT 4.0 operating system) running LSM510 software (Zeiss, Oberkochen, Germany). To synchronize imaging with electrical recordings, a TTL pulse generated by the confocal scanner at the beginning of the scan protocol was recorded on a digital tape recorder (DTR-1204, Biologic Science Instruments, Claix, France), simultaneously with whole-cell current. Image processing was carried out using an Indy workstation (Silicon Graphic, Inc., Mountain View, CA, U.S.A.) with custom routines written in IDL (Research Systems, Inc., Boulder, CO, U.S.A.), and only fluorescence signal in the absence of detectable movement/contractile response was analysed.
Flash photolysis
InsP3 was rapidly and uniformly released within the cell, as described previously (Zholos et al., 1994). Briefly, light emitted from a xenon arc lamp as a flash of approximately 1 ms duration was filtered (300–380 nm) and focused for photolysis of ‘caged' InsP3. The capacitor was charged to 75 V.
Solutions
The basic external solution in which cationic current was recorded consisted of (in mM): CsCl 120, glucose 12, N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid (HEPES) 10, pH adjusted to 7.4 with CsOH (total Cs+ 124 mM). In the experiments without [Ca2+]i buffering, 2.5 mM CaCl2 and 1.2 mM MgCl2 were added to prevent passive Ca2+ store depletion. Pipettes were filled with the following solution (in mM): CsCl 80, adenosine 5′-triphosphate magnesium salt (ATP) 1, creatine 5, GTP 1, D-glucose 5, HEPES 10, 1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid (BAPTA) 10, CaCl2 4.6 (calculated [Ca2+]i=100 nM), pH adjusted to 7.4 with CsOH (total Cs+ 124 mM). GTP was omitted when GTPγS was added to the pipette solution to activate mICAT directly. In the experiments without [Ca2+]i buffering, BAPTA and CaCl2 were omitted and either fluo-3 (pentapotassium salt) (50 or 100 μM as indicated) or ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA; 50 μM) were added to the pipette solution. CsCl 40 mM was also added to the pipette solution to keep the total Cs+ concentration at 124 mM.
In flash photolysis experiments, the external solution contained (in mM): NaCl 120, KCl 6, CaCl2 2.5, MgCl2 1.2, glucose 12, HEPES 10, pH adjusted to 7.4 with NaOH (this solution was also used to keep the cells prior to carbachol or GTPγS applications as well as to wash out the agonist). Pipette solution had the following composition (in mM): KCl 130, MgATP 1, creatine 5, D-glucose 20, EGTA 0.05, caged InsP3 0.03, HEPES 10, pH adjusted to 7.4 with NaOH. In order to block K+ currents, K+ was replaced by Cs+ and Na+ in the pipette and external solutions, respectively. In Na+-free external solution, Na+ was substituted by N-methyl-D-glucamine (NMDG)+ and TEAOH was used to adjust pH.
1-[6-([(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino)hexyl]-1H-pyrrole-2,5-dione (U-73122), 1-[6-([(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino)hexyl]-2,5-pyrrolidinedione (U-73343), aristolochic acid and 1-oleoyl-2-acetyl-sn-glycerol (OAG) were dissolved in dimethyl sulphoxide (DMSO) to prepare stock solutions (2, 2, 20 and 10 mM, respectively) and then diluted to the required concentration with bathing solution. Since DMSO slightly inhibited mICAT itself, it was added to the external solution first, followed by drug administration at constant DMSO concentration. Thus, in most cases, DMSO (1 : 800 v v−1) was applied simultaneously with carbachol but, in some cells using carbachol and in all cells using GTPγS to activate mICAT, it was applied at a maximum response (these moments are indicated by arrows).
Data analysis
Cationic conductance activation curves were fitted by the Boltzman equation in the following form:
where G is the cationic conductance at potential V; Gmax is the maximal cationic conductance; V1/2 is the potential at which G=0.5Gmax and k is the slope factor of the activation curve.
Inhibition concentration–effect curves were constructed by plotting normalized mICAT amplitude against the blocker concentration on a semi-logarithmic scale. They were fitted by the following equation:
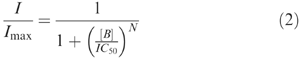 |
where I is mICAT amplitude at a given concentration of the blocker, [B], Imax is the control amplitude of mICAT, IC50 is the concentration required to produce a current equal to 50% of Imax and N is the slope factor of the inhibition curve.
The data were analysed and plotted using MicroCal Origin software (MicroCal Software, Inc., Northampton, MA, U.S.A.). Values are given as the means±s.e.m.; n represents the number of cells tested. To determine the statistical significance of differences between the means, a t-test was used. Differences were judged to be statistically significant when the two-tailed P-value was less than 0.05.
Chemicals used
Collagenase (type 1A), ATP (magnesium salt), GTP (lithium salt), GTPγS (tetralithium salt), creatine, HEPES, BAPTA, EGTA, D-myo-inositol 1,4,5,-tris-phosphate (InsP3), U-73122, U-73343, O-tricyclo[5.2.1.02,6]dec-9-yl dithiocarbonate potassium salt (D-609), aristolochic acid, OAG, DMSO, NMDG and carbamylcholine chloride (carbachol) were obtained from Sigma Chemical Co., Poole, Dorset, U.K. Myo-inositol 1,4,5-triphosphate,P4(5)-1-(2-nitrophenyl)ethyl ester, sodium salt (caged InsP3), cyclopiazonic acid (CPA), ryanodine and thapsigargin were obtained from Calbiochem-Novabiochem (U.K.) Ltd, Beeston, Nottingham, U.K. Parafluoro-hexahydro-sila-difenidol hydrochloride (p-F-HHSiD) was obtained from Research Biochemicals Inc. (Natick, MA, U.S.A.). Fluo-3 (pentapotassium salt) was purchased from Molecular Probes Europe BV, Leiden, The Netherlands. All other chemicals were from BDH Laboratory Supplies (AnalaR grade), Pool, U.K.
Results
cAMP has no effect on mICAT
Agents which raise cAMP did not affect mICAT recorded using conventional whole-cell patch-clamp configuration (e.g., [Ca2+]i ‘clamped' at 100 nM). Thus, 8-Br-cAMP at 10 μM had no effect on either GTPγS-induced (n=5) or 100 μM carbachol-induced (n=6) current in the presence of 1 mM GTP in the pipette solution (Figure 1b, c). Since these are conditions of maximal activation of mICAT as well as maximal prevention of its desensitization (Zholos & Bolton, 1996), similar tests were performed at low (1 μM) or half-maximal (10 μM) agonist concentration with or without intracellular GTP. There was no effect on the currents evoked by 1, 10 or 100 μM carbachol with (n=5) or without (n=8) GTP in the pipette solution (e.g., Figure 1a). Also, increasing 8-Br-cAMP to 100 μM (n=3) or applying 1 μM isoproterenol (n=5) had no effect on mICAT evoked by 10 μM carbachol (Figure 1d). Thus, cAMP did not affect mICAT at least under conditions when modulation of intracellular free Ca2+ concentration was prevented.
Figure 1.

No role of cAMP in mICAT generation in ileal myocytes. (a, b) mICAT responses at −40 mV to carbachol and GTPγS applications. 8-Br-cAMP was applied at 10 μM, as indicated by the horizontal bars. (c, d) Steady-state I–V relationships in two different cells in control and after 8-Br-cAMP (10 μM) or isoproterenol (1 μM) were applied, as shown by the grey lines. The horizontal dotted lines in this and all the subsequent figures indicate zero current level.
PLCβ affects mICAT in Ca2+-independent manner: weak [Ca2+]i buffering
The role of PLCβ in mICAT activation was investigated using a potent blocker of PLC, U-73122, which has been shown to inhibit PLC at a low micromolar concentration in a number of tissues (Bleasdale et al., 1990; Smith et al., 1990; Thompson et al., 1991; Smallridge et al., 1992; Yule & Williams, 1992), and thus it is one of the most widely used PLC inhibitors in functional studies (Helliwell & Large, 1997; Wang & Kotlikoff, 2000; Cho et al., 2001; Stemkowski et al., 2002; Lee et al., 2003). IICR facilitated mICAT as Ca2+ waves and concurrent mICAT oscillations were observed in fluo 3-loaded myocytes when G-proteins were gradually activated by cell dialysis with GTPγS (Figure 2c, d) (compare to Pacaud & Bolton, 1991; Komori et al., 1993; Kohda et al., 1996).
Figure 2.

Cationic current responses to intracellular GTPγS in control (a) and their inhibition by U-73122 (b, c) under conditions when intracellular Ca2+ was lightly buffered (a, c – 50 μM fluo-3; b – 50 μM EGTA in the pipette solution). The holding potential was −50 mV. (d) Corresponding time-course plot of the normalized intensity of fluo-3 fluorescence averaged within the total confocal optical slice (Δz<1.2 μm) of the myocyte. A time series of 227 x–y confocal images of fluo-3 fluorescence was acquired (at a rate of 496 ms per frame) simultaneously with recording of whole-cell cationic current (shown in (c)). The fluorescence intensity was normalized to the average fluorescence intensity in eight images, showing the most uniform and least intense fluorescence among the series. Note ‘whole-cell' oscillations in [Ca2+]i associated with oscillations of mICAT.
Typically, the response to intracellular application of GTPγS was as illustrated in Figure 2a, c, that is, the amplitude of mICAT oscillations and accompanying [Ca2+]i oscillations increased with time following break-through, eventually giving rise to a sustained mICAT as [Ca2+]i oscillations fused to produce a high intracellular Ca2+ level (Figure 2d). U-73122 at 2.5 μM inhibited this sustained mICAT strongly (n=10; Figure 2c), although the effect on [Ca2+]i at this concentration was usually small (Figure 2d). Since GTPγS application is known to deplete Ca2+ stores in these cells (Komori et al., 1992) and as [Ca2+]i was not appreciably reduced by U-73122 while mICAT was almost abolished, it appeared that Ca2+ store depletion or some other aspects of Ca2+ metabolism were not involved in this inhibition. In the remaining cells, either mICAT oscillations were observed (without a substantial sustained component), which were abolished by U-73122 (n=2; one example is shown in Figure 2b), or a large sustained current rapidly developed following break-through, which was also blocked by 2.5 μM U-73122 (n=3; data not shown).
Similar results were obtained for carbachol-induced current. U-73122 at 2.5 μM abolished the mICAT oscillations usually seen at lower agonist concentrations (5 or 10 μM; n=8), as well as the more sustained responses evoked by 50 μM carbachol (n=3; data not shown). Often, the inhibition was not accompanied by a change in the fluo-3 fluorescence signal, although in some cases the latter declined to the resting level in parallel with mICAT decline (compare panels (a) and (b) in Figure 3). In the latter case, mICAT inhibition could only partially be attributed to [Ca2+]i decline, since even 30 nM [Ca2+]i is not without effect on these cationic channels (Tsytsyura et al., 2000).
Figure 3.

mICAT (top) and corresponding normalized fluo-3 fluorescence signal acquired in the line-scan mode at 127 Hz (bottom) in two different cells (a, b), both exposed to 10 μM carbachol. The holding potential was −50 mV. Fluo-3 concentration was 100 μM. The fluorescence intensity in the images was normalized to the average fluorescence intensity during the first 1300 lines in each image, and grey scale-coded, as indicated by the bars (F/F0). The plots below the images show the time course of the normalized fluorescence averaged along the whole scan line (in (a) and (b) 12 and 18 μm, respectively).
PLCβ is involved in mICAT Ca2+-independent regulation: strong [Ca2+]i buffering
The above results suggested that PLC inhibition could result in mICAT depression in a Ca2+-independent manner. To test this further, we have ‘clamped' [Ca2+]i at 100 nM using 10 mM BAPTA and removed divalent cations from the external solution. This Ca2+ concentration, which is close to the physiological resting level, was found to be advantageous to study mICAT properties, since at lower [Ca2+]i (e.g., 30 nM) the current was much smaller while at higher [Ca2+]i (e.g., 500 nM) the signal-to-noise ratio was not improved significantly, but strong Ca2+-dependent desensitization of the current developed instead (Tsytsyura et al., 2000). Tests using confocal [Ca2+]i imaging in the line-scan mode (the line positioned just below the membrane) confirmed that there was no measurable fluorescence signal change even with three times lower buffering capacity (e.g., 3.3 mM BAPTA/1.518 mM CaCl2; n=3).
Under these conditions, steady mICAT with slow desensitization (Zholos & Bolton, 1996) was generated in response to 50 μM carbachol application or cell dialysis with GTPγS; the latter was used to explore the possibilities that the inhibition produced by U-73122 occurred upstream (e.g., receptor/G-protein coupling) or downstream of G-protein activation. In both cases, mICAT was inhibited nearly completely at 2.5 μM U-73122 (Figure 4a, b). The inhibition developed with a time constant of 16.3±2.7 s (n=10) and was irreversible.
Figure 4.

Inhibition of mICAT under conditions when [Ca2+]i was ‘clamped' at 100 nM using 10 mM BAPTA/Ca2+ buffer. mICAT was activated by 50 μM carbachol (a) or cell dialysis with 200 μM of GTPγS (b), and DMSO at 1 : 800 v v−1 was administered before U-73122 application (as shown by arrows) to account for its small inhibitory effect. Vertical deflections on the current trace in (b) correspond to the moments when the I–V curves were measured by the voltage ramp protocol at 30 s intervals. Superimposed dotted lines in (a, b) show single exponentials fitted to the data points during mICAT inhibition by U-73122, with the time constants indicated. (c) U-73122 was applied cumulatively at ascending concentrations, as indicated at the top, to evaluate the concentration dependence of its effect. (d) Mean relative carbachol- and GTPγS-evoked current amplitude measured at −50 mV (n=7 for both) plotted against U-73122 concentration on a semilogarithmic scale. Data points were fitted by the Hill equation (equation (2)) with an IC50 value of 0.64 μM and Hill slope of 1.8 for carbachol-evoked current and an IC50 value of 0.50 μM and Hill slope of 1.9 for GTPγS-evoked current.
Increasing U-73122 concentration cumulatively (0.1, 0.3, 1 and 3 μM; see Figure 4c as an example) showed that at −40 mV carbachol- and GTPγS-induced currents were inhibited with IC50 values of 0.60±0.07 μM (n=7) and 0.51±0.05 μM (n=7), respectively, as shown in Figure 4d. The difference in these averaged best-fit values was not statistically significant (P=0.32). The Hill slopes were also similar: 1.82±0.06 for carbachol- and 1.99±0.06 for GTPγS-evoked mICAT (P=0.07). Note that the smooth curves that approximate the average data points in Figure 4d have slightly different parameters compared to those averaged from individual cells.
Effects of PLC inhibitors on biophysical properties of mICAT: GTPγS-evoked current
We have further studied the changes of mICAT biophysical properties by monitoring its steady-state I–V relationship at 30 s intervals during the drug's action (as seen by the vertical deflections on the trace in Figure 4b). From the onset of cell dialysis with GTPγS, mICAT gradually develops as GDP molecules bound to the α-subunits of G proteins are gradually replaced by GTPγS, resulting in a slow accumulation of the activated G-proteins (since GTPγS binding is almost irreversible). Associated with this there were characteristic changes of the I–V curve: (i) current developed and saturated much faster at positive potentials; (ii) the linear portion of the I–V curve was extended in the negative range, resulting in a shift of the peak of the I–V curve towards more negative potentials; while (iii) the curve became less U-shaped since the extent of the current deactivation decreased at negative potentials (see Figure 5a, traces a–c were measured during mICAT development, as indicated by the corresponding letters in Figure 4b). These observations are identical to those seen when the carbachol concentration was raised. We have previously attributed them to the negative shift of the activation curve although the underlying mechanism remains unknown (Zholos & Bolton, 1994).
Figure 5.

Changes in mICAT biophysical properties during the action of U-73122. (a) Steady-state I–V relationships measured in the experiment of Figure 4b, at the moments indicated by the corresponding letters which were selected to allow comparison of mICAT voltage dependence at the beginning of the GTPγS action and when the current became nearly fully inhibited (traces a, e), as well as at about half-maximal activation and half-maximal inhibition (traces b, d). (b) The activation curves in control as well as 20 and 50 s after U-73122 application were fitted by the Boltzman equation (equation (1); superimposed smooth lines) with potentials of half-maximal activation of −70 (control), −61 (20 s) and −55 mV (50 s). Maximal conductance was reduced from 43 to 24 and 14 nS, respectively, whereas the slope factor remained unchanged (−16 mV). (c) mICAT was activated by 50 μM carbachol and its I–V relationship was measured before and during cumulative applications of U-73122 at ascending concentrations, as indicated by the numbers (in μM) near each trace. (d) The analysis was similar to that shown in (b). The V1/2 value shifted progressively from −77 mV in control to –74, –64, and −57 mV when concentration of the PLC blocker was increased, whereas the slope factor remained the same (−19 mV). At 3 μM U-73122, the parameters could not be determined, as the current at negative potentials was completely blocked. Note, however, that at this high concentration the difference current was inward at potentials positive to about 50 mV (c). This was because the outward current in the presence of carbachol and 3 μM of U-73122 became smaller than the outward current before carbachol application (e.g., background current subtraction produces a net inward current at positive potentials). These channels can open spontaneously, particularly at very positive potentials, likely because some background receptor activation and receptor-G-protein coupling exist in nonstimulated cells, as we have found that the outward current could also be inhibited by atropine (50 nM) or intracellular application of GDP-βS (2 mM) (not shown). Thus, U-73122 at 3 μM inhibits not only the agonist-activated current, but also this spontaneous channel activity.
Superimposed I–V relationships measured during mICAT inhibition after U-73122 application are also shown in Figure 5a for comparison (traces d and e). It can be seen that PLC inhibition reversed the changes due to gradual G protein activation, so that the I–V curves measured early after breakthrough, or at steady-state inhibition (traces a and e), or at about half-maximal activation and half-maximal inhibition (traces b and d), were closely similar.
Further analysis was done by converting I–V relationships into the conductance curves (by dividing current amplitudes at each potential by the driving force at the same potential) and fitting these by the Boltzman equation. This revealed that, during the inhibition, the maximal cationic conductance, Gmax, was significantly suppressed (from 39.4±6.6 nS in control to 11.4±3.3 nS in the presence of U-73122 (n=6); P=0.0035), while the activation curve shifted positively by 22.8±1.8 mV (n=6) when the shift was measured in individual cells (e.g., from −80.0±3.7 mV in control to −57.2±4.9 mV with U-73122 (n=6); P=0.004) (Figure 5b; traces in control and at 20 and 50 s after 2.5 μM U–73122 are superimposed). However, the slope factor of the curve remained unchanged (−25.4±1.8 mV before and −23.0±1.7 mV (n=6) after U-73122 application; P=0.355).
The changes in the activation curves during the onset of GTPγS action and during the inhibition produced by U-73122 were accompanied by similar changes in mICAT kinetics. Thus, shortly after breakthrough with GTPγS in the pipette solution (e.g., at 30 s), the current upon a voltage step from −40 to −120 mV increased instantaneously, but rapidly declined to a smaller steady-state level (τ=20.2±6.2 ms). However, when the conductance was fully activated (e.g., 3–4 min later), the time constant increased to 97.0±15.2 ms, but it decreased again to 21.1±9.5 ms when the current was inhibited by the PLC blocker (n=6).
In contrast to the action of U-73122, U-73343, a weak inhibitor of PLC often used as a negative control (Bleasdale et al., 1990; Smith et al., 1990; Smallridge et al., 1992; Helliwell & Large, 1997; Stemkowski et al., 2002), had no or very slight effect at 2.5 μM on GTPγS-evoked current (Figure 6c).
Figure 6.

U-73343 inhibited mICAT activated by carbachol (b), but not the current induced by direct activation of G-proteins by GTPγS, bypassing muscarinic receptors (c). The inhibition produced by U-73343, but not that evoked by U-73122, could be relieved by increasing carbachol concentration (compare (a) and (b)). In (a, b), DMSO was applied simultaneously with carbachol, whereas in (c) the moment of its application is shown by the arrow. Vertical deflections in (c) are produced by voltage ramps. D-609, an inhibitor of phosphatidylcholine-specific PLC (Ki=5–10 μM), at 100 μM had no or a very small effect (<15% inhibition) on mICAT (d).
Carbachol-evoked current
Similar effects were observed when the U-73122 concentration was increased in a cumulative manner to 3 μM in the presence of 50 μM carbachol (Figure 5c). The more negative the membrane potential, the earlier and more pronounced was the inhibition. The cationic conductance was suppressed and the potential of half-maximal activation shifted positively by 22±5 mV (n=3) (Figure 5d; P=0.82 compared to the effect on the GTPγS-evoked current). These changes of the parameters of the activation curve were similar to those produced by desensitization (Zholos & Bolton, 1996), but opposite to those produced by increasing agonist concentration (Zholos & Bolton, 1994).
Control experiments were done using U-73343, an inactive analog of U-73122. In contrast to the lack of its effect on mICAT activated by GTPγS, U-73343 inhibited mICAT evoked by external carbachol application. However, in several aspects, the inhibition was clearly different from the inhibition by U-73122. First, at a concentration of 2.5 μM U-73343, it developed more slowly (τ=29.3±4.5 s, n=5; P=0.02), and it was reversible upon the drug's withdrawal. Second, increasing carbachol concentration to 1 mM could prevent the action of U-73343 (n=3) but not U–73122 (n=3) (Figure 6a, b), thus suggesting that the former could act as a competitive inhibitor of the muscarinic receptors.
The inability of U-73343 to affect GTPγS-evoked current excludes the possibility that this compound (and by the same argument the structurally related U-73122) blocks cationic channels directly, while competition experiments using increasing carbachol concentrations suggest that U-73343 may serve as a competitive inhibitor of the muscarinic receptors. However, the latter effect does not seem to contribute significantly to the action of U-73122, since the inhibition of mICAT caused by this compound, unlike that with U-73343, was neither reversible (Figure 4a) nor surmountable (Figure 6a). Moreover, U-73122 was less potent against carbachol-induced than against GTPγS-evoked current (Figure 4d), while the opposite would be expected from the combined inhibitory action of U-73122 on PLC and muscarinic receptors.
U-73122 is also a weak blocker of phospholipase A2 (PLA2). However, aristolochic acid, a potent selective inhibitor of PLA2, at 100 μM had no effect on carbachol-induced mICAT, while in the same cells the current could be blocked by U-73122 (n=3; data not shown). A possible involvement of other phospholipases in mICAT regulation was tested using D-609, inhibitor of phosphatidylcholine-specific PLC (Ki=5–10 μM). At 100 μM, D-609 had no or a very small effect (<15% inhibition) on carbachol- or GTPγS-induced mICAT (Figure 6d). Thus, phosphatidylinositol-specific phospholipase C (PI-PLC) is the likely link for activated G-proteins in the generation of mICAT in ileal myocytes. Since U-73122 inhibits the coupling of G-protein to PI-PLC (Bleasdale et al., 1990; Thompson et al., 1991; Yule & Williams, 1992), this appears to be the underlying mechanism.
Mechanisms of PLC-dependent activation of mICAT: no role of Ca2+ store depletion
Previous studies have shown that cell dialysis with high-EGTA solution (e.g., 40 mM), which is expected to completely deplete Ca2+ stores, did not evoke cationic currents in ileal myocytes. Moreover, under the conditions of such abnormally low [Ca2+]i, acetylcholine also failed to activate mICAT (Inoue & Isenberg, 1990b). [Ca2+]i was thus postulated to have some permissive role in the cationic channel opening. Therefore, to test the hypothetical link between the filling status of the Ca2+ stores and mICAT generation, further tests were needed, whereby [Ca2+]i would be allowed to rise to, or at least remain at, some level sufficiently high to enable cationic channel opening.
In K+-filled myocytes, InsP3 rapidly released within the cell from its inactive caged precursor by flash photolysis evoked a large Ca2+-activated K+ current, which was of up to 1 nA in size at −50 mV (Figure 7a, left trace; n=19), thus suggesting an efficient Ca2+ release. Since, when applied at intervals, InsP3- and 10 mM caffeine-induced responses were similar in size (Zholos et al., 1994), it appeared that the Ca2+ store was efficiently emptied by flash-released InsP3. However, under K+-free conditions (K+ in the pipette and external solution was replaced by Cs+ and Na+, respectively) and the absence of carbachol or GTPγS, there was only a small inward current in response to flash of the same intensity (Figure 7a, right trace). Its average amplitude at −50 mV was 12.1±2.0 pA (n=8). This was cationic current, as it became somewhat larger in Cs+ containing external solution (on average by 47%; n=4); it was not affected when [Cl−]o was reduced to 17 mM (aspartate substitution; n=7) (data not shown), but it was abolished when external Na+ was replaced with NMDG+ (Figure 7b). Similarly, no substantial current developed in cells treated with thapsigargin (TG; 10–20 min at 100 nM, n=5, data not shown).
Figure 7.

No role of Ca2+ store depletion in mICAT generation. (a) Left trace shows a typical response to flash-released InsP3 in K+-filled ileal cell (weakly buffered with 50 μ) held at −50 mV. The application of UV flashes is indicated by the up-triangles. A typical response to the same flash intensity when K+ was replaced (with Cs+ in the pipette and Na+ in the external solution) is shown on the right. (b) Mean relative photolysis InsP3-induced current amplitude plotted against membrane potential (voltage steps of 50 ms duration were applied from 20 to −60 mV, with a 20 mV increment) before and after external Na+ removal (substituted by NMDG+). Current amplitude in Na+-containing solution at 20 mV was normalized as 1.0 (the average value was 19.9±3.4 pA; n=16). (c) mICAT evoked by carbachol at cumulatively increasing concentrations, as indicated at the top in control (internal [Ca2+]i clamped to 100 nM), and following 3 min pretreatment in a Ca2+-free solution containing 30 μM CPA, 10 μM ryanodine and 10 mM caffeine. During mICAT measurements, caffeine was removed. Note a small steady inward current appearing following Ca2+ store depletion (its mean value was 44±26 pA; n=8). (d) Mean normalized concentration–response data points were fitted by the Hill equation with the following parameters: the EC50 values of 5.3 and 8.4 μM; Hill slopes of 1.23 and 1.15 before and after Ca2+ store depletion, respectively. mICAT amplitude at 300 μM carbachol before Ca2+ store depletion was normalized as 1.0 (this corresponded to 796±90 pA (n=8).
Thus, Ca2+ store release alone, even under conditions when [Ca2+]i is allowed to rise, activates only a small cationic current which is about two orders of magnitude smaller than the carbachol- or GTPγS-induced mICAT. We have also performed Ca2+ store depletion while measuring mICAT in response to carbachol under conditions when [Ca2+]i was clamped at 100 nM using 10 mM BAPTA buffer. This efficiently prevents any Ca2+-dependent modulation of cationic channels. In these experiments, agonist curves were generated by applying carbachol cumulatively at ascending concentrations, rather than applying carbachol at a single concentration, as this would allow us to detect more subtle changes in the signal transduction (e.g., a possible sensitizing effect). A typical example is illustrated in Figure 7c. After establishing the carbachol concentration–response relationship, the cell was kept in a Ca2+-free solution containing CPA (30 μM), ryanodine (10 μM) and caffeine (10 mM) for 3–4 min before the second agonist curve was generated (this was done in the continuous presence of CPA and ryanodine, but caffeine was removed since it is known to block these cationic channels, as reported by Chen et al., 1993). Again, the inward current which developed during the Ca2+ store depletion protocol was small (notice a small downward trace displacement in Figure 7c, right; the average value was 44±26 pA, n=8) compared to the average maximal response at 300 μM carbachol of 796±90 pA, n=8 (normalized as 1.0 in Figure 7d). Moreover, there was no significant change of the agonist curve (EC50 value of 5.3 μM in control vs 8.4 μM after Ca2+ store depletion for the fitted curves in Figure 7d; Hill slopes were 1.23 and 1.15, respectively). Maximal response was not potentiated but declined to some extent, which was indistinguishable from the usual desensitization (compare to Zholos & Bolton, 1996; 1997). The averaged EC50 values obtained from individual cells were 6.5±0.6 μM (control) and 11.1±1.7 μM (after Ca2+ store depletion) (n=8; paired t-test, P>0.05). The efficiency of the Ca2+ store depletion protocol was verified by confocal fluorescence imaging of [Ca2+]i in combination with flash photolysis of caged InsP3 – it was observed that treating the cells with CPA alone (e.g., at the same concentration of 30 μM over comparable period of time) completely abolished the Ca2+ response to InsP3.
It can thus be concluded that Ca2+ store depletion neither appreciably activates these cationic channels itself nor affects signal transduction between muscarinic receptor activation and channel opening when [Ca2+]i is clamped at 100 nM.
Inhibitory action of U-73122 on mICAT is not related to InsP3 and DAG production
PI-PLC accelerates the hydrolysis of membrane phosphoinositides (e.g., phosphatidylinositol 4,5-bisphosphate, PIP2), producing two important intracellular second messengers, InsP3 and DAG, which could thus mediate the effect of PLC on mICAT. The cells were dialysed with 1 μM InsP3 by adding it to the pipette solution for 3 min before carbachol was applied, followed by U-73122 application, as shown in Figure 8a. The inhibition of mICAT was not affected (n=3). In rabbit portal vein myocytes, noradrenaline-evoked cationic current, which has some common features with the muscarinic cationic current in ileal myocytes, can be directly activated by OAG, a synthetic cell-permeable DAG analog (Helliwell & Large, 1997; Albert & Large, 2001). In ileal myocytes, OAG (20 μM) applied externally evoked only a small, less than 20 pA, inward current (Figure 8b and inset), and did not affect the current after it was activated by carbachol (Figure 8c). The inhibition of mICAT by U-73122 was also unaffected (Figure 8b,c).
Figure 8.

InsP3 (a; applied in the pipette) and OAG (b, c; applied in the external solution) have no effect on mICAT or its inhibition by U-73122. In the experiment shown in (a), carbachol was applied 5 min after breakthrough with 1 μM InsP3 in the pipette solution. (b, c) OAG was applied in the external solution. In the inset in (b), a magnified segment of the current trace is shown on the same time scale. Holding potential was −40 mV throughout.
Functional relevance of M3 receptor activation
These results imply that the M3/PLC signalling link is important for both cationic channel opening (e.g., maximal conductance and hence the number of active channels depend on PLC activation) and their voltage dependence (e.g., stronger depolarization is required to open the same proportion of channels when this link is disabled) (Figure 5). Inhibition of the muscarinic receptors by an M3-selective antagonist p-F-HHSiD at low, 10−7 M, concentration (compare to pA2=6.7; Zholos & Bolton, 1997) mimicked the effect of U-73122 at 0.1 μM (compare Figures 9a and 5c and Figure 5c). The inhibition was stronger at negative potentials, resulting in a positive shift of the activation curve by an average of 14±1 mV (n=3) (Figure 9a, inset).
Figure 9.

M3 selective antagonist p-F-HHSiD mimics the action of U-73122. (a) I–V relationships of mICAT evoked by 50 μM carbachol in control and in the presence of 100 nM of the antagonist. The activation curves plotted in the inset were fitted by the Boltzman relation (equation (1)), which showed a positive shift of the activation curve by about 13 mV (in the fitted curves half-maximal activation occurred at −81.0 and −67.7 mV). Note that the effect at this low antagonist concentration was similar to that produced by a low U-73122 concentration (e.g., Figure 5c): there was little inhibition of the current at positive potentials, while at −120 mV the current was suppressed by 67%. (b) When the negative shift of the activation curve during the onset of carbachol action was inhibited by p-F-HHSiD (added 2 min prior to the second carbachol application to allow for equilibration), mICAT was inhibited, but its activation kinetics became much more rapid. Holding potential was −80 mV. (c) The superimposed normalized traces were fitted by single exponential functions with the time constants indicated. Note that the onset of mICAT in control was sigmoidal rather than exponential; therefore, the time constant gives the lower limit of the activation rate. Mathematical modelling can reproduce this behaviour based on the time dependence of the activation curve negative shift, but a more detailed description is beyond of the scope of this paper. Notably, this sigmoidal part disappears when the V1/2 shift is blocked by p-F-HHSiD.
The shift of the activation curve during the action of U-73122 or p-F-HHSiD was of particular interest, since increasing the agonist concentration not only opens these channels but also modulates the voltage range of their activation in the opposite manner (Zholos & Bolton, 1994). This latter process is likely to be physiologically relevant for determining the rates of mICAT activation – as the activation curve shifts negatively during the onset of the agonist action, this is expected to increase the current amplitude at the same holding potential. Since this effect becomes more pronounced at hyperpolarized potentials (simply because a larger shift of the V1/2 value would be needed), the following tests were done at −80 mV. mICAT was induced by 50 μM carbachol application, followed by the agonist wash-out and cell incubation with the antagonist (at 0.5 μM for 2 min) and subsequent carbachol application in the presence of p-F-HHSiD (Figure 9b). The current was inhibited, but at the same time its activation became considerably faster: the time constant decreased from 7.61±1.45 s in control to 2.12±0.13 s in the presence of the M3 antagonist (n=5; P<0.006) (compare superimposed normalized current traces in Figure 9c).
Thus, this putative slow intracellular regulatory process, which links PI-PLC activation to mICAT, not only determines the size of the response, but also modulates mICAT kinetics.
Discussion
cAMP/PKA pathway
The fact that mICAT generation is accompanied by the inhibition of adenylyl cyclase (an M2 effect) may imply that cAMP inhibits cationic channels. However, our results do not support this, which is consistent with the lack of correlation between various muscarinic agonist potencies to inhibit adenylyl cyclase and to induce mICAT in the longitudinal smooth muscle of guinea-pig small intestine (Okamoto et al., 2002). Similarly, oxotremorine-M contractile responses were not significantly altered by 1 μM isoproterenol (Ostrom & Ehlert, 1998). Consistent with the present results, we also found that inhibition of Gi or Gs proteins by antibodies did not alter mICAT response in these cells (Yan et al., 2003).
PLC pathway
Our present results call for a review of the existing hypotheses explaining the involvement of M2 and M3 muscarinic receptors in mICAT generation in visceral smooth muscles. When PI-PLC was inhibited by U-73122 in the presence of weak [Ca2+]i buffering, mICAT inhibition was accompanied by a substantial reduction of [Ca2+]i (e.g., Figure 3a), consistent with the classical views that the M3/Gq/11/PLC link is only important in raising [Ca2+]i, which in turn potentiates mICAT due to the strong Ca2+ dependence of these cationic channels (for a review, see Bolton et al., 1999). One of the reported nonspecific actions of relatively high concentrations of U-73122 (e.g., 5 μM) is to increase [Ca2+]i and InsP3 levels (Smallridge et al., 1992). Indeed, occasionally, [Ca2+]i increased slightly following U-73122 application (e.g., Figures 2d and 3b; probably nonspecific action, see above), but mICAT was still invariably suppressed. Thus, these processes are not necessarily related. These observations prompted us to perform the tests at strongly buffered [Ca2+]i, whereby any Ca2+-dependent changes of the cationic channel gating could be prevented. Under these conditions, U-73122 also abolished mICAT (Figures 4, 5, 8), thus suggesting that a link via PLC upstream of IICR is crucially important in opening cationic channels.
Recently, it was found in cardiac myocytes that IK,ACh could be inhibited by U-73122 irrespective of G-protein activation, for example, channel activity induced by Na+ ions was also suppressed (Cho et al., 2001). As there was no evidence of a direct channel block, the authors postulated the interference with PIP2-channel interaction as the most likely mechanism. The role of Gβγ and Na+ in stabilizing the interactions of PIP2 with the KACh channel to generate IK,ACh is well established, but at present no data are available for a similar mechanism of cationic channel opening, and thus the role of PIP2 in mediating the inhibitory effect of U-73122 on mICAT remains to be established. Notably, the inhibition of KACh channel was voltage-independent and the IC50 values for both blockers were similar and significantly lower (0.12–0.16 μM; Cho et al., 2001) than the values reported for PLC inhibition by U-73122 (1–4 μM; Bleasdale et al., 1990; Smith et al., 1990; Thompson et al., 1991). The IC50 value for mICAT inhibition (0.5–0.6 μM; Figure 4d) is closer to that expected to produce a significant inhibition of PI-PLC. Moreover, it was nearly the same for both carbachol- and GTPγS-evoked currents.
Identical changes of mICAT biophysical properties (Figure 5b, d), closely similar kinetics (Figure 4a, b) and concentration dependence of the inhibitory action of U-73122 (Figure 4d) on both carbachol- and GTPγS-evoked currents are several lines of evidence, which taken together strongly suggest that the inhibitor blocks both currents via the same process. Since its inactive counterpart, U-73343, did not affect GTPγS-induced current, the site of U-73122 action is likely to be PLC, and since GTPγS activates G-proteins directly, our findings are in agreement with the postulated site of U-73122 action at the interface of the G-protein/PLC system (Bleasdale et al., 1990; Thompson et al., 1991; Yule & Williams, 1992). Moreover, the specificity of the action of U-73122 was further confirmed by the findings that inhibition of the muscarinic receptors by an M3-selective antagonist p-F-HHSiD at low, 10−7 M, concentration mimicked its action (Figure 9a).
If U-73122 acted simply as a channel blocker, its action would have to be strongly voltage-dependent, increasing with membrane hyperpolarization (Figure 5) in a manner indistinguishable from the intrinsic voltage dependence of the cationic conductance (since the Boltzman slope of the activation curve remained unchanged when mICAT was suppressed). Also, such presumed channel blockade should have the properties similar to those of mICAT desensitization (Zholos & Bolton, 1996), but exactly opposite to those seen when agonist concentration is increased (Zholos & Bolton, 1994) or GTPγS acts to activate mICAT (Figure 5a). Thus, we consider that a channel block by U-73122 is a highly unlikely possibility. Nevertheless, mICAT inhibition by U-73343 raised some doubts and therefore further tests were performed, which showed that it could act as a competitive antagonist of the muscarinic receptors (Figure 6b). This effect must be added to the growing list of other actions of this drug. This action seems a general one, since in the mouse urinary bladder carbachol-evoked contraction was also competitively antagonized by U-73343 (Dr Jörg Wegener, Institut für Pharmakologie und Toxikologie, München, Germany; personal communication).
There is compelling evidence of the importance of the M2-Gi/Go link in mICAT generation. Previous studies have shown that Pertussis toxin treatment abolishes this current in ileal and gastric cells (Inoue & Isenberg, 1990a; Komori et al., 1992; Pucovsky et al., 1998; Rhee et al., 2000). These results suggested that the M2 receptor subtype was involved, as it couples preferentially to Gi/Go proteins, and this coupling is selectively blocked by Pertussis toxin. Later on, using antibodies, the G proteins involved in the signal transduction were pinpointed as Go in gastric and ileal cells (Kim et al., 1998a; Yan et al., 2003; in the latter study, the role of Gβγ subunit was also excluded) or both Gi and Go in tracheal myocytes (Wang et al., 1997). Parallel pharmacological studies have consistently established the importance of the M2 receptor subtype (Bolton & Zholos, 1997; Kang et al., 1997; Wang et al., 1997; Zholos & Bolton, 1997; Komori et al., 1998).
However, in recent years, it also became increasingly evident that the M3 subtype stimulation is also very important for mICAT generation. Although this is agreed in general, the underlying molecular mechanisms remain unknown, while the particulars of the M3-mediated effect on mICAT differ. Thus, in tracheal cells, stimulation of M2 receptors alone was insufficient to activate mICAT, unless there was a parallel rise of [Ca2+]i associated with the M3 receptor activation (Wang et al., 1997). In terms of the underlying mechanisms, such M3 receptor involvement has the most straightforward interpretation: Ca2+ release via the M3/Gq/11/PLC/InsP3 pathway potentiates cationic channel opening, which is a prominent feature of these channels (Inoue & Isenberg, 1990b; Pacaud & Bolton, 1991). In guinea-pig gastric cells, when Ca2+ was present in the external solution and [Ca2+]i was not buffered, inhibition of the M3-subtype reduced the sensitivity of the M2-mediated response to the agonist, without affecting the maximal response (Rhee et al., 2000). Finally, and perhaps most strikingly, when [Ca2+]i was ‘clamped' at 100 nM in the absence of external Ca2+ (that is under conditions when any change in [Ca2+]i was prevented), the M3-subtype inhibition still strongly suppressed the maximal response without affecting significantly the sensitivity to the agonist (Bolton & Zholos, 1997; Zholos & Bolton, 1997) and, under the same conditions, significant correlation of muscarinic agonist potencies between the M2/mICAT and M3/PLC/InsP3 systems was found (Okamoto et al., 2002).
Thus, based on these observations, we have previously proposed that some ‘permissive' mechanism is associated with the M3-subtype stimulation, which operates independently of [Ca2+]i elevation. It is interesting that, resembling the nonsurmountable action of U-73122 (Figure 6a), we have earlier found that M3 receptor antagonists also acted to suppress the mICAT response noncompetitively. It should be noted that M3-selective antagonists even at high concentrations produced less than complete inhibition of mICAT, while PLC inhibition abolished the response. Also, we have recently found that Gαq/Gα11 antibodies, which are expected to inhibit the M3/PLC system (though complete inhibition using the antibodies is difficult to achieve), did not affect mICAT in ileal myocytes (Yan et al., 2003). These apparently contradictory results can be explained on the assumption that the G-proteins associated with the M2 subtype can also couple to PI-PLC, as shown in many tissues including longitudinal muscle from the small intestine of the guinea-pig, where Pertussis toxin treatment reduced inositol phospholipid hydrolysis by about 50% (Prestwich & Bolton, 1995). Thus, crosslinkage of the M2 and M3 receptor subtypes to the same G-proteins may explain the above contradictions; when PLC is inhibited by U—73122, nearly complete inhibition of mICAT results.
Our present results considerably extend the knowledge of the processes involved in this permissive action of M3 receptor activation. We tested several plausible mechanisms such as Ca2+ store depletion, as well as production of InsP3 and DAG as possible intermediates. However, none of these was found relevant, and thus the signal transduction is apparently limited to the upstream events such as PLC activation itself, or perhaps PIP2 hydrolysis on its own, irrespective of InsP3 or DAG accumulation. Interestingly, ATP inhibition of M current in sympathetic neurones is similarly PLC-dependent, but does not involve InsP3 or two important targets of diacylglycerol, PKC and Ras (Stemkowski et al., 2002). Notably, Ca2+ store depletion and OAG induced only tiny inward currents in ileal cells, which were about two orders of magnitude smaller than mICAT at maximally effective agonist concentration. In tracheal myocytes after Ca2+ store depletion, methacholine failed to activate the cationic current (Wang et al., 1997), while OAG did not induce any current and did not affect histamine- or methacholine-activated currents (Wang & Kotlikoff, 2000). In contrast, in portal vein myocytes, noradrenaline-evoked cationic current could be directly activated by OAG (Helliwell & Large, 1997; Albert & Large, 2001). The amplitude of this OAG-induced current in ileal and portal vein myocytes is similar, but since the maximal response to the agonist in ileal cells is much larger, this pathway cannot make any significant contribution to mICAT generation.
One of the most interesting and significant results of this study is the observation that PI-PLC inhibition can reverse the action of GTPγS in such a manner that the I–V relationships during mICAT development and during PLC inhibition were closely similar (Figure 5a). Formally, this could be interpreted as a voltage-dependent effect of the PLC blocker, since the inhibition at negative potentials began earlier and at lower U-73122 concentrations compared to the positive range (Figures 5a, c). However, in our previous studies, we established two principal mechanisms of mICAT activation: an increase in the maximal cationic conductance reflecting the increasing number of active channels and a negative shift of the activation curve, indicative of the changes in the gating properties of the activated channels (Zholos & Bolton, 1994). At that time, nothing was known about the convergence of several signalling pathways to open this channel. Thus, assuming that an activated G-protein directly gates this channel, such effects were explained by possible binding of several activated G-proteins to the same channel. Analysing the I–V relationships in a similar manner, we found that the process, which could appear as a voltage-dependent inhibition by U-73122, was in fact a positive shift of the cationic conductance activation curve (Figure 5b, d). These observations provide the first insight into the molecular mechanisms underlying the modulation of the voltage range of mICAT activation. Similar observations were made using an M3-selective antagonist p-F-HHSiD (Figure 9). It thus became evident that PLC activation not only determines the size of the response but also modulates the voltage sensitivity of cationic channels, and through this process it can strongly modulate the kinetics of mICAT (Figure 9).
The mechanisms of the interactions between PLC and cationic channel remain unclear. If indeed this cationic channel is a homo- or a heteromultimer of TRP proteins (Walker et al., 2001), then the problem of identifying the mechanism(s) of its opening has a much wider significance, as the mechanisms of activation and functions of TRP proteins remain generally unclear (Clapham et al., 2001). Interestingly, in the Drosophila phototransduction cascade, signalling depends on key elements which include TRP channel protein, PLCβ, PKC and calmodulin (Hardie & Raghu, 2001), and the same elements are important for mICAT generation is smooth muscles (Kim et al., 1995; 1998b; Ahn et al., 1997 and present study). It now appears that the signal transduction underlying mICAT is very complex, as signals from two different muscarinic receptor subtypes converge on the same channel protein, and each receptor subtype has a permissive role.
Acknowledgments
This work was supported by The Wellcome Trust grant number 062926 (TBB and AVZ) and equipment grant 042293 (to TBB). D.V. Gordienko is a Wellcome Trust Senior Lecturer (grant number 060659).
Abbreviations
- ATP
adenosine 5′-triphosphate magnesium salt
- BAPTA
1,2-bis(2-aminophenoxy)-ethane-N,N,N′,N′-tetraacetic acid
- [Ca2+]i
ionized intracellular Ca2+ concentration
- CPA
cyclopiazonic acid
- D-609
O-tricyclo[5.2.1.02,6]dec-9-yl dithiocarbonate potassium salt
- DAG
1,2-diacyl-sn-glycerol
- DMSO
dimethyl sulphoxide
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- GTP
guanosine 5′-triphosphate lithium salt
- GTPγS
guanosine 5′-O-(3-thiotriphosphate)
- HEPES
N-2-hydroxyethylpiperazine-N′-2-ethanesulphonic acid
- IICR
InsP3-induced Ca2+ release
- InsP3
D-myo-inositol 1,4,5,-trisphosphate
- I–V relationship
current–voltage relationship
- mAChR
muscarinic acetylcholine receptor
- mICAT
muscarinic receptor cationic current
- NMDG
N-methyl-D-glucamine
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- p-F-HHSiD
parafluoro-hexahydro-sila-difenidol hydrochloride
- PIP2
phosphatidylinositol 4,5-bisphosphate
- PLA2
phospholipase A2
- PLC
phospholipase C
- U-73122
1-[6-([(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino)hexyl]-1H-pyrrole-2,5-dione
- U-73343
1-[6-([(17β)-3-methoxyestra-1,3,5(10)-trien-17-yl]amino)hexyl]-2,5-pyrrolidinedione
References
- AHN S.C., KIM S.J., SO I., KIM K.W. Inhibitory effect of phorbol 12,13 dibutyrate on carbachol-activated nonselective cationic current in guinea-pig gastric myocytes. Pflugers Arch. 1997;434:505–507. doi: 10.1007/s004240050429. [DOI] [PubMed] [Google Scholar]
- ALBERT A.P., LARGE W.A. Comparison of spontaneous and noradrenaline-evoked non-selective cation channels in rabbit portal vein myocytes. J. Physiol. 2001;530:457–468. doi: 10.1111/j.1469-7793.2001.0457k.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLEASDALE J.E., THAKUR N.R., GREMBAN R.S., BUNDY G.L., FITZPATRICK F.A., SMITH R.J., BUNTING S. Selective inhibition of receptor-coupled phospholipase C-dependent processes in human platelets and polymorphonuclear neutrophils. J. Pharmacol. Exp. Ther. 1990;255:756–768. [PubMed] [Google Scholar]
- BOLTON T.B., PRESTWICH S.A., ZHOLOS A.V., GORDIENKO D.V. Excitation–contraction coupling in gastrointestinal and other smooth muscles. Annu. Rev. Physiol. 1999;61:85–115. doi: 10.1146/annurev.physiol.61.1.85. [DOI] [PubMed] [Google Scholar]
- BOLTON T.B., ZHOLOS A.V. Activation of M2 muscarinic receptors in guinea-pig ileum opens cationic channels modulated by M3 muscarinic receptors. Life Sci. 1997;60:1121–1128. doi: 10.1016/s0024-3205(97)00056-8. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P. Muscarinic receptors – characterization, coupling and function. Pharmacol. Ther. 1993;58:319–379. doi: 10.1016/0163-7258(93)90027-b. [DOI] [PubMed] [Google Scholar]
- CAULFIELD M.P., BIRDSALL N.J.M. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 1998;50:279–290. [PubMed] [Google Scholar]
- CHEN S., INOUE R., ITO Y. Pharmacological characterization of muscarinic receptor-activated cation channels in guinea-pig ileum. Br. J. Pharmacol. 1993;109:793–801. doi: 10.1111/j.1476-5381.1993.tb13644.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHO H., YOUM J.B., RYU S.Y., EARM Y.E., HO W.K. Inhibition of acetylcholine-activated K+ currents by U73122 is mediated by the inhibition of PIP2-channel interaction. Br. J. Pharmacol. 2001;134:1066–1072. doi: 10.1038/sj.bjp.0704347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CLAPHAM D.E., RUNNELS L.W. The TRP ion channel family. Nat. Rev. Neurosci. 2001;2:387–396. doi: 10.1038/35077544. [DOI] [PubMed] [Google Scholar]
- EGLEN R.M., HEGDE S.S., WATSON N. Muscarinic receptor subtypes and smooth muscle function. Pharmacol. Rev. 1996;48:531–565. [PubMed] [Google Scholar]
- GORDIENKO D.V., ZHOLOS A.V., BOLTON T.B. Membrane ion channels as physiological targets for local Ca2+ signalling. J. Microsc. 1999;196:305–316. doi: 10.1046/j.1365-2818.1999.00599.x. [DOI] [PubMed] [Google Scholar]
- HARDIE R.C., RAGHU P. Visual transduction in Drosophila. Nature. 2001;413:186–193. doi: 10.1038/35093002. [DOI] [PubMed] [Google Scholar]
- HELLIWELL R.M., LARGE W.A. α1-adrenoceptor activation of a non-selective cation current in rabbit portal vein by 1,2-diacyl-sn-glycerol. J. Physiol. 1997;499:417–428. doi: 10.1113/jphysiol.1997.sp021938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INOUE R., ISENBERG G. Acetylcholine activates nonselective cation channels in guinea pig ileum through a G protein. Am. J. Physiol. 1990a;258:C1173–C1178. doi: 10.1152/ajpcell.1990.258.6.C1173. [DOI] [PubMed] [Google Scholar]
- INOUE R., ISENBERG G. Intracellular calcium ions modulate acetylcholine-induced inward current in guinea-pig ileum. J. Physiol. 1990b;424:73–92. doi: 10.1113/jphysiol.1990.sp018056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- INOUE R., OKADA T., ONOUE H., HARA Y., SHIMIZU S., NAITOH S., ITO Y., MORI Y. The transient receptor potential protein homologue TRP6 is the essential component of vascular α1-adrenoceptor-activated Ca2+-permeable cation channel. Circ. Res. 2001;88:325–332. doi: 10.1161/01.res.88.3.325. [DOI] [PubMed] [Google Scholar]
- INOUE R., WANIISHI Y., YAMADA K., ITO Y. A possible role of tyrosine kinases in the regulation of muscarinic receptor-activated cation channels in guinea pig ileum. Biochem. Biophys. Res. Commun. 1994;203:1392–1397. doi: 10.1006/bbrc.1994.2339. [DOI] [PubMed] [Google Scholar]
- JUNG S., STROTMANN R., SCHULTZ G., PLANT T.D. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am. J. Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- KANG T.M., KIM S.J., RHEE P.L., RHEE J.C., KIM K.W. Carbachol activates a nonselective cation current through M2 muscarinic receptor subtype in guinea pig gastric smooth muscle. J. Auton. Nerv. Syst. 1997;65:146. [Google Scholar]
- KIM S.J., AHN S.C., SO I., KIM K.W. Role of calmodulin in the activation of carbachol-activated cationic current in guinea-pig gastric antral myocytes. Pflugers Arch. 1995;430:757–762. doi: 10.1007/BF00386173. [DOI] [PubMed] [Google Scholar]
- KIM Y.C., KIM S.J., KANG T.M., SUH S.H., SO I., KIM K.W. Effects of myosin light chain kinase inhibitors on carbachol-activated nonselective cationic current in guinea-pig gastric myocytes. Pflugers Arch. 1997;434:346–353. doi: 10.1007/s004240050407. [DOI] [PubMed] [Google Scholar]
- KIM Y.C., KIM S.J., SIM J.H., CHO C.H., JUHNN Y.S., SUH S.H., SO I., KIM K.W. Suppression of the carbachol-activated nonselective cationic current by antibody against α subunit of Go protein in guinea-pig gastric myocytes. Pflugers Arch. 1998a;436:494–496. doi: 10.1007/s004240050663. [DOI] [PubMed] [Google Scholar]
- KIM Y.C., KIM S.J., SIM J.H., JUN J.Y., KANG T.M., SUH S.H., SO I., KIM K.W. Protein kinase C mediates the desensitization of CCh-activated nonselective cationic current in guinea-pig gastric myocytes. Pflugers Arch. 1998b;436:1–8. doi: 10.1007/s004240050597. [DOI] [PubMed] [Google Scholar]
- KOHDA M., KOMORI S., UNNO T., OHASHI H. Carbachol-induced [Ca2+]i oscillations in single smooth muscle cells of guinea-pig ileum. J. Physiol. 1996;492:315–328. doi: 10.1113/jphysiol.1996.sp021311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMORI S., KAWAI M., PACAUD P., OHASHI H., BOLTON T.B. Oscillations of receptor-operated cationic current and internal calcium in single guinea-pig ileal smooth muscle cells. Pflugers Arch. 1993;424:431–438. doi: 10.1007/BF00374905. [DOI] [PubMed] [Google Scholar]
- KOMORI S., KAWAI M., TAKEWAKI T., OHASHI H. GTP-binding protein involvement in membrane currents evoked by carbachol and histamine in guinea-pig ileal muscle. J. Physiol. 1992;450:105–126. doi: 10.1113/jphysiol.1992.sp019118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOMORI S., UNNO T., NAKAYAMA T., OHASHI H. M2 and M3 muscarinic receptors couple, respectively, with activation of nonselective cationic channels and potassium channels in intestinal smooth muscle cells. Jpn. J. Pharmacol. 1998;76:213–218. doi: 10.1254/jjp.76.213. [DOI] [PubMed] [Google Scholar]
- LEE Y.M., KIM B.J., KIM H.J., YANG D.K., ZHU M.H., LEE K.P., SO I., KIM K.W. TRPC5 as a candidate for the nonselective cation channel activated by muscarinic stimulation in murine stomach. Am. J. Physiol. 2003;284:G604–G616. doi: 10.1152/ajpgi.00069.2002. [DOI] [PubMed] [Google Scholar]
- OKAMOTO H., PRESTWICH S.A., ASAI S., UNNO T., BOLTON T.B., KOMORI S. Muscarinic agonist potencies at three different effector systems linked to the M2 or M3 receptor in longitudinal smooth muscle of guinea-pig small intestine. Br. J. Pharmacol. 2002;135:1765–1775. doi: 10.1038/sj.bjp.0704642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OSTROM R.S., EHLERT F.J. M2 muscarinic receptors inhibit forskolin- but not isoproterenol-mediated relaxation in bovine tracheal smooth muscle. J. Pharmacol. Exp. Ther. 1998;286:234–242. [PubMed] [Google Scholar]
- PACAUD P., BOLTON T.B. Relation between muscarinic receptor cationic current and internal calcium in guinea-pig jejunal smooth muscle cells. J. Physiol. 1991;441:477–499. doi: 10.1113/jphysiol.1991.sp018763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRESTWICH S.A., BOLTON T.B. G-protein involvement in muscarinic receptor-stimulation of inositol phosphates in longitudinal smooth muscle from the small intestine of the guinea-pig. Br. J. Pharmacol. 1995;114:119–126. doi: 10.1111/j.1476-5381.1995.tb14915.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUCOVSKY V., ZHOLOS A.V., BOLTON T.B. Muscarinic cation current and suppression of Ca2+ current in guinea pig ileal smooth muscle cells. Eur. J. Pharmacol. 1998;346:323–330. doi: 10.1016/s0014-2999(98)00059-4. [DOI] [PubMed] [Google Scholar]
- RHEE J.C., RHEE P.L., PARK M.K., SO I., UHM D.Y., KIM K.W., KANG T.M. Muscarinic receptors controlling the carbachol-activated nonselective cationic current in guinea pig gastric smooth muscle cells. Jpn. J. Pharmacol. 2000;82:331–337. doi: 10.1254/jjp.82.331. [DOI] [PubMed] [Google Scholar]
- SMALLRIDGE R.C., KIANG J.G., GIST I.D., FEIN H.G., GALLOWAY R.J. U-73122, an aminosteroid phospholipase C antagonist, noncompetitively inhibits thyrotropin-releasing hormone effects in GH3 rat pituitary cells. Endocrinology. 1992;131:1883–1888. doi: 10.1210/endo.131.4.1396332. [DOI] [PubMed] [Google Scholar]
- SMITH R.J., SAM L.M., JUSTEN J.M., BUNDY G.L., BALA G.A., BLEASDALE J.E. Receptor-coupled signal transduction in human polymorphonuclear neutrophils: effects of a novel inhibitor of phospholipase C-dependent processes on cell responsiveness. J. Pharmacol. Exp. Ther. 1990;253:688–697. [PubMed] [Google Scholar]
- STEMKOWSKI P.L., TSE F.W., PEUCKMANN V., FORD C.P., COLMERS W.F., SMITH P.A. ATP-inhibition of M current in frog sympathetic neurons involves phospholipase C but not Ins P3, Ca2+, PKC, or Ras. J. Neurophysiol. 2002;88:277–288. doi: 10.1152/jn.2002.88.1.277. [DOI] [PubMed] [Google Scholar]
- THOMPSON A.K., MOSTAFAPOUR S.P., DENLINGER L.C., BLEASDALE J.E., FISHER S.K. The aminosteroid U-73122 inhibits muscarinic receptor sequestration and phosphoinositide hydrolysis in SK-N-SH neuroblastoma cells. A role for Gp in receptor compartmentation. J. Biol. Chem. 1991;266:23856–23862. [PubMed] [Google Scholar]
- TSYTSYURA Y.D., ZHOLOS A.V., SHUBA M.F., BOLTON T.B. Effects of intracellular Ca2+ on muscarinic cationic current in guinea pig ileal smooth muscle cells. Neurophysiology. 2000;32:236–237. [Google Scholar]
- WALKER R.L., HUME J.R., HOROWITZ B. Differential expression and alternative splicing of TRP channel genes in smooth muscles. Am. J. Physiol. 2001;280:C1184–C1192. doi: 10.1152/ajpcell.2001.280.5.C1184. [DOI] [PubMed] [Google Scholar]
- WANG Y.X., FLEISCHMANN B.K., KOTLIKOFF M.I. M2 receptor activation of nonselective cation channels in smooth muscle cells: calcium and Gi/G0 requirements. Am. J. Physiol. 1997;273:C500–C508. doi: 10.1152/ajpcell.1997.273.2.C500. [DOI] [PubMed] [Google Scholar]
- WANG Y.X., KOTLIKOFF M.I. Signalling pathway for histamine activation of non-selective cation channels in equine tracheal myocytes. J. Physiol. 2000;523:131–138. doi: 10.1111/j.1469-7793.2000.t01-3-00131.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAN H.-D., OKAMOTO H., UNNO T., TSYTSYURA YA.D., PRESTWICH S.A., KOMORI S., ZHOLOS A.V., BOLTON T.B. Effects of G protein-specific antibodies and Gβγ subunits on the muscarinic receptor-operated cation current in guinea-pig ileal smooth muscle cells. Br. J. Pharmacol. 2003;139:605–615. doi: 10.1038/sj.bjp.0705289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YULE D.I., WILLIAMS J.A. U73122 inhibits Ca2+ oscillations in response to cholecystokinin and carbachol but not to JMV-180 in rat pancreatic acinar cells. J. Biol. Chem. 1992;267:13830–13835. [PubMed] [Google Scholar]
- ZHOLOS A.V., BOLTON T.B. G-protein control of voltage dependence as well as gating of muscarinic metabotropic channels in guinea-pig ileum. J. Physiol. 1994;478:195–202. doi: 10.1113/jphysiol.1994.sp020242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOLOS A.V., BOLTON T.B. A novel GTP-dependent mechanism of ileal muscarinic metabotropic channel desensitization. Br. J. Pharmacol. 1996;119:997–1012. doi: 10.1111/j.1476-5381.1996.tb15770.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOLOS A.V., BOLTON T.B. Muscarinic receptor subtypes controlling the cationic current in guinea-pig ileal smooth muscle. Br. J. Pharmacol. 1997;122:885–893. doi: 10.1038/sj.bjp.0701438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOLOS A.V., KOMORI S., OHASHI H., BOLTON T.B. Ca2+ inhibition of inositol trisphosphate-induced Ca2+ release in single smooth muscle cells of guinea-pig small intestine. J. Physiol. 1994;481:97–109. doi: 10.1113/jphysiol.1994.sp020421. [DOI] [PMC free article] [PubMed] [Google Scholar]