

Abstract
The contribution of nitric oxide (NO) and peroxynitrite (PN) to inflammation in a zymosan-induced (1 mg, intra-articular, i.art.) rat model of arthritis was assessed by histopathology and by measuring the glycosaminoglycan (GAG) content of the articular cartilage.
Progression of the chronic synovitis in zymosan-induced arthritis (ZYA) was associated with increased nitrite and nitrotyrosine (3-NT) levels in the joint exudates that paralleled a progressive loss of the GAG content. An increase in 3-NT was also observed after i.art. PN.
The nonselective nitric oxide synthase (NOS) inhibitor L-NG-nitroarginine methyl ester (25–75 mg kg−1day−1) or the selective inducible NOS inhibitor aminoguanidine (50–100 mg kg−1day−1) given 1 h before (prophylactic) or 3 days after (therapeutic) injection of the zymosan ameliorated the synovitis, but worsened the GAG loss, as measured at the end of the experiment (day 7).
The PN scavenger uric acid (100–250 mg kg−1 i.p. four times daily) given prophylactically until the end of the experiment (day 14), in a dose compatible with its PN scavenging activity, significantly decreased both the synovitis and the GAG loss.
In conclusion, PN formation is associated with cartilage damage in addition to proinflammatory activity in ZYA. NOS inhibitors and a PN scavenger were able to reduce the cellular infiltration, while displaying opposite effects on cartilage homeostasis either by enhancing or ameliorating the damage, respectively.
Keywords: Inflammation, rheumatoid arthritis, nitric oxide, cartilage, peroxynitrite, arthritis, zymosan, uric acid, antioxidants
Introduction
NO is synthesized by NOS from L-arginine, and three isoforms of NOS have been described. Endothelial (NOS1) and neuronal (NOS3) isoforms are constitutive (cNOS), whereas NOS2 is the inducible isoform (iNOS) (Moncada et al., 1991). The increased production of NO has been linked to both protective and proinflammatory mechanisms associated with tissue damage in inflammatory disease (Clancy et al., 1988). The ability of NO to promote tissue damage in arthritis was demonstrated by studies in the early 1990s, where the inflammatory changes observed in arthritis models were attenuated after administration of NOS inhibitors (Ialenti et al., 1993; Stefanovic-Racic et al., 1994). Furthermore, an increased level of NO in synovial tissue was associated with the progression of streptococcal cell wall (SCW)-induced arthritis, and this was attenuated by the administration of a nonselective NOS inhibitor (McCartney-Francis et al., 1993). However, these protective effects of NOS inhibitors have not been universally observed in arthritis models. The administration of either the nonselective NOS inhibitor NG-methyl-L-arginine or the selective iNOS inhibitor N-iminoethyl-L-lysine (L-NIL) during ongoing antigen-induced arthritis (AIA) was not associated with the anti-inflammatory properties of these drugs that have been previously observed when they have been given as a prophylactic treatment (Stefanovic-Racic et al., 1995; Fletcher et al., 1998). Furthermore, a second iNOS inhibitor, aminoguanidine (AG), had neither a prophylactic nor a therapeutic effect (Stefanovic-Racic et al., 1995). These results have led to the suggestion that NOS inhibitors may not be effective in established arthritis. More recent findings have revealed that the administration of L-NIL exacerbated SCW-induced arthritis (McCartney-Francis et al., 2001) and that NO plays a protective role in the acute phases of AIA in the iNOS knockout mouse (Veihelmann et al., 2001).
The deleterious effects of NO might be linked, at least in part, to the reactive nitrogen species generated following NO release (Pryor & Squadrito, 1995). During inflammatory conditions, large amounts of NO and the superoxide (O2−) anion are produced, leading to the formation of a strong oxidant, the peroxynitrite anion (PN). The proposed cytotoxic properties of PN include protein nitration, lipid peroxidation, inhibition of cellular metabolic pathways and signal transduction mechanisms, and DNA strand breakages (Beckman, 1996). The formation of 3-nitrotyrosine from PN activity in proteins has routinely been used as a biological marker for PN formation in vivo (van der Vliet et al., 1996), although it is acknowledged that 3-nitrotyrosine can also be produced via non-PN-mediated pathways. Detection of 3-nitrotyrosine in atherosclerotic plaques (Beckman et al., 1994), in the serum and synovial fluid of rheumatoid arthritis patients (Kaur & Halliwell, 1994), and in rejected human kidney allografts (Macmillan-Crow et al., 1996) are some of the examples given as evidence of the association of PN formation with human disease states. In keeping with this finding, recent in vitro data have shown that NO is not damaging to either osteoblasts or chondrocytes in vitro when the generation of PN is inhibited (Da Rocha & De Brum-Fernandes, 2002; Del Carlo & Loeser, 2002).
Zymosan, a polysaccharide from yeast cell walls, produces a severe and erosive synovitis (Keystone et al., 1989; Gegout et al., 1994) that is also associated with hyperalgesia (Tonussi & Ferreira, 1992; Rocha et al., 1999). We have recently shown that the prophylactic, but not therapeutic, administration of NOS inhibitors is antinociceptive in this model. In contrast, NO donors provided analgesia when given therapeutically (Rocha et al., 2002).
Thus, the results of recent studies suggest that the therapeutic administration of NOS inhibitors is not universally associated with an anti-inflammatory effect in animal models of arthritis. Most techniques involving experimental arthritis utilize either clinical articular indices or histopathological scores to evaluate the severity of the arthritis. These assays, in addition to interobserver bias, act as a global measure of whole joint edema or cell influx into the synovial membrane, rather than of specific events in the joint cavity. Using the zymosan-induced arthritis (ZYA) model, we are able to measure cell influx and release of inflammatory mediators into the synovial fluid (Rocha et al., 1999,2002) and the glycosaminoglycan (GAG) content of the articular cartilage. GAGs are the building blocks of the proteoglycans (Sledge, 1993). Quantification of GAG in the joint exudate has been previously used as an indicator of joint damage in arthritis (Bensouyad et al., 1990; Sharif et al., 1996; Ishimaru et al., 2001). In the present study, we determined the GAG content of the articular cartilage of the femoral condyles as an index of joint lesion. This has allowed us to discriminate between the inflammatory events and the structural joint damage. Our present data show that inhibition of NOS generation is able to decrease the synovitis, without protecting the articular cartilage loss associated with ZYA. By comparison, the administration of uric acid (UA), a PN scavenger, in addition to reducing the inflammatory parameters, also prevents the loss of articular cartilage. The results provide important in vivo evidence of the multiple roles that NO and its reactive oxygen species play in the arthritic joint.
Methods
Chemicals
Most agents were purchased from either Sigma Chemical Company, St Louis, U.S.A. or Amersham Pharmacia Biotech, Bucks, U.K. Other agents were acquired as follows: avidin-biotinylated HRP complex, diaminobenzidine, donkey anti-rabbit immunoglobulin biotinylated antibody from DAKO, Cambridgeshire, U.K.; rabbit polyclonal anti-3-nitrotyrosine antibody from Upstate Biotechnology Incorporated, Lake Placid, NY, U.S.A. Chondroitin 4-sulfate (C4), chondroitin 6-sulfate (C6), heparan sulfate, and chondroitinase ABC from Seikagaku Kogyo Co., Tokyo, Japan.
Induction of the ZYA and drug treatments
Male Wistar rats (180–220 g) bred in house with free access to food and water were used for all experiments. All experiments were designed to minimize animal suffering and to use the minimum number associated with valid statistical evaluation. Animals received an intra-articular (i.art.) injection of 1 mg zymosan (50 μl total volume), dissolved in sterile saline, into both knee joints. Control (C) animals received saline i.art. Animals were injected at the time point required for the experimental protocol. For example, in Figure 1a, rats were treated at 21, 14, 7, 4 days, 6, 3, or 1 h before they were killed, in order to analyze the response at the required time points. The NOS inhibitors were either injected 30 min prior to the induction of the arthritis (prophylactic intervention) or 3 days after arthritis induction (therapeutic intervention), and then daily. Groups of rats received either L-NG-nitroarginine methyl ester (L-NAME 25–75 mg kg−1 i.p.) or AG (50–100 mg kg−1 i.p.) until the rats were killed under terminal anesthesia with chloral hydrate (400 mg kg−1), and exsanguinated at 7 days after arthritis induction. The PN scavenger UA (100 mg kg−1 or 250 mg kg−1 i.p.) was administered four times daily, starting 30 min prior to the induction of the arthritis (prophylactic intervention), in order to maintain significantly raised serum UA levels, as compared to vehicle-injected animals, and animals were killed after 14 days. These UA doses were based on a previous study reporting significant increase in UA levels using the same doses, leading to amelioration of an experimental allergic encephalomyelitis (EAE) model. This effect was attributed to a PN scavenging activity of UA (Hooper et al., 1998). The side effects of drug treatment were assessed by visual appearance of rodent behavior and fluctuation of body weight. PN was injected i.art. in one series of experiments, in order to determine whether the PN presence in the joint cavity is associated with 3-nitrotyrosine formation. Animals received an injection of 200 nmol of PN, decomposed PN or relevant solvent controls into their right hind knee joints −96, −24, −6, −3, or −1 h before they were killed.
Figure 1.

Detection of nitrite and nitrate in synovial fluid exudates and measurement of articular cartilage damage over 1 h–21 days after induction of zymosan arthritis. Zymosan was injected i.art. into both knee joints and samples collected after termination of the experiment at the times indicated. Control (C) animals received only saline i.art. and the sample was taken after 7 days; (a) Shows the release of NO, as determined by measurement of NO2− and NO3− released into the joint exudates of rats. (b) Shows joint damage, as determined by measurement of the GAG chondroitin sulfate content, in articular cartilage samples of rats. Results are expressed as the mean±s.e.m. of values for each group of six animals. *P<0.05 compared to C.
Pharmacokinetic analysis of UA serum levels
Groups of animals received i.p. injections of UA at either 100 or 250 mg kg−1 doses, and the serum UA levels were assessed in the peripheral blood 1, 3, 6, 12, and 24 h later, using a quantitative enzymatic assay (Roche Diagnostics™, São Paulo, Brasil).
Collection of the synovial exudates and of the synovial membranes
The joints were washed twice with 0.2 ml saline containing 10 mM EDTA, and the synovial exudates were collected for determination of total and differential cell counts using a Neubauer chamber and stained smears, respectively. After centrifugation (500 g 10 min−1), the supernatant was stored at −70°C until further use. The synovial membranes were surgically excised, paraffin-embedded and routinely processed for staining with hematoxylin & eosin (H&E).
Determination of NO production
The Griess reaction was used as an indirect assay of NO production to determine the total nitrite and nitrate as a measure of the degree of NO production. Total NO2− and NO3− levels were determined after conversion of NO3− in the synovial exudates supernatants (0.08 ml) to NO2− by incubation with 0.01 ml nitrate reductase from Aspergillus species (1 U ml−1) and 0.01 ml NADPH (1 mM) for 30 min at 37°C. The NO2− levels were determined spectrophotometrically at 540 nm by comparing the absorbance of 0.1 ml sample after adding 0.1 ml Griess reagent (sulfanilic acid (1% w v−1) and N-(1-naphythyl)ethylenediamine (0.1 w v−1) in 5% phosphoric acid) to that of a NaNO2 (1–100 μM) standard.
Determination of the GAG content of the articular cartilage
Proteolytic digestion and isolation of the GAG: The distal femoral extremities were removed and the articular cartilage was excised with a surgical blade. The cartilage was dried overnight (80°C) and the dry weight was recorded. The dry cartilage was kept in 10 volumes of acetone until further analysis. The dried samples were later subjected to total protease digestion followed by isolation of GAG, as described previously (Dietrich & Dietrich, 1972).
Identification and characterization of GAG: Samples (5 μg) were run on a 0.6% agarose gel in a 0.05 M 1,3-diaminopropane buffer, pH 9.0 for 90 min at 100 V, as described previously (Jacques et al., 1968; Dietrich & Dietrich, 1972).
Characterization of the GAG: To further confirm the identification of the GAG, the purified samples were subjected to enzymatic degradation using chondroitinase ABC isolated from P. vulgaris and heparitinases from F. heparinum, as described previously (Jeronimo et al., 1994).
Quantification of the GAG: After the enzymatic digestion, the samples were applied to a Whatman paper (isobutyric: 1.25 M ammonium hydroxide (5 : 3 v v−1) solvent) for 8 h. After drying, the di, tetra or hexasaccharides were identified and quantitated by densitometry (525 nm). Samples were compared to C4 and C6 standards subjected to the same protocol. Data were expressed as μg GAG mg−1 of dried cartilage.
Preparation of authentic PN
The original light yellow solution of PN was stored for no more than 1 week upon arrival, at −80°C in NaOH 0.3 M, as supplied by the manufacturer (Cayman Chem. Co., U.S.A.). Immediately before use, the PN concentration was checked by measuring absorbance at 302 nm (extinction coefficient; ɛ=1670 M−1 cm−1) using 0.3 M NaOH as blank. Working solutions of PN were made up in 50 μl aliquots of 0.3 M NaOH. Control experiments included injection of 0.3 M NaOH, as well as inactive PN.
Detection of protein nitration
The competitive ELISA used was modified from that of Khan et al. (1998). BSA nitrated with tetranitromethane and containing 4–5 nitrotyrosine residues per protein was made up in 50 mM carbonate buffer (pH 9.0) and coated onto 96-well plates (7.6 pmol 0.1 ml well−1). After incubation for 2 h at 37°C and washing with 10 mM PBS, 0.5% ovalbumin in PBS was added for 2 h at 37°C. After three washes using PBS with Tween 20 (0.05%, pH 7.4), the plates were aspirated, covered, and stored at 4°C until required. Samples (0.02–0.5 mg protein 0.1 ml−1) or standard nitrated BSA (0.01–150 pmol 0.1 ml−1) were added to duplicate wells, and incubated for 2 h at 37°C with 0.1 ml primary rabbit polyclonal antinitrotyrosine antibody (1 : 30,000). After three washes using 10 mM PBS with 0.05% Tween 20 (pH 7.4) to remove any unfixed antibody, sequential 1 h incubations (and washes) were then performed with 0.1 ml biotinylated donkey anti-rabbit IgG (1 : 5000) and an avidin-biotinylated horseradish peroxidase complex. After further washing, color development was initiated by addition of 0.1 ml substrate (2.2 mM o-phenylene-diamine in 0.03% sodium perborate) and was allowed to develop for up to 10 min at room temperature and terminated by the addition of 0.05 ml sulfuric acid (2.5 M). Antibody binding was determined from the absorbance at 492 nm. The detection limit was 0.2 pmol 0.1 ml−1.
Histopathological analysis
The H&E-stained synovial membranes were semiquantitatively evaluated for cell influx, fibrosis, angiogenesis, edema, necrosis, and hemorrhage, giving a (0–3) score grade for each of these parameters. Absence of any of these alterations was given a zero (0) score (naïve animals), whereas pronounced cell influx and edema, marked angiogenesis (obtained by counting the number of microvessels per high power field), extensive fibrosis with prominent necrotic areas, and hemorrhage would receive a maximum score (18). These observations were made by an experienced pathologist who was blinded to the treatment protocol (Melo, DN).
Statistics
Unless otherwise stated, the effects of the different treatments were compared to nontreated (NT) ZYA that consisted of animals that received just the zymosan i.art. and the drug vehicle given systemically. Results are expressed as mean±s.e.m. Histological data are expressed as medians. To compare the differences between means, we used one-way ANOVA followed by Tukey's test. To compare medians, we used the Kruskal–Wallis test. P<0.05 was considered significant.
Results
Effect of zymosan on nitrite levels in synovial exudates and articular GAG content
The amount of nitrite detected in the joint exudates, measured at various time points up to 21 days after zymosan administration, is shown in Figure 1a. The maximum levels of nitrite measured are observed soon (3–6 h) after i.art. injection of zymosan, as previously observed (Rocha et al., 2002). These results are now extended to show that they remain significantly raised, although to a lesser extent, for the whole 21 days, when compared to saline-injected joints. The articular cartilage damage is shown in Figure 1b. Zymosan induced a significant reduction in the GAG content after 7 days, and this remained low up to 21 days after injection of the zymosan.
Effect of L-NAME and AG on zymosan-induced nitrite levels and articular GAG content
The NOS inhibitors caused a lack of ‘well being' in the arthritic rats, that was associated with loss of body weight. Thus, the experiment was terminated after 7 days. The administration of either L-NAME (25–75 mg kg−1 i.p.) or AG (50–100 mg kg−1 i.p.) significantly inhibited the nitrite release into the joint exudates, as compared to NT rats (Figure 2a). The inhibition occurred regardless of whether the compounds were administered prophylactically or therapeutically. By comparison, L-NAME and AG did not reverse the loss of GAG content, whether injected either prophylactically or therapeutically (Figure 2b). In fact, a significant (P<0.05) increase in the loss of GAG was observed, with the exception of the animals that received the higher dose of L-NAME (75 mg kg−1), as compared to NT animals.
Figure 2.

Effect of systemic prophylactic or therapeutic administration of NOS inhibitors on NO release and articular joint damage 7 days after induction of zymosan arthritis. (a) Shows the release of NO, as determined by measurement of NO2− and NO3− released into the joint exudates of rats. (b) Shows joint damage as determined by measurement of the GAG chondroitin sulfate content, in articular cartilage samples of rats. Either L-NAME (25 or 75 mg kg−1 i.p.), or AG (50 or 100 mg kg−1 i.p.) were injected 30 min before (prophylactic administration, dotted bars), or 3 days after and then daily (therapeutic administration, hatched bars) after induction of arthritis with zymosan. Non-treated (NT) ZYA rats were given saline i.p. 30 min prior to zymosan, in order to act as vehicle control. Control (C) animals received only saline i.art. and no systemic treatment. Results are expressed as the mean±s.e.m. of values for each group of six animals. #P<0.05 compared to C rats; *P<0.05 compared to NT rats.
Effect of the PN scavenger UA on zymosan-induced nitrite levels and articular GAG content
UA has been used as a PN scavenger previously (Hooper et al., 1998,2000), and was used in this study to investigate its effect on articular inflammation and damage. In initial experiments to determine pharmacokinetics, we found that, 30 min after a single i.p. injection of UA, serum levels were significantly raised, returning to baseline levels by 6 h (Figure 3). Thus, UA was given as a four-times daily dose regimen (every 6 h) of either 100 or 250 mg kg−1, in order to enable it to act as an in vivo scavenger of PN. The effect of UA on nitrite levels released into the joint exudates or assessed in the peripheral blood in ZYA after 14 days is shown in Figures 4a and b, respectively. It was possible to extend the experiment as compared to the groups treated with the NOS inhibitors, as the rats that received UA appeared clinically improved when compared with the NT rats. An inhibition of nitrite levels in the joint exudates, but not in the peripheral blood, was seen with both doses of UA. Moreover, systemic administration of UA significantly reversed the GAG loss from the articular cartilage, measured at day 14, as compared to NT rats (Figure 4c). A representative gel electrophoresis of the treatments is shown in Figure 5.
Figure 3.
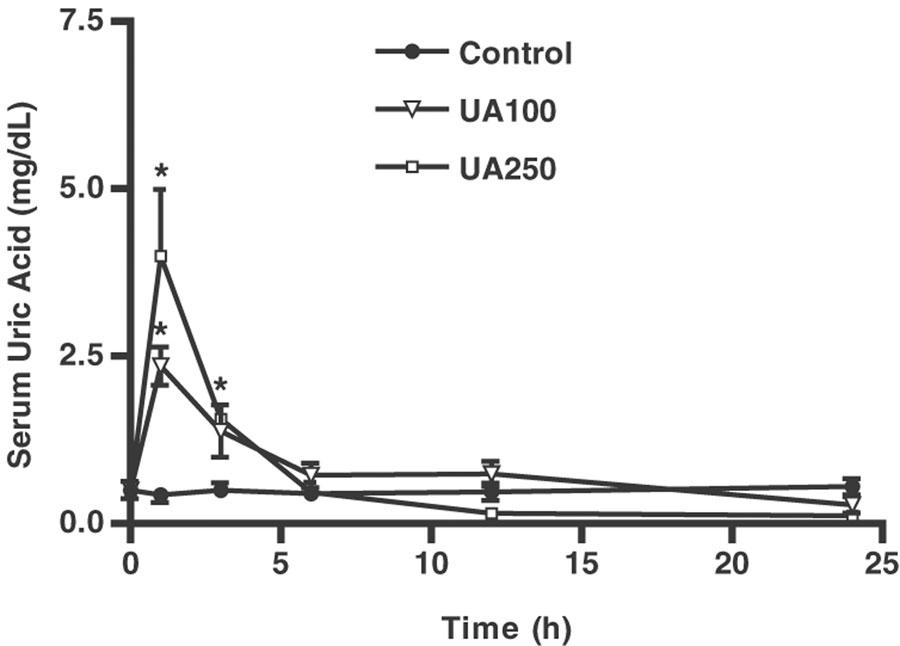
Serum UA levels after systemic administration of UA. Groups of rats received i.p. injection of either 100 or 250 mg kg−1 of UA and the serum UA levels were assessed in the peripheral blood 1, 3, 6, 12, and 24 h later. Control (C) rats received only saline (i.p.). Results are expressed as the mean±s.e.m. of values for each group of six animals. *P<0.05 compared to C rats at the same time point.
Figure 4.

Effect of UA administration on articular and systemic NO production and articular joint damage 14 days after induction of zymosan arthritis. The production of NO, as determined by measurement of NO2− and NO3−, is shown in (a) joint exudates and (b) peripheral blood. The joint damage was determined by measurement of the GAG content, in articular cartilage samples of rats, as shown in (c). Zymosan was injected i.art. into both knee joints and the NO release in exudates (as NO2− and NO3−) or peripheral blood was measured at 14 days after zymosan. UA (100 or 250 mg kg−1 i.p.) was injected 30 min before zymosan and four times daily, until sacrifice, at 14 days. NT rats received zymosan i.art., but were given (i.p.) saline in order to act as vehicle control. Control (C) animals received only saline i.art. and no systemic treatment. Results are expressed as the mean±s.e.m. of values for each group of six animals. #P<0.05 compared to C rats; *P<0.05 compared to NT rats.
Figure 5.

Representative gel electrophoresis of the GAGs extracted from the articular cartilage after NOS inhibitors or UA administration in zymosan arthritis. Zymosan was injected i.art. into both knee joints and the articular cartilage damage was evaluated by measuring its GAG content at 7 or 14 days after zymosan. L-NAME (25 mg kg−1) or AG (50 mg kg−1) was injected 30 min before zymosan and daily, until sacrifice, at 7 days. UA (100 mg kg−1 i.p.) was injected 30 min before zymosan and four times daily, until sacrifice, at 14 days. GAG content in extracts was determined by densitometry (525 nm), using the standards for comparison. M – standard GAG mixture with 5 μg containing equal amounts of C4, C6, and HS; C – animals that received only saline i.art.; ABC – sample from C animals that was subjected to enzymatic digestion using chondroitinase ABC; NT – animals that received saline (i.p.) after zymosan injection killed 7 days later; L-NAME 25 – animals treated with L-NAME (25 mg kg−1 i.p.) and killed 7 days after zymosan injection; AG 50 – animals treated with AG (50 mg kg−1 i.p.) and killed 7 days after zymosan injection; UA 100 and UA 250 – animals treated with with UA (100 or 250 mg kg−1 i.p.) and killed 14 days after zymosan injection, respectively.
Assessment of 3-nitrotyrosine content in the joint and the effect of UA
In order to confirm that nitrated protein formation is associated with the production of PN in the joint, we measured the levels of 3-nitrotyrosine in the synovial fluids after the injection of authentic PN into the joint. Figure 6a illustrates that a single injection of a 200 nmol solution of authentic PN leads to increased levels of 3-nitrotyrosine in the synovial fluids, as compared to animals that received either the vehicle (C) or inactive PN (i.art). Zymosan (i.art.) induced a significant increase in nitrated proteins into the rat knee joint, as previously published in abstract form (Greenacre et al., 2001) and as shown in Figure 6b. The levels remained elevated for at least 14 days. The increase in 3-nitrotyrosine levels was substantially inhibited by treatment with the 100 mg kg−1 dose of UA and reduced by the 250 mg kg−1 dose, as shown in Figure 6c. The NOS inhibitor L-NAME also decreased 3-nitrotyrosine levels, as measured at 6 h in the synovial exudates of zymosan arthritic rats(Figure 6c).
Figure 6.

Measurement of 3-nitrotyrosine levels assessed as nitrated BSA equivalents in synovial fluid extracts. (a) Shows the effect of PN injection (200 nmol, i.art.). Control (C) animals received only saline i.art. A solution of PN allowed to decompose completely before injection i.art. was used as inactive PN to exclude the influence of contaminants (nitrate, nitrite, etc.) present in the original PN solution; both saline and inactive PN were injected into naïve joints. (b) Shows the effect of zymosan (i.art.) with 3-nitrotyrosine levels detected at different times, as shown, after induction of zymosan-arthritis. Control (C) animals received only saline i.art. (c) Shows the effect of UA and L-NAME (LN) administration on the 3-nitrotyrosine levels in the joint exudates of rats with ZYA. Zymosan was injected i.art. into both knee joints and the 3-nitrotyrosine levels were detected either at 6 h (NT6 h) or 14 days (NT14d) after zymosan injection, using ELISA. UA (100 or 250 mg kg−1 i.p.) was injected 30 min before zymosan and four times daily, until the end of the experiment, at 14 days. L-NAME 75 mg kg−1 i.p. was injected 30 min before the zymosan and animals were killed 6 h later. Non-treated (NT) rats were given (i.p.) saline and zymosan i.art. and killed 6 h (NT6 h) or 14 days later (NT14d). Control (C) animals received only saline i.art. with no systemic treatment. Results are expressed as the mean±s.e.m. of values for each group of six animals. #P<0.05 compared to C rats; *P<0.05 compared to NT6h; **P<0.05 compared to NT14d.
Effect of NOS inhibitors and UA on inflammatory cell accumulation and histopathology
At 7 days after injection of zymosan, the synovial membranes displayed an intense cell infiltration, mostly due to lymphocytes and macrophages. There was also marked angiogenesis, together with edema and areas of necrosis. At day 14, the lymphocytes and macrophage cell influx were still observed but the necrotic and edematous areas were scarce, with angiogenesis no longer detected, while marked fibrosis was seen (data not shown). Table 1 shows the effect of the administration of the NOS inhibitors L-NAME and AG on the synovial changes, as demonstrated by histopathology, at day 7. There was a statistically significant amelioration, as compared to the NT rats, mostly due to a reduction of the cell influx in the groups that received the NOS inhibitors (P<0.05). The administration of either L-NAME or AG, whether prophylactically or therapeutically, significantly inhibited the cell influx into the joint cavities, as measured at day 7, compared to NT rats, as shown in Table 1. The effect of UA on the cellular influx into the synovia and into the joint exudates is also shown in Table 1. The results show that both doses of UA significantly reduced the inflammatory cell influx into the synovium, as compared to samples from NT rats. The effect of UA on the histopathological changes was mainly manifested as a significant reduction of the lymphocytes and macrophages numbers in the synovium, as compared to NT rats (P<0.05). A representative histological appearance showing a marked reduction in the synovial cell influx in both L-NAME- and UA-treated rats as compared to a NT-rat is shown in Figure 7. It should be noted that none of the groups achieved the maximum possible score, since some alterations, for example, angiogenesis, were prominent at 7 days, while others, for example, fibrosis, occurred in later phases (14 days).
Table 1.
Zymosan-induced arthritis – effect of NOS inhibitors and a peroxynitrite scavenger given prophylactically (P) or therapeutically (T) on the cell influx and synovitis
| Treatment | Cells (× 103 mm−3) | Histopathology |
|---|---|---|
| Control (C) | 28.1±3.1 | 0.0 (0–0) |
| NT (7 days) | 1446.4±396.3# | 8.0 (6–9)# |
| L-NAME 25 (P) | 1552.5±272# | 4.0 (3–5)#, * |
| L-NAME 75 (P) | 619.4±195.2#, * | 7.0 (6–7)# |
| AG 50 (P) | 490.0±105.8#, * | 4.0 (3–5)#, * |
| AG 100 (P) | 496.9±113.9#, * | 4.5 (3–7)#, * |
| L-NAME 25 (T) | 467.5±97.3#, * | 4.5 (4–6)#, * |
| L-NAME 75 (T) | 453.1±88.8#, * | 6.0 (4–7)#, * |
| AG 50 (T) | 330.6±77.7#, * | 5.0 (3–5)#, * |
| AG 100 (T) | 337.5±100.4#, * | 4.0 (3–6)#, * |
| NT (14 days) | 664.2±157.2# | 6.0 (5–8)# |
| UA 100 (P) | 247.7±45.2#, * | 5.0 (3–7)#, * |
| UA 250 (P) | 234.4±70.2#, * | 4.0 (3–7)#, * |
Rats were given zymosan (i.art.). The cell influx was assessed in the synovial exudates and the synovitis was assessed using a histopathological score, either 7 or 14 days after zymosan. Experiments with the NOS inhibitors L-NAME (mg kg−1) and AG (mg kg−1) were analyzed after 7 days of arthritis and those with the peroxynitrite scavenger UA (mg kg−1) after 14 days. NT rats were given (i.p.) saline and zymosan i.art. Control (C) animals received only saline i.art. with no systemic treatment. Results are expressed as the mean±s.e.m. for cell counts and median (range) for histopathological scores of values for each group of six animals.
P<0.05 compared to NT rats.
P<0.05 compared to C rats, using ANOVA followed by Tukey's test to compare the means and Kruskal–Wallis to compare the medians.
Figure 7.

Representative histopathology of the synovia after L-NAME or UA administration in zymosan arthritis. Zymosan was injected i.art. into both hind knee joints and the synovial membranes were excised at 7 or 14 days later. L-NAME (75 mg kg−1) was injected 30 min before zymosan and daily, until sacrifice, at 7 days. UA (100 mg kg−1 i.p.) was injected 30 min before zymosan and four times daily, until sacrifice, at 14 days. (a) Control animals received just saline i.art.; (b) non-treated (NT) animals received zymosan i.art. and saline i.p., killed 7 days later; (c, d) L-NAME- and UA-treated animals, respectively, showing a marked reduction in the cell influx, as compared to the NT rat. H&E staining (original × 100 magnification).
Discussion
This study, using a zymosan model of arthritis, has revealed that the total nitrite levels in synovial fluid remain raised throughout the 3-week experimental period, although the highest levels were observed during the acute phase after induction of arthritis. Meanwhile, measurement of GAG levels, as an indicator of cartilage damage, was unaltered for the first 4 days, but then associated with a significant and sustained loss of cartilage over the subsequent 17 days. Histopathological analysis of the cellular composition of both the joint tissue and synovial fluid revealed the ongoing inflammatory changes associated with synovitis. The use of nonselective and inducible NOS inhibitors demonstrated a general anti-inflammatory effect in the synovium. However, the NOS inhibitors increased the damage to the cartilage. These data have led us to conclude that inhibition of NOS is not protective in ZYA. Thus, it is suggested that endogenous NO, derived from iNOS, has a protective role during the ongoing joint inflammation and that this can override the anti-inflammatory effects of iNOS inhibitors. NO can be metabolized, depending on circumstance, to a range of reactive agents including PN. We have investigated the effect of an established PN-scavenging agent, UA, on the ZYA. UA suppressed the inflammatory parameters, while also being able to reverse the cartilage damage, an effect that was associated with a reduction in 3-nitrotyrosine levels in the inflamed joint.
A previous study demonstrated that NOS inhibitors were only antiarthritic when given prophylactically, rather than therapeutically, in adjuvant arthritis (Stefanovic-Racic et al., 1995). In our model, both NOS inhibitors (L-NAME and AG), whether given prophylactically or therapeutically, caused a significant reduction of the cellular influx into the synovial membranes and cavities. However, we have previously reported that NOS inhibitors, despite reducing nitrite levels in the joints, are analgesic in zymosan arthritis only when given as a prophylactic treatment (Rocha et al., 2002). Indeed, the animals treated with the iNOS inhibitors appeared clinically disadvantaged compared with animals that received zymosan alone, failing to recover from the weight loss and displaying reduced mobility (data not shown). This led us to terminate the experiment at 7 days after induction, rather than the 14 days that made it possible to study in the UA-treated rats. We believe that this lack of ‘well-being' could be derived from systemic side effects secondary to the effect of the NOS inhibitors on joint inflammation. Thus, the conclusion drawn from these data is that NOS inhibition, though reducing the cell influx, does not protect from the articular cartilage lesion that occurs in the chronic phase of the zymosan arthritis. This leads to a second consideration that, if this is so, NO must exert a protective effect to maintain cartilage homeostasis during arthritis. We propose that iNOS-derived NO produced during the inflammatory response plays an essential role in maintaining cartilage metabolism in the arthritic joint, possibly related to additional metabolic requirements. This is, to our knowledge, the first demonstration of an in vivo protective role for NO on articular cartilage during experimental arthritis. In accordance with these data, we have previously shown that NO is essential to the proliferation and differentiation of human osteoblasts in vitro and PN scavenging is protective to these cells (Da Rocha & De Brum-Fernandes, 2002). It was also recently reported that NO by itself is protective to chondrocytes under oxidative stress in vitro, while reactive oxygen species, including PN, promote chondrocyte death (Del Carlo & Loeser, 2002).
Several studies have shown that the inhibition of iNOS has been associated with a deterioration of arthritis in animal models. The administration of the selective iNOS inhibitor L-NIL worsened the SCW-induced arthritis in rats (McCartney-Francis et al., 2001). These authors suggested that the failure of L-NIL to protect from the articular damage in that study could be due to the inability of L-NIL to suppress NO production from the cNOS isoforms (McCartney-Francis et al., 2001). However, our results do not concur with this hypothesis, as L-NAME had similar consequences as L-NIL. In addition, iNOS knockout mice subjected to AIA display more edema and leukocyte infiltration in the acute phases (Veihelmann et al., 2001), indicating an in vivo anti-inflammatory effect of iNOS-derived NO in murine experimental arthritis. This again was not observed in our studies, as both NOS inhibitors attenuated the inflammatory cell accumulation.
Our results are in keeping with the possibility that the consequences of NO formation could lead to multiple mechanisms with respect to cartilage degradation in arthritis. While NO by itself would be essential for cartilage homeostasis, NO produced in large amounts could indirectly facilitate cartilage breakdown as a consequence of the formation of PN or other reactive nitrogen species. PN is well known to induce lipid peroxidation with a range of functional consequences at the enzyme and cellular level (Dowling et al., 1997; Greenacre & Ischiropoulos, 2001; Greenacre et al., 2002). We explored whether PN generation could be detected in the joint, by injecting PN i.art. and then assaying the synovial fluid extract for nitrated proteins (3-nitrotyrosine formation). In our hands, in the joint, levels of 3-nitrotyrosine were high 30 min–1 h after injection, and then quickly returned to basal levels, indicating that 3-nitrotyrosine is rapidly cleared from the synovial fluid after formation. This efficient clearance of 3-nitrotyrosine is most probably due to the high synovial blood flow (Sledge, 1993). Significantly increased levels of 3-nitrotyrosine were detected after the onset of ZYA throughout the 14-day experimental period, although maximal levels were observed at 6 h after ZYA. We have recently shown that, in rat skin, intradermal zymosan administration provokes 3-nitrotyrosine formation that is neutrophil dependent (Greenacre et al., 2002). In ZYA, there is a similar close correlation between neutrophil influx and both nitrite and 3-nitrotyrosine release at 6 h (Greenacre et al., 2001). Our present results extend these observations showing that nitrite and 3-nitro tyrosine levels are significantly increased at 24 h or 14 days of zymosan arthritis. As PN-induced 3-nitrotyrosine was cleared rapidly from the joint, the local initial accumulation of 3-nitrotyrosine probably does not account for the increased levels seen at the later time points. Rather, a continual generation of nitrated proteins from infiltrating mononuclear phagocytes is more likely.
Our first experiments aimed to determine a treatment regime via which serum UA levels could be maintained at levels compatible with its PN scavenging activity. The serum levels of this natural antioxidant are significantly lower in rodents than in humans (Usuda et al., 1998). These animals rapidly metabolize UA to allantoin, using the urate-oxidase enzyme, which is not active in humans (Keilin, 1959; Kock et al., 1992). It has been proposed that the mechanism that resulted in the inactivation of the urate-oxidase gene was an evolutionary advantage to humans, due to the antioxidant properties of UA (Yeldandi et al., 1991; Wu et al., 1992). UA significantly reduced the cell influx both into the synovial fluid and in the inflamed synovium. However, contrary to the results with the NOS inhibitors, the administration of UA provoked a clinical amelioration of the animals, with faster weight recovery as compared to NT rats (data not shown). Thus, we were able to prolong this experimental series until 14 days after injection of the zymosan. UA also significantly reversed the GAG loss of the articular cartilage, restoring it to basal levels, an effect that was associated with a reduction of 3-nitrotyrosine levels to a greater extent than was achieved with the NOS inhibitor L-NAME, at 6 h of arthritis. These data, to the best of our knowledge, are the first to demonstrate that a PN scavenger acts to protect from the articular cartilage damage in an experimental arthritis model. This effect is most likely to be due to an in vivo PN scavenging effect of UA as, in vitro, UA prevented damage to renal tubular epithelial cells exposed to ischemia–reperfusion, an effect that was associated with a specific PN scavenging activity of UA (Wangsiripaisan et al., 1999). Raising serum UA levels in rats in vivo to equivalent human levels, as attempted in the present study, was protective in animals subjected to hemorrhagic shock (Tsukada et al., 2000), and also in the EAE model (Hooper et al., 1998). In the present study, treatment with UA reduced nitrite levels in the synovial exudates. However, serum nitrite levels, which are significantly increased in the rats subjected to the zymosan arthritis, were not altered by UA administration (as occurs with NOS inhibitors (data not shown).
It has been shown in vitro that UA has no effect on the nitrite production by LPS-stimulated macrophages (Hooper et al., 1998). Indeed, it has been shown in vitro that UA and PN reaction yields an endothelium-independent vasorelaxing nitrated UA derivative (Skinner et al., 1998). During inflammation, this compound could ameliorate local blood flow. However, in addition to being temperature and light dependent, this reaction is yet to be demonstrated in vivo.
Apart from being able to scavenge PN, UA also scavenges singlet oxygen, peroxyl and hydroxyl radicals, ozone, and hypochlorous acid (Whiteman et al., 2002). Therefore, the possibility exists that the effect of UA in the present study could be due to its scavenging effect on other substances and this cannot be ruled out at this moment. In addition, nitryl chloride, formed from the reaction of nitrite and hypochlorous acid, has been shown to contribute to 3-nitrotyrosine formation (Eiserich et al., 1998). However, exposure of human HEpG2 hepatoma and SW 1353 chondrosarcoma cells to hypochlorous acid and nitrite did not result in 3-nitrotyrosine formation, as it happened when PN was added to the culture medium (Whiteman et al., 2003). Also, neutrophils and monocytes behave differently regarding nitrotyrosine formation, since protein nitration in human leukocytes was reported to be predominantly mediated by PN and a myeloperoxidase inhibitor could even increase nitration in these cells (Galinanes & Matata, 2002). In leukocyte-rich inflammatory reactions in mice, both myeloperoxidase and eosinophil peroxidase appeared not to contribute to nitrotyrosine formation (Brennan et al., 2002). Hence, we should consider that other reactive nitrogen species apart from PN may be involved in nitrotyrosine formation in our system. However, this possibility does not invalidate our conclusion that UA, probably through its reactive nitrogen species scavenging activity, was protective to the joint cartilage in zymosan arthritis.
Previous authors have also shown that UA administration leads to a decreased expression of the iNOS isoenzyme in the inflamed spinal cord in the rat EAE model (Hooper et al., 2000; Spitsin et al., 2000). Additionally, reactive oxygen and nitrogen species could influence cell accumulation in inflammation in the central nervous system through interactions with cytokines (Merrill & Murphy, 1997). The mechanisms via which UA acted in an anti-inflammatory manner in ZYA are unknown. We have shown that PN increases microvascular permeability in the rat skin (Ridger et al., 1997), but increased microvascular permeability is not considered to play an important role in the chronic phase of ZYA (Rocha et al., 1999).
In vitro data have shown that bicarbonate can effectively alter the PN scavenging activity of UA (Whiteman et al., 2002). Though i.art. bicarbonate levels are comparable to the concentrations used in vitro in that study, it has not been shown that this bicarbonate effect happens in vivo. During inflammatory synovitis, the i.art. milieu is rendered acidic due, at least partially, to a relative hypoxic state, with slower blood flow and, thus, clearance (Harris Jr, 1993). Therefore, the possible influence of i.art. bicarbonate levels on the scavenging activity of UA in our in vivo model remains to be tested.
Low serum UA levels have been linked in a negative manner with disease severity in multiple sclerosis patients (Hooper et al., 1998). By comparison, gout is known to be closely associated with hyperuricemia. Interestingly, the coexistence of both gout and multiple sclerosis is rarely seen. Indeed, the analysis of >20,000,000 patient records has shown that gout and multiple sclerosis are almost mutually exclusive, suggesting that hyperuricemia could be protective for multiple sclerosis (Hooper et al., 1998). Similar to multiple sclerosis, although both rheumatoid arthritis and gout are two of the most prevalent human inflammatory arthropathies, the coexistence of these diseases is rarely seen (Kelley & Schumacher, 1993).
In summary, our results demonstrate that, although treatment with NOS inhibitors was associated with an anti-inflammatory effect, cartilage degradation was accelerated in ZYA. Thus, we propose that NO can play an essential protective role in cartilage metabolism in the inflamed joint, in experimental arthritis. However, the arthritis was also associated with the detection of nitrated proteins in synovial fluid extracts, both after administration of PN and induction of ZYA. The protective effect of NO is therefore intriguing, considering that PN is formed by the reaction between NO and O2−. The time profiles produced in this study indicate the potential of reactive species, such as PN, to be generated in the arthritic joint during both acute and chronic experimental phases. Indeed, we demonstrate that PN scavenging can be protective in zymosan arthritis, as treatment with UA led to an inhibition of articular cartilage damage. As both NOS inhibitors and UA protect against cartilage degradation, we propose that, while NO can act in a protective manner in experimental arthritis, we also consider that NO, as a consequence of reactive oxygen species generation, promotes joint damage in experimental arthritis induced by zymosan in the rat. The realization of the involvement of multiple mechanisms related to cartilage degradation in ZYA is indicative of the need for further investigation that may lead to the design of potential therapeutic alternatives in inflammatory arthropathies.
Acknowledgments
This research was partially supported by the Fundação Cearense de Apoio ao Desenvolvimento Científico e Tecnológico (FUNCAP) and by the Arthritis Research Campaign of Great Britain. We are grateful to Denise Nunes Oliveira from the Department of Pathology, Faculty of Medicine, Federal University of Ceará (Fortaleza, Brazil) for the histopathological analysis and Ismar Paiva from the Department of Biochemistry, Federal University of Rio Grande do Norte (Natal, Brazil) for technical assistance in the GAG assay.
Abbreviations
- AG
aminoguanidine
- AIA
antigen-induced arthritis
- ANOVA
one-way analysis of variance
- C4
chondroitin 4-sulfate
- C6
chondroitin 6-sulfate
- cNOS
constitutive nitric oxide synthase
- EAE
experimental allergic encephalomyelitis
- ELISA
enzyme-linked immunosorbent assay
- GAG
glycosaminoglycans
- H&E
hematoxylin & eosin
- i.art.
intra-articular
- iNOS
inducible nitric oxide synthase
- i.p.
intraperitoneal
- L-NAME
L-NG-nitroarginine methyl ester
- L-NIL
N-iminoethyl-L-lysine
- LPS
lipopolysaccharide
- NO
nitric oxide
- NOS
nitric oxide synthase
- NT
nontreated
- OVA
ovalbumin
- s.c.
subcutaneous
- PN
peroxynitrite
- SCW
streptococcal cell wall
- UA
uric acid
- ZYA
zymosan-induced arthritis
References
- BECKMAN J.S. Oxidative damage and tyrosine nitration from peroxynitrite. Chem. Res. Toxicol. 1996;9:836–844. doi: 10.1021/tx9501445. [DOI] [PubMed] [Google Scholar]
- BECKMAN J.S., YE Y.Z., ANDERSON P.G., CHEN J., ACCAVITTI M.A., TARPEY M.M., WHITE C.R. Extensive nitration of protein tyrosines in human atherosclerosis detected by immuno histochemistry. Biol. Chem. Hoppe-Seyler. 1994;375:81–88. doi: 10.1515/bchm3.1994.375.2.81. [DOI] [PubMed] [Google Scholar]
- BENSOUYAD A., HOLLANDER A.P., DULARAY B., BEDWELL A.E., COOPER R.A., HUTTON C.W., DIEPPE P.A., ELSON C.J. Concentrations of glycosaminoglycans in synovial fluids and their relation with immunological and inflammatory mediators in rheumatoid arthritis. Ann. Rheum. Dis. 1990;49:301–307. doi: 10.1136/ard.49.5.301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRENNAN M.L., WU W., FU X., SHEN Z., SONG W., FROST H., VADSETH C., NARINE L., LENKIEWICZ E., BORCHERS M.T., LUSIS A.J., LEE J.J., LEE N.A., ABU-SOUD H.M., ISCHIROPOULOS H., HAZEN S.L. A tale of two controversies: defining both the role of peroxidases in nitrotyrosine formation in vivo using eosinophil peroxidase and myeloperoxidase-deficient mice, and the nature of peroxidase-generated reactive nitrogen species. J. Biol. Chem. 2002;277:17415–17427. doi: 10.1074/jbc.M112400200. [DOI] [PubMed] [Google Scholar]
- CLANCY P.M., AMIN A.R., ABRAMSON S.B. The role of nitric oxide in inflammation and immunity. Arthritis Rheum. 1988;41:1141–1151. doi: 10.1002/1529-0131(199807)41:7<1141::AID-ART2>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- DA ROCHA F.A.C., DE BRUM-FERNANDES A.J. Evidence that peroxynitrite affects human osteoblasts proliferation and differentiation. J. Bone Miner. Res. 2002;17:434–442. doi: 10.1359/jbmr.2002.17.3.434. [DOI] [PubMed] [Google Scholar]
- DEL CARLO M.J., LOESER R.F. Nitric oxide-mediated chondrocyte cell death requires the generation of additional reactive oxygen species. Arthritis Rheum. 2002;46:394–403. doi: 10.1002/art.10056. [DOI] [PubMed] [Google Scholar]
- DIETRICH C.P., DIETRICH S.M. Simple micro method for identification of heparin and other acidic mucopolysaccharides from mammalian tissues. Anal. Biochem. 1972;46:209–218. doi: 10.1016/0003-2697(72)90413-7. [DOI] [PubMed] [Google Scholar]
- DOWLING P., HUSAR W., MENNONA J., DONNENFELD H., COOK S., SIDHU M. Cell death and birth in multiple sclerosis brain. J. Neurol. Sci. 1997;149:1–11. doi: 10.1016/s0022-510x(97)05213-1. [DOI] [PubMed] [Google Scholar]
- EISERICH J.P., HRISTOVA M., CROSS C.E., JONES A.D., FREEMAN B.A., HALLIWELL B., VAN DER VLIET A. Formation of nitric oxide-derived inflammatory oxidants by myeloperoxidase in neutrophils. Nature. 1998;391:393–397. doi: 10.1038/34923. [DOI] [PubMed] [Google Scholar]
- FLETCHER D.S., WIDMER W.R., LUELL S., CHRISTEN A., OREVILLO C., SHAH S., VISCO D. Therapeutic administration of a selective inhibitor of nitric oxide synthase does not ameliorate the chronic inflammation and tissue damage associated with adjuvant-arthritis in rats. J. Pharmacol. Exp. Ther. 1998;284:714–721. [PubMed] [Google Scholar]
- GALINANES M., MATATA B.M. Protein nitration is predominantly mediated by a peroxynitrite-dependent pathway in cultured human leucocytes. Biochem. J. 2002;367:467–473. doi: 10.1042/BJ20020825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GEGOUT P., GILLET P., CHEVRIER D., GUINGAMP C., TERLAIN B., NETTE R.P. Characterization of zymosan-induced arthritis in the rat: effects on joint inflammation and cartilage metabolism. Life Sci. 1994;55:321–326. doi: 10.1016/0024-3205(94)00771-3. [DOI] [PubMed] [Google Scholar]
- GREENACRE A.S., ISCHIROPOULOS H. Tyrosine nitration: localisation, quantification, consequences for protein function and signal transduction. Free Radic. Res. 2001;34:541–581. doi: 10.1080/10715760100300471. [DOI] [PubMed] [Google Scholar]
- GREENACRE S.A., BEZERRA M.M., ROCHA F.A.C., BRAIN S.D. Correlation between neutrophil accumulation and protein nitration in zymosan-induced inflammation in the rat. J. Physiol. 2001;531:S222. [Google Scholar]
- GREENACRE S.A., ROCHA F.A.C., RAWLINGSON A., MEINERIKANDATHEVAN S., POSTON R.N., RUIZ E., HALLIWELL B., BRAIN S.D. Protein nitration in cutaneous inflammation in the rat: essential role of inducible nitric oxide synthase and polymorphonuclear leukocytes. Br. J. Pharmacol. 2002;36:985–994. doi: 10.1038/sj.bjp.0704798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRIS E.D., JREtiology and pathogenesis of rheumatoid arthritis Textbook of Rheumatology 1993Philadelphia: WB Saunders Co; 833–873.ed. Kelly, W.N., Harris Jr, E.D., Ruddy, S. & Sledge, C.B. 4th edn. pp [Google Scholar]
- HOOPER D.C., SCOTT G.S., ZBOREK A., MIKHEEVA T., KEAN R.B., KOPROWSKI H., SPITSIN S.V. Uric acid, a peroxynitrite scavanger, inhibits CNS inflammation, blood–CNS barrier permeability changes and tissue damage in a mouse model of multiple sclerosis. FASEB J. 2000;14:691–698. doi: 10.1096/fasebj.14.5.691. [DOI] [PubMed] [Google Scholar]
- HOOPER D.C., SPITSIN S., KEAN R.B., CHAMPION J.M., DICKSON G.M., CHAUDHRY I., KOPROWSKI H. Uric acid, a natural scavenger of peroxynitrite in experimental allergic encephalomyelitis and multiple sclerosis. Proc. Natl. Acad. Sci. U.S.A. 1998;95:675–680. doi: 10.1073/pnas.95.2.675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- IALENTI A., MONCADA S., DI ROSA M. Modulation of adjuvant arthritis by endogenous nitric oxide. Br. J. Pharmacol. 1993;110:701–706. doi: 10.1111/j.1476-5381.1993.tb13868.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ISHIMARU J.I., OGI N., MIZUNO S., GOSS A.N. Quantitation of chondroitin-sulfates, disaccharides and hyaluronan in normal, early and advanced osteoarthritic sheep temporomandibular joints. Osteoarthritis Cartilage. 2001;9:365–370. doi: 10.1053/joca.2000.0397. [DOI] [PubMed] [Google Scholar]
- JACQUES L.B., BALLIEUX R.E., DIETRICH C.P., KAVANAGH L.W. A microelectrophoresis method for heparin. Can. J. Physiol. Pharmacol. 1968;46:351–360. doi: 10.1139/y68-055. [DOI] [PubMed] [Google Scholar]
- JERONIMO S.M., SALES A.O., FERNANDES M.Z., MELO F.P., SAMPAIO L.O., DIETRICH C.P., NADER H.B. Glycosaminoglycan structure and content differ according to the origins of human tumors. Braz. J. Med. Biol. Res. 1994;27:2253–2258. [PubMed] [Google Scholar]
- KAUR H., HALLIWELL B. Evidence for nitric oxide-mediated oxidative damage in chronic inflammation. Nitrotyrosine in serum and synovial fluid from rheumatoid patients. FEBS Lett. 1994;350:9–12. doi: 10.1016/0014-5793(94)00722-5. [DOI] [PubMed] [Google Scholar]
- KEILIN J. The biological significance of uric acid and guanine excretion. Biol. Rev. 1959;34:265–296. [Google Scholar]
- KELLEY W.N., SCHUMACHER H.R., JRGout Textbook of Rheumatology 19932Philadelphia: WB Saunders Co; 1291–1296.ed. Kelly, W.N., Harris Jr, E.D., Ruddy, S. & Sledge, C.B.Vol [Google Scholar]
- KEYSTONE E.C., SCHORLEMMER H.U., POPE C., ALLISON A.C. Zymosan-induced arthritis: a model of chronic proliferative arthritis following activation of the alternative pathway of complement. Arthritis Rheum. 1989;20:1396–1401. doi: 10.1002/art.1780200714. [DOI] [PubMed] [Google Scholar]
- KHAN J., BRENNAND D.M., BRADLEY N., GAO B., BRUCKDORFER R., JACOBS M., BRENNAN D.M. 3-Nitrotyrosine in the proteins of human plasma determined by an ELISA method. Biochem. J. 1998;330:795–801. doi: 10.1042/bj3300795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOCK R., DELVOUX B., GREILING H. A high-performance liquid chromatographic method for the determination of hypoxanthine, xanthine, uric acid and allantoin in serum. Eur. J. Clin. Chem. Biochem. 1992;31:303–310. doi: 10.1515/cclm.1993.31.5.303. [DOI] [PubMed] [Google Scholar]
- MACMILLAN-CROW L.A., CROW J.P., KERBY J.D., BECKMAN J.S. Nitration and inactivation of manganese superoxide dismutase in chronic rejection of human renal allografts. Proc. Natl. Acad. Sci. U.S.A. 1996;93:11853–11858. doi: 10.1073/pnas.93.21.11853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARTNEY-FRANCIS N., ALLEN J.B., MIZEL D.E., ALBINA J.E., XIE Q.W., NATHAN C.F., WAHL S.M. Supression of arthritis by an inhibitor of nitric oxide synthase. J. Exp. Med. 1993;178:749–754. doi: 10.1084/jem.178.2.749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCARTNEY-FRANCIS N.L., SONG X., MIZEL D.E., WAHL S.M. Selective inhibition of inducible nitric oxide synthase exacerbates erosive joint disease. Immunology. 2001;166:2734–2740. doi: 10.4049/jimmunol.166.4.2734. [DOI] [PubMed] [Google Scholar]
- MERRILL J., MURPHY S. Inflammatory events at the blood–brain barrier: regulation of adhesion molecules, cytokine and chemokines by reactive nitrogen and oxygen species. Brain Behav. Immunity. 1997;11:245–263. doi: 10.1006/brbi.1997.0496. [DOI] [PubMed] [Google Scholar]
- MONCADA S., PALMER R.M.J., HIGGS E.A. Nitric oxide: physiology, pathophysiology and pharmacology. Pharmacol. Rev. 1991;43:1109–1142. [PubMed] [Google Scholar]
- PRYOR W.A., SQUADRITO G.L. The chemistry of peroxynitrite: a product from the reaction of nitric oxide and superoxide. Am. J. Physiol. 1995;268:L699–L722. doi: 10.1152/ajplung.1995.268.5.L699. [DOI] [PubMed] [Google Scholar]
- RIDGER V.C., GREENACRE S.A., HANDY R.L., HALLIWELL B., MOORE P.K., WHITEMAN M., BRAIN S.D. Effect of peroxynitrite on plasma extravasation, microvascular blood flow and nociception in the rat. Br. J. Pharmacol. 1997;122:1083–1088. doi: 10.1038/sj.bjp.0701498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROCHA F.A.C., ARAGÃO A.G.M., OLIVEIRA R.C., POMPEU M.M.L., VALE M.R., RIBEIRO R.A. Periarthritis promotes gait disturbance in zymosan-induced arthritis in rats. Inflamm. Res. 1999;48:485–490. doi: 10.1007/s000110050491. [DOI] [PubMed] [Google Scholar]
- ROCHA J.C.S., PEIXOTO M.E., JANCAR S., CUNHA F.Q., RIBEIRO R.A., ROCHA F.A.C. Dual effect of nitric oxide in articular inflammatory pain in zymosan-induced arthritis in rats. Br. J. Pharmacol. 2002;136:588–596. doi: 10.1038/sj.bjp.0704755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHARIF M., OSBORNE D.J., MEADOWS K., WOODHOUSE S.M., COLVIN E.M., SHEPSTONE L., DIEPPE P.A. The relevance of chondroitin and keratan sulphate markers in normal and arthritic synovial fluid. Br. J. Rheumatol. 1996;35:951–957. doi: 10.1093/rheumatology/35.10.951. [DOI] [PubMed] [Google Scholar]
- SKINNER K.A., WHITE C.R., PATEL R., TAN S., BARNES S., KIRK M., DARLEY-USMAR V., PARKS D.A. Nitrosation of uric acid by peroxynitrite. Formation of a vasoactive nitric oxide donor. J. Biol. Chem. 1998;273:24491–24497. doi: 10.1074/jbc.273.38.24491. [DOI] [PubMed] [Google Scholar]
- SLEDGE C.B.Biology of the joint Textbook of Rheumatology 19931Philadelphia: WB Saunders Co; 1–21.ed. Kelly, W.N., Harris Jr, E.D., Ruddy, S. & Sledge, C.B. Vol [Google Scholar]
- SPITSIN S.V., SCOTT G.S., KEAN R.B., MIKHEEVA T., HOOPER D.C. Protection of myelin basic protein immunized mice from free-radical mediated inflammatory cell invasion of the central nervous system by the natural peroxynitrite scavenger uric acid. Neurosci. Lett. 2000;292:137–141. doi: 10.1016/s0304-3940(00)01446-4. [DOI] [PubMed] [Google Scholar]
- STEFANOVIC-RACIC M., MEYERS K., MESCHTER C., COFFEY J.W., HOFFMAN R.A., EVANS C.H. N-monomethyl arginine, an inhibitor of nitric oxide synthase, suppresses the development of adjuvant arthritis in rats. Arthritis Rheum. 1994;37:1062–1069. doi: 10.1002/art.1780370712. [DOI] [PubMed] [Google Scholar]
- STEFANOVIC-RACIC M., MEYERS K., MESCHTER C., COFFEY J.W., HOFFMAN R.A., EVANS C.H. Comparison of the nitric oxide synthase inhibitors methylarginine and aminoguanidine as prophylactic and therapeutic agents in rat adjuvant arthritis. J. Rheumatol. 1995;22:1922–1928. [PubMed] [Google Scholar]
- TONUSSI C.R., FERREIRA S.H. Rat knee-joint incapacitation test: an objective screen for central and peripheral analgesics. Pain. 1992;48:421–427. doi: 10.1016/0304-3959(92)90095-S. [DOI] [PubMed] [Google Scholar]
- TSUKADA K., HASEGAWA T., TSUTSUMI S., KATOH H., KUWANO H., MIYAZAKI T., YAMAMOTO Y. Effect of uric acid on liver injury during hemorrhagic shock. Surgery. 2000;127:439–446. doi: 10.1067/msy.2000.104486. [DOI] [PubMed] [Google Scholar]
- USUDA N., REDDY M.K., HASHIMOTO T., RAO M.S., REDDY J.K. Tissue specificity and species differences in the distribution of urate oxidase in peroxisomes. Lab. Invest. 1998;58:100–111. [PubMed] [Google Scholar]
- VAN DER VLIET A., EISERICH J.P., KAUR H., CROSS C.E., HALLIWELL B. Nitrotyrosine as biomarker for reactive nitrogen species. Methods Enzymol. 1996;269:175–184. doi: 10.1016/s0076-6879(96)69019-3. [DOI] [PubMed] [Google Scholar]
- VEIHELMANN A., LANDES J., HOFBAUER A., DORGER M., REFIOR H.J., MESSMER K., KROMBACH F. Exacerbation of antigen-induced arthritis in inducible nitric oxide synthase-deficient mice. Arthritis Rheum. 2001;44:1420–1427. doi: 10.1002/1529-0131(200106)44:6<1420::AID-ART237>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- WANGSIRIPAISAN A., GENGARO P.E., NEMENOFF R.A., LING H., EDELSTEIN C.L., SCHRIER R.W. Effect of nitric oxide donors on renal tubular epithelial cell–matrix adhesion. Kidney Int. 1999;55:2281–2288. doi: 10.1046/j.1523-1755.1999.00484.x. [DOI] [PubMed] [Google Scholar]
- WHITEMAN M., KETSAWATSAKUL U., HALLIWELL B. A reassessment of the peroxynitrite scavenging activity of uric acid. Ann. N.Y. Acad. Sci. 2002;962:242–259. doi: 10.1111/j.1749-6632.2002.tb04072.x. [DOI] [PubMed] [Google Scholar]
- WHITEMAN M., SIAU J.L., HALLIWELL B. Lack of tyrosine nitration by hypochlorous acid in the presence of physiological concentrations of nitrite. Implications for the role of nitryl chloride in tyrosine nitration in vivo. J. Biol. Chem. 2003;278:8380–8384. doi: 10.1074/jbc.M211086200. [DOI] [PubMed] [Google Scholar]
- WU X.W., MUZNY D.M., LEE C.C., CATSKEY C.T. Two independent mutational events in the loss of urate oxidase during hominid evolution. J. Mol. Evol. 1992;34:78–84. doi: 10.1007/BF00163854. [DOI] [PubMed] [Google Scholar]
- YELDANDI A.V., YELDANDI V., KUMAR S., MURTHY C.V., WANG X.D., ALVARES K., RAO M.S., REDDY J.K. Molecular evolution of the urate oxidase-encoding gene in hominid primates: nonsense mutation. Gene. 1991;109:281–284. doi: 10.1016/0378-1119(91)90622-i. [DOI] [PubMed] [Google Scholar]