

Abstract
Cardiac hypertrophy is a homeostatic response to elevated afterload. Na+/H+ exchanger (NHE) inhibition reduces the hypertrophic response in animal models of left ventricular hypertrophy (LVH) and myocardial infarction. We examined the effect of chronic treatment with cariporide, a selective inhibitor of Na+/H+ exchanger isoform 1 (NHE-1), on left ventricular (LV) systolic and diastolic function under pressure overload conditions.
Male CD-1 mice were randomized to receive either a control diet or an identical diet supplemented with 6000 p.p.m. of cariporide. Cardiac pressure overload was induced by thoracic aortic banding. LV dimension and systolic and diastolic function were assessed in sham and banded mice by echocardiography and cardiac catheterization 2 and 5 weeks after surgery. Histological analysis was also performed.
After 2 weeks of pressure overload, the vehicle-treated banded mice (Veh-Bd) had enhanced normalized LV weight (about +50%) and normal chamber size and function, whereas cariporide-treated banded mice (Car-Bd) showed a preserved contractility and systolic function despite a marked attenuation of LVH. Diastolic function did not differ significantly among groups. After 5 weeks, the Veh-Bd developed LV chamber enlargement and systolic dysfunction as evidenced by a 16% increase in LV end-diastolic diameter, a 36% decrease in myocardial contractility, and a 26% reduction in percent fractional shortening. In contrast, Car-Bd showed an attenuated increase in LV mass, normal chamber size, and a maintained systolic function. A distinct histological feature was that in banded mice, cariporide attenuated the development of cardiomyocyte hypertrophy but not the attendant myocardial fibrosis.
In conclusion, the results of the present study indicate that (i) the hypertrophic response to pressure overload is dependent on NHE-1 activity, and (ii) at the 5-week stage, banding-induced deterioration of LV performance is prevented by NHE-1 inhibition.
Keywords: Pressure overload, Na+/H+ exchanger, cardiac remodeling, systolic function
Introduction
Hypertension-related left ventricular hypertrophy (LVH) strongly enhances the risk of future cardiovascular events and death, so that preventing LVH is a major goal in the clinical management of hypertension.
Previous studies have indicated that cardiac hypertrophy is associated with an increase in intracellular sodium (Na[i]) (Jelicks & Siri, 1995; Gray et al., 2001; Pogwizd et al., 2003; Verdonck et al., 2003). Conversely, an increase in Na[i] (produced by, e.g. inhibiting Na+/K+-ATPase with ouabain or enhancing extracellular sodium concentration) can promote cardiomyocyte hypertrophy (Peng et al., 1996; Gu et al., 1998). Furthermore, a recent study from our laboratory showed that agents interfering with fast sodium channel function (e.g. d-propranolol or disopyramide) display antihypertrophic activity in pressure-overloaded rat hearts (Marano et al., 2002). Taken together, these observations suggest that the mechanisms regulating cellular sodium homeostasis are involved in the cardiac hypertrophic response secondary to pressure overload.
The cardiac sodium/hydrogen exchanger, in addition to its role in intracellular pH regulation, is a pathway for Na+ influx (Frelin et al., 1984) and its pharmacological blockade should promote a decrease of Na[i] levels. The activation of the cardiac sodium ion/hydrogen ion exchanger isoform 1 (NHE-1) has been implicated in the cardiomyocyte hypertrophic process, inasmuch as (a) in pressure-overloaded hearts, both NHE-1 mRNA levels and Na[i] concentration are significantly elevated compared to control, nonoverloaded hearts (Takewaki et al., 1995), (b) the activity of the antiporter is augmented by hypertrophic factors such as α1-adrenergic activation (Yokoyama et al., 1998) and angiotensin II (Gunasegaram et al., 1999), and (c) cariporide, a newly developed NHE-1-specific inhibitor (Scholz et al., 1995), inhibits the hypertrophy of cardiac myocytes in DOCA/salt rats (Fujisawa et al., 2003) and in response to stretch as well as to β1-adrenergic receptor (β1-AR) stimulation (Yamazaki et al., 1998; Engelhardt et al., 2002). Additionally, it has been shown that cariporide was able (i) to attenuate compensatory right ventricular hypertrophy following pulmonary vascular injury (Chen et al., 2001), (ii) to inhibit the development of pathological remodeling after myocardial infarction (Kusumoto et al., 2001), and (iii) to regress both cardiomyocyte hypertrophy and interstitial fibrosis in the spontaneously hypertensive rats (Camilion De Hurtado et al., 2002; Cingolani et al., 2003). Suppression of the compensatory myocardial hypertrophy might be expected to induce systolic dysfunction more readily because chronic pressure overload-induced LVH is thought to develop as an adaptive response by which LV wall stress is moderated and LV function is supported. However, the consequences of NHE-1 inhibition on systolic function and cardiac contractility in pressure-overloaded animals have not been investigated.
Therefore, the aim of the present study was to evaluate the effect of chronic treatment with cariporide on systolic and diastolic LV function and remodeling after thoracic aortic banding (TAB). This is a standard maneuver that reproducibly induces LV pressure overload, development of LVH and transition to LV dilation and dysfunction within a few weeks after surgery (Esposito et al., 2002), in adult mice.
Methods
Animal preparation
CD-1 mice (12-week-old male, 28–30 g) were shipped by Charles River (Calco, Como, Italy) and maintained in accordance with EC guidelines (86/609/EEC) and with the Guide for the Care and Use of Laboratory Animals (NIH 85-23, revised 1996). Mice were randomly assigned to four groups: sham surgery plus control diet; sham surgery plus cariporide diet (containing 6000 p.p.m. of cariporide); aortic banding (see below) plus control diet; aortic banding plus cariporide diet.
Experimental protocol
Animals were maintained for 2 and 5 weeks after surgery and were then anesthetized with isoflurane in order to perform hemodynamic measurements. In the animals allocated to cariporide treatment, the drug was administered starting 10 days before banding surgery to allow plasma levels to reach steady-state values (Kusumoto et al., 2001). Although circulating cariporide levels were not determined in our study, it was shown (Engelhardt et al., 2002) that mice treated with a dosage similar to ours had cariporide levels of 2.5±0.3 μmol l–1, able to inhibit NHE-1 activity by more than 50% in isolated cardiomyocytes (Scholz et al., 1995).
Mouse model of LV pressure overload
The development of LVH was induced by banding the thoracic aorta. Each mouse was anesthetized with a mixture of ketamine (100 mg kg–1) and xylazine (5 mg kg–1), orally intubated with a 24-gauge tubing under direct laryngeal visualization, and ventilated with a tidal volume of 200 μl and a respiratory rate of 110 breaths min–1. The chest was then opened at the second intercostal space. TAB was performed by placing a ligature on the thoracic aorta between the innominate artery and left common carotid artery along with a 25-gauge needle using a 6-0 nylon suture. After the suture was secured, the needle was removed, the chest was closed, and the pneumothorax evacuated. The mortality rate for TAB was 3.5% (4/114) during the surgical procedure and 14.5% (16/110) after 24 h. The perioperative deaths could be attributed to aortic rupture or acute heart failure. A concurrent group of mice was subjected to a sham operation, in which an identical surgical procedure was performed but the ligature was not tightened. The total perioperative mortality was 0% for sham-operated mice. To measure pressure aortic gradient, both right- and left-carotid arteries were cannulated by a 1.4-F, four-electrode pressure–volume Millar catheter and stretched P50 tubing, respectively. The Millar catheter was connected to the pressure–conductance unit (model MPCU-200, Millar, U.S.A.), whereas the other catheter was connected to a P23 Statham pressure transducer.
Echocardiography
At 2 and 5 weeks after banding, hemodynamic analyses were performed in mice intubated and anesthetized with isoflurane (1% in 100% oxygen). Echocardiographic examination was performed with an Esaote SIM 7000 Challenge (Esaote Biomedica, Firenze, Italy) equipped with a 10 MHz imaging transducer. After good-quality 2D short-axis images of the left ventricle were obtained, M-mode freeze frames were printed on common echocardiographic paper and digitized. End-systolic (left ventricular end-systolic diameter, LVESD) and end-diastolic (left ventricular end-diastolic diameter, LVEDD) left ventricular internal diameters, and posterior-wall end-diastolic (PWTd) and end-systolic (PWTS) thickness were measured by an image-analysis system (Metamorph, Universal Image Corporation, PA, U.S.A.). Percent fractional shortening was calculated as
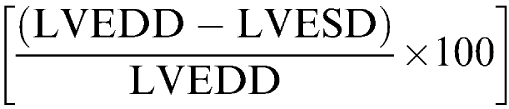 |
End-systolic wall stress was estimated as follows:
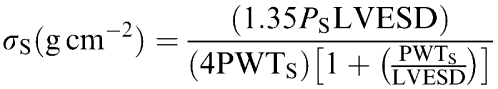 |
where Ps is the peak left ventricular systolic pressure (LVSP) (in mmHg) and 1.35 is the conversion factor from mmHg to grams per centimeter squared. LVESD and PWTs were measured from the M-mode echocardiogram. These measurements were made at end-systole, as defined by the time point corresponding to the smallest cavity dimension.
Hemodynamic analysis
To measure arterial blood pressure and to obtain LV pressure–volume curves, a 1.4-F, four-electrode pressure–volume catheter (model SPR-839, Millar Instruments, U.S.A.) was inserted into the right carotid artery, and retrogradely advanced through the ascending aorta into the LV under isoflurane anesthesia. The pressure–volume catheter was connected to an MPCU-200 unit (Millar Instruments, U.S.A.). Correct catheter positioning was confirmed by online visualization of the pressure–volume loops. Conductance-derived pressure–volume data were analyzed with IOX software (version 1594, EMKA, France). The end-systolic point of pressure–volume loops was computed by an iterative method and end-systolic elastance (Ees) was determined from end-systolic pressure–volume relationships (ESPVRs). To change the cardiac preload, inferior caval occlusion was produced over 3 s. Opening the thorax induced a drop in LV pressure in all groups. All steady-state and caval occlusion pressure–volume loops were acquired, while pulmonary ventilation was temporarily suspended. The data were recorded as a series of 8–10 pressure–volume loops.
Histological analysis
At the end of the in vivo experiment, animals were deeply anesthetized, the heart was removed and the left ventricle was separated from the other chambers, weighed and normalized by body weight (BW) to determine LVH. For histological assessment, the LV was fixed with 10% buffered formalin solution and embedded in paraffin. LV sections, 4 μm thick, were cut and stained with hematoxylin and eosin for the measurement of myocyte cross-sectional area or by the sirius red/picric acid method to determine LV fibrosis by quantitative morphometry (Morphometric, Universal Imaging Corporation, PA, U.S.A.). Myocyte mean cross-sectional area was calculated by measuring the dimensions of no less than 50 cells per section (four or five sections of each left ventricle). The visual fields were accepted for quantitative analysis if cross-sections of myocytes were present, nuclei were visible, and their cellular membranes were intact. Cardiomyocyte width was assessed by marking the borders of the cells. The extent of interstitial LV fibrosis was calculated in five fields randomly selected from a section by measuring picrosirius red-stained fibrosis area and dividing by the total myocardium area (four or five sections of each left ventricle). Perivascular fibrosis was estimated as the ratio of collagen-stained fibrosis surrounding the vessel wall to the total vessel area (15–20 coronary arteries for each animal). Areas of reparative fibrosis were not considered in the analysis.
Data analysis
Group means±s.e. were calculated for all relevant variables. Statistical comparisons were performed by ANOVA followed by the Neumann–Keuls test. The limit of statistical significance was set at P<0.05.
Results
To investigate the effect of cariporide on pressure overload-induced myocardial responses, mice were subjected to aortic banding. Experimental measurements of cardiac hypertrophy and function were taken at earlier (2 weeks) and later (5 weeks) time points following surgery. To make sure that the effects of cariporide were not mediated through indirect hemodynamics effects, we measured the pressure gradient between the right- and left-carotid arteries and the LVSP in both treated and untreated banded groups 2 weeks after surgery. The administration of cariporide did not significantly influence the pressure gradient across the aortic banding (vehicle-treated banded mice, Veh-Bd, 50±3 mmHg; cariporide-treated banded mice, Car-Bd, 49±4 mmHg). Also, the LVSP was not different in the untreated banded group (151±5 mmHg) vs the cariporide-treated banded group (149±4 mmHg).
Effect of cariporide on cardiac hypertrophy
After 2 weeks from surgery, the left ventricular weight (left ventricular weight-to-body weight ratio, LVW/BW) of banded mice was significantly increased (47%) compared to sham controls (Table 1, Veh groups). In contrast, cariporide-treated banded animals showed a much lower enhancement of ventricular weight (15%) (Table 1, Car groups), indicating that the inhibition of the cardiac NHE-1 can markedly attenuate the development of LVH. There were no significant differences in LVW/BW ratio between cariporide-treated (Car-Sh) and vehicle-treated sham mice (Veh-Sh), indicating that the drug has no intrinsic effects on heart weight in the absence of pressure overload (Table 1). Likewise, there were no effects of cariporide on the overall BW gain (Table 1).
Table 1.
Body weight and LV hypertrophic parameters in sham and banded mice 2 and 5 weeks after aortic surgery
| 2 Weeks | 5 Weeks | |||||||
|---|---|---|---|---|---|---|---|---|
| Veh-Sh | Veh-Bd | Car-Sh | Car-Bd | Veh-Sh | Veh-Bd | Car-Sh | Car-Bd | |
| (n=10) | (n=11) | (n=9) | (n=12) | (n=7) | (n=9) | (n=7) | (n=10) | |
| BW (g) | 32.3±1.1 | 32.4±1.1 | 32.3±1.2 | 32.5±1.3 | 35.4±0.6 | 35.7±0.5 | 35.9±0.3 | 35.5±0.4 |
| LVW/BW (mg g−1) | 3.2±0.1 | 4.7±0.2† | 3.3±0.1 | 3.8±0.2* | 3.4±0.3 | 5.6±0.2† | 3.3±0.3 | 4.3±0.2* |
| CSA (μm2) | 102±4 | 152±8† | 96±5 | 122±3* | 110±8 | 177±9† | 108±5 | 145±4* |
BW, body weight; LVW/BW, left ventricular weight-to-body weight ratio; CSA, cardiomyocyte cross-sectional area. Veh-Sh: vehicle-treated, sham-operated; Veh-Bd: vehicle-treated, banded; Car-Sh: cariporide-treated; Car-Bd: cariporide-treated, banded.
n=number of experimental points; values are expressed as mean±s.e., *P<0.05 vs sham-operated groups
P<0.05 vs all other groups.
To analyze how this antihypertrophic effect of cariporide is reflected by a change in the myocardium structure, the hearts of both treated and untreated animals were subjected to histological evaluation. Morphometric analysis of cross-sections of left ventricular tissue revealed a significant increase (49%) in cardiomyocyte cross-sectional area (CSA) in the hearts of banded animals compared to controls, but this increase was significantly smaller (19%) under cariporide treatment (Table 1). Cariporide thus appears to reduce the enhancement of the mean cellular area that accounts for the early hypertrophic response. Again, no significant differences were observed between sham mice groups (Table 1). In all groups, there were minimal and nonsignificant changes in collagen ventricular deposition and perivascular fibrosis 2 weeks after surgery (data not shown).
After 5 weeks from surgery, the enhancement of ventricular weight induced by aortic banding was larger, both in nontreated (65%) and cariporide-treated (30%) animals, but the inhibitory effect of the antiporter blocker remained clearly evident, and to an extension similar to that observed at 2 weeks (Table 1).
Morphometric analysis at 5 weeks confirmed a significant decrease in cardiomyocyte hypertrophic response in Car-Bd hearts compared with Veh-Bd ones (CSA, Table 1). However, it also revealed an increased perivascular and interstitial collagen deposition in both banded groups (Figure 1, a–c). There was no difference in the intensity of collagen staining between cariporide treatment and controls, suggesting that cariporide has no influence on LV collagen deposition under pressure overload conditions.
Figure 1.

Effect of banding with and without cariporide treatment on perivascular and interstitial myocardial collagen in mice 5 weeks after surgery. Bar graphs show myocardial perivascular (a) and interstitial (b) fibrosis in left ventricles. Augmented perivascular and interstitial fibrosis is apparent in banded groups (Veh-Bd and Car-Bd). Representative photomicrographs of cross-sections of coronary arteries are shown (c). Picrosirius red-stained sections (× 200) outline collagen (arrows). Veh-Sh, vehicle-treated, sham-operated; Veh-Bd, vehicle-treated, banded; Car-Sh, cariporide-treated, sham-operated; Car-Bd, cariporide-treated, banded. *P<0.05 vs sham groups.
Effects of cariporide on LV remodeling and cardiac function
To measure how cariporide affected ventricular performance, echocardiography and cardiac catheterization were performed both after 2 and 5 weeks from aortic banding surgery.
Following 2 weeks of chronic pressure overload, the enlargement of cardiac mass in Veh-Bd was primarily accounted for by concentric hypertrophy. In fact, the relative wall thickness (RWT, calculated as ratio between posterior-wall end-diastolic thickness and end-diastolic diameter) was significantly increased (Table 2, Veh-Bd group), whereas the systolic wall stress (σs) remained normal (Veh-Sh, 76±5 g cm–2; Veh-Bd, 78±7 g cm–2), presumably because of the adequate increase in the RWT.
Table 2.
Invasive hemodynamics and echocardiographic parameters in sham and banded mice 2 and 5 weeks after aortic surgery
| 2 Weeks | 5 Weeks | |||||||
|---|---|---|---|---|---|---|---|---|
| Veh-Sh | Veh-Bd | Car-Sh | Car-Bd | Veh-Sh | Veh-Bd | Car-Sh | Car-Bd | |
| (n=10) | (n=11) | (n=9) | (n=12) | (n=7) | (n=9) | (n=7) | (n=10) | |
| HR (bpm) | 412±8 | 390±7 | 411±9 | 398±8 | 395±7 | 399±8 | 401±8 | 392±11 |
| LV SP (mmHg) | 102±2 | 151±5* | 98±3 | 149±4* | 99±4 | 147±3* | 101±4 | 148±2* |
| LVEDP (mmHg) | 5.1±0.9 | 5.9±0.7 | 6.6±1.1 | 5.3±0.9 | 5.2±1.1 | 11.1±2.1† | 6.4±1.2 | 6.3±2.4 |
| LVEDD (mm) | 3.8±0.2 | 3.9±0.1 | 3.8±0.1 | 3.9±0.2 | 3.9±0.1 | 4.5±0.2† | 3.9±0.2 | 4.0±0.1 |
| RWT (mm mm−1) | 0.14±0.02 | 0.20±0.02† | 0.13±0.03 | 0.15±0.02 | 0.14±0.01 | 0.17±0.02* | 0.13±0.02 | 0.17±0.03* |
| LV FS (%) | 47±3 | 46±2 | 47±3 | 48±3 | 46±3 | 34±2 | 45±1 | 45±2 |
| Ees (mmHg μl−1) | 2.8±0.4 | 2.9±0.3 | 3.1±0.2 | 3.0±0.3 | 2.8±0.3 | 1.8±0.3† | 2.9±0.2 | 2.8±0.3 |
| (n=6) | (n=7) | (n=7) | (n=6) | (n=5) | (n=5) | (n=6) | (n=6) | |
HR, heart rate; LVSP, left ventricular systolic pressure; LVEDP, left ventricular end-diastolic pressure; LVEDD, left ventricular end-diastolic diameter; RWT, posterior-wall thickness-to-left ventricular end-diastolic ratio; LV FS, left ventricular fractional shortening; Ees, end-systolic elastance. Other abbreviations as in Table 1. n=number of experimental points; values are expressed as mean±s.e.
P<0.05 vs sham-operated groups
P<0.05 vs all other groups.
In contrast, cariporide-treated banded animals displayed a larger σs value (Car-Bd, 94±6 g cm–2), likely reflecting the attenuated increase of wall thickness exerted by the drug treatment. Remarkably, despite the blunting of the hypertrophic response and the resultant increase in the systolic wall stress, cariporide-treated banded animals exhibited preserved LV contractility and systolic function. These parameters were evaluated as Ees, a load-independent index of myocardial contractility, and left ventricular fractional shortening (LV FS), respectively (Table 2). Both were identical in cariporide-treated and untreated sham animals. Moreover, as also shown in Table 2, the maintained ventricular performance in cariporide-treated banded animals cannot be explained by a possible effect of the drug on preload (left ventricular end-diastolic pressure, LVEDP), afterload (LVSP), or heart rate (HR), that is, by indirectly affecting the hemodynamic loading. Also, diastolic function (evaluated as LVEDP) did not significantly differ among experimental groups (Table 2).
After 5 weeks of surgery, signs of cardiac dysfunction induced by pressure overload became evident. Animals not treated with cariporide (Veh-Bd in Table 2) exhibited a 16% increase in ventricular end-diastolic dimension (LVEDD), 36% decrease in myocardial contractility (Ees), and a 26% reduction in percent fractional shortening (%FS), as shown in Table 2. These changes were accompanied by a 100% increase in LVEDP compared with the sham control group (Table 2). Cariporide appeared quite effective in contrasting such a decline in heart performance, since echocardiograms revealed the preservation of systolic function evaluated as %FS in the Car-Bd (compare Veh-Bd and Car-Bd groups in Table 2). This is also shown by ESPVRs recorded in Car-Bd. The slopes (Ees) computed from such relationships were significantly steeper in the Car-Bd than in the Veh-Bd (Table 2), which indicate intact LV contractility. Representative ESPVRs recorded at 5 weeks are presented in Figure 2.
Figure 2.

Representative pressure–volume relationships of vehicle-sham (a), cariporide-treated banded (b), and vehicle-banded (c) mice 5 weeks after surgery. End-systolic pressure was plotted with end-systolic volume to determine the end-systolic pressure–volume relationship (ESPVR). Changes in hemodynamic load were obtained by transitory caval occlusion (see text for details). Slope of the regression line (ESPVR) is ventricular end-systolic elastance (Ees), a load-independent index of cardiac contractility.
Discussion
In the present study, we demonstrate that cariporide treatment has two major effects on the hearts of mice subjected to pressure overload. One effect is inhibition of the hypertrophic response, which is explained by a reduced enlargement of myocardiocyte size. This is in agreement with previously documented effects in in vitro as well as in vivo studies (Yamazaki et al., 1998; Camilion de Hurtado et al., 2002; Engelhardt et al., 2002), and extend to the mouse model of aortic banding-induced LVH. In the mouse, the inhibitory effect was present both at early and delayed stages after surgery and was of similar magnitude at these time points. This suggests that cariporide treatment may slow down the rate of hypertrophy development rather than block the maximal extent of the phenomenon.
The second effect is that cariporide can prevent (or delay) the pathological remodeling and the deterioration of cardiac systolic function at least up to 5 weeks of pressure overload. This protective influence on cardiac function occurs despite the lack of effect of cariporide on the cardiac fibrosis, observable only after 5 weeks of surgery.
The antihypertrophic effect of cariporide is intriguing. In fact, despite the marked blunting of the adaptive cardiac hypertrophic response to mechanical overload, Car-Bd have a systolic function as effective as that of control mice in which a much larger LV mass is developed. These findings suggest at least two possible explanations: (i) one is that in the earlier stages, there is no proportionality between the degree of hypertrophic response and extent of compensation. In other words, the left ventricle may have the potential to cope with an increased afterload even without becoming hypertrophic. Similar findings were reported for other inhibitors of cardiac hypertrophy, such as propranolol (Marano et al., 2002), cyclosporin (Hill et al., 2000), or were inferred from the study of mice deficient in catecholamine synthesis (Esposito et al., 2002). However, controversy exists as to whether the abolition of hypertrophy has detrimental effects on cardiac function. Some studies (Meguro et al., 1999; Oie et al., 2000) found that pharmacological inhibition of the LVH response induces a more rapid decline in LV systolic function and progression to failure, whereas according to other authors (Hill et al., 2000; Esposito et al., 2002), the abolition of LVH is associated with a preserved systolic function. Thus the functional implications of pressure overload-induced LVH are still a matter of debate. (ii) Another explanation is that inhibition of NHE-1 may enhance myocardiocyte performance so that a smaller degree of enlargement is sufficient to achieve the same extent of compensation. The exact mechanism by which NHE-1 inhibition prevents pressure overload-induced cardiac hypertrophy and preserves LV systolic function under hemodynamic overload conditions, remains to be elucidated. Changes in the intracellular concentrations of sodium ion might be involved. Sodium has been shown to exert hypertrophic effects in cultured cardiac myocytes and a high Na+ diet induces cardiac enlargement in hypertensive and normotensive rats (Frohlich et al., 1993; Gu et al., 1998). The amount of Na+ entering through the Na+/H+ exchanger (NHE) accounts for approximately half the total Na+ cellular influx and the intracellular Na+ was found to be increased in pressure-overloaded hearts (Frelin et al., 1984; Gray et al., 2001). Additional evidence indicates that NHE-dependent sodium influx is a major contributor to hypertrophy produced by various agonists, including α1-adrenergic agonists, endothelin-1, or phorbol esters, all of which act via the activation of various protein kinase C (PKC) isoforms. NHE-1 inhibition attenuates both the hypertrophy and the PKC activation (Hayasaki-Kajiwara et al., 1999). An increase in intracellular sodium, via the activation of cardiac Na+/Ca2+ exchanger, may reduce intracellular calcium transients and the activation of calcium-dependent signaling molecules such as PKC and transcription factors. As a consequence, the beneficial effects of NHE-1 inhibition could be due to both changes in myocyte energy handling and prevention of cytosolic Ca2+ overload through reduction of Na+ influx.
The protective effect of cariporide on the derangement of cardiac function induced by prolonged pressure overload is important and may have important therapeutic implications. However, also in this case the underlying mechanism is unknown. We have shown in this study that cariporide treatment contrasts the progression toward LV chamber dilation and dysfunction. We also showed that cariporide does not act by reducing arterial blood pressure, HR, or LV filling, that is, its effect is independent of any attenuation in cardiac mechanical overload. Again, this suggests that cariporide can directly inhibit the cardiomyocyte remodeling process.
Our observations integrate and extend those from previous studies, in which the beneficial effects of NHE-1 inhibition on cardiomyocyte hypertrophy were observed in nonischemic regions of the myocardium either in a postmyocardial infarction model (Kusumoto et al., 2001), or in response to enhanced β1-AR expression (Engelhardt et al., 2002). Thus cariporide effects can be observed both in ischemic and nonischemic tissues. However, attenuation of the increase in myocyte size was not accompanied by a significant attenuation of pressure overload-related interstitial fibrosis. This suggests that at least in the earlier stages, the myocyte and the nonmyocyte cellular components of myocardial tissue may grow independent of each other and that the mechanisms responsible for the growth of either component may be different. In this regard, our data are in keeping with the observations of Camilion de Hurtado et al. (2002) in the SHR model, but are at variance with those from Engelhardt et al. (2002) in the cardiac hypertrophic transgenic mouse with overexpression of the β1-AR, in which cariporide was shown to affect both cardiomyocyte hypertrophic and the interstitial fibrotic processes. The discrepancy between their data and our data may be related to the fact that different models of heart failure were used. In our experimental model, LVH develops as a response to mechanical overload rather than as a result of genome manipulation as is the case for the β1-AR transgenic mouse (Engelhardt et al., 2002). On the other hand, it is to be considered that in the transgenic mice the treatment period was longer than in our banded mice and that this might also be a relevant factor considering that the regressions of myocyte hypertrophy and collagen accumulation have different time course (Cingolani et al., 2003).
The preserved cardiac function 5 weeks after aortic banding could reflect a positive inotropic effect or hemodynamic effects of cariporide. However, in cariporide-treated mice, the drug affected neither cardiac contractility nor hemodynamic indices such as preload, afterload, or HR. Taken together, these findings suggest that the contribution of hemodynamic or inotropic effects to the preserved cardiac function can be excluded. In experimental models of cardiac hypertrophy and heart failure, cardiomyocyte apoptosis causes the progression of compensated myocardial hypertrophy to LV dilation and dysfunction (Condorelli et al., 1999). At present, we cannot exclude the fact that the preserved cardiac function 5 weeks after aortic banding was a result of reduced cardiomyocyte loss.
In conclusion, we demonstrated in a mouse model of chronic pressure overload that inhibiting the cardiac NHE-1 by cariporide administration opposes the development of LVH independent of any reduction in cardiac hemodynamic load, and that this treatment favorably affects the progression to LV pathological remodeling and systolic dysfunction.
Acknowledgments
This work was supported in part by the Ministry of Health Grants ICS 030.6/RF00-49 and 1AH/FW. We thank Aventis Pharma (Frankfurt, Germany) for supplying the experimental diet.
Abbreviations
- BW
body weight
- Car
cariporide
- Car-Bd
cariporide-treated, banded mice
- Car-Sh
cariporide-treated, sham-operated mice
- CSA
cardiomyocyte cross-sectional area
- Ees
end-systolic elastance
- HR
heart rate
- LVEDD
left ventricular end-diastolic diameter
- LVEDP
left ventricular end-diastolic pressure
- LV FS
left ventricular fractional shortening
- LVSP
left ventricular systolic pressure
- LVW/BW
left ventricular weight-to-body weight ratio
- NHE-1
Na+/H+ exchanger isoform 1
- RWT
posterior-wall thickness-to-left ventricular end-diastolic diameter ratio
- σs
end-systolic left ventricular wall stress
- Veh-Sh
vehicle-treated, sham-operated mice
- Veh-Bd
vehicle-treated, banded mice
References
- CAMILION DE HURTADO M.C., PORTIANSKY E.L., PEREZ N.G., REBOLLEDO O.R., CINGOLANI H.E. Regression of cardiomyocyte hypertrophy in SHR following chronic inhibition of the Na+/H+ exchanger. Cardiovasc. Res. 2002;53:862–868. doi: 10.1016/s0008-6363(01)00544-2. [DOI] [PubMed] [Google Scholar]
- CHEN L., GAN X.T., HAIST J.V., FENG Q., LU X., CHAKRABARTI S., KARMAZYN M. Attenuation of compensatory right ventricular hypertrophy and heart failure following monocrotaline-induced pulmonary vascular injury by the Na+–H+ exchange inhibitor cariporide. J. Pharmacol. Exp. Ther. 2001;298:469–476. [PubMed] [Google Scholar]
- CINGOLANI H.E., REBOLLEDO O.R., PORTIANSKY E.L., PEREZ N.G., CAMILION DE HURTADO M.C. Regression of hypertensive myocardial fibrosis by Na+/H+ exchange inhibition. Hypertension. 2003;41:373–377. doi: 10.1161/01.hyp.0000051502.93374.1c. [DOI] [PubMed] [Google Scholar]
- CONDORELLI G., MORISCO C., STASSI G., NOTTE A., FARINA F., SGARAMELLA G., DE RIENZO A., RONCARATI R., TRIMARCO B., LEMBO G. Increased cardiomyocyte apoptosis and changes in proapoptotic and antiapoptotic genes bax and bcl-2 during left ventricular adaptations to chronic pressure overload in the rat. Circulation. 1999;99:3071–3078. doi: 10.1161/01.cir.99.23.3071. [DOI] [PubMed] [Google Scholar]
- ENGELHARDT S., HEIN L., KELLER U., KLAMBT K., LOHSE M.J. Inhibition of Na+/H+ exchange prevents hypertrophy, fibrosis, and heart failure in β1-adrenergic receptor transgenic mice. Circ. Res. 2002;90:814–819. doi: 10.1161/01.res.0000014966.97486.c0. [DOI] [PubMed] [Google Scholar]
- ESPOSITO G., RAPACCIUOLO A., NAGA PRASAD S.V., TAKAOKA H., THOMAS S.A., KOCH W.J., ROCKMAN H.A. Genetic alterations that inhibit in vivo pressure-overload hypertrophy prevent cardiac dysfunction despite increased wall stress. Circulation. 2002;105:85–92. doi: 10.1161/hc0102.101365. [DOI] [PubMed] [Google Scholar]
- FRELIN C., VIGNE P., LAZDUNSKI M. The role of the Na+/H+ exchange system in cardiac cells in relation to the control of the internal Na+ concentration. A molecular basis for the antagonistic effect of ouabain and amiloride on the heart. J. Biol. Chem. 1984;259:8880–8885. [PubMed] [Google Scholar]
- FROHLICH E.D., CHIEN Y., SESOKO S., PEGRAM B.L. Relationship between dietary sodium intake, hemodynamics, and cardiac mass in SHR and WKY rats. Am. J. Physiol. 1993;264:R30–R34. doi: 10.1152/ajpregu.1993.264.1.R30. [DOI] [PubMed] [Google Scholar]
- FUJISAWA G., OKADA K., MUTO S., FUJITA N., ITABASHI N., KUSANO E., ISHIBASHI S. Na/H exchange isoform 1 is involved in mineralocorticoid/salt-induced cardiac injury. Hypertension. 2003;41:493–498. doi: 10.1161/01.HYP.0000056769.73726.E5. [DOI] [PubMed] [Google Scholar]
- GRAY R.P., MCINTYRE H., SHERIDAN D.S., FRY C.H. Intracellular sodium and contractile function in hypertrophied human and guinea-pig myocardium. Pflugers Arch. 2001;442:117–123. doi: 10.1007/s004240000512. [DOI] [PubMed] [Google Scholar]
- GU J.W., ANAND V., SHEK E.W., MOORE M.C., BRADY A.L., KELLY W.C., ADAIR T.H. Sodium induces hypertrophy of cultured myocardial myoblasts and vascular smooth muscle cells. Hypertension. 1998;31:1083–1087. doi: 10.1161/01.hyp.31.5.1083. [DOI] [PubMed] [Google Scholar]
- GUNASEGARAM S., HAWORTH R.S., HEARSE D.J., AVKIRAN M. Regulation of sarcolemmal Na+/H+ exchanger activity by angiotensin II in adult rat ventricular myocytes: opposing actions via AT1 vs. AT2 receptors. Circ. Res. 1999;85:919–930. doi: 10.1161/01.res.85.10.919. [DOI] [PubMed] [Google Scholar]
- HAYASAKI-KAJIWARA Y., KITANO Y., IWASAKI T., SHIMAMURA T., NAYA N., IWAKI K., NAKAJIMA M. Na+ influx via Na+/H+ exchange activates protein kinase C isozymes δ and ε in cultured neonatal rat cardiac myocytes. J. Mol. Cell. Cardiol. 1999;31:1559–1572. doi: 10.1006/jmcc.1999.0993. [DOI] [PubMed] [Google Scholar]
- HILL J.A., KARIMI M., KUTSCHKE W., DAVISSON R.L., ZIMMERMAN K., WANG Z., KERBER R.E., WEISS R.M. Cardiac hypertrophy is not a required compensatory response to short-term pressure overload. Circulation. 2000;101:2863–2869. doi: 10.1161/01.cir.101.24.2863. [DOI] [PubMed] [Google Scholar]
- JELICKS L.A., SIRI F.M. Effects of hypertrophy and heart failure on [Na+]i in pressure-overloaded guinea pig heart. Am. J. Hypertens. 1995;8:934–943. doi: 10.1016/0895-7061(95)00219-F. [DOI] [PubMed] [Google Scholar]
- KUSUMOTO K., HAIST J.V., KARMAZYN M. Na+/H+ exchange inhibition reduces hypertrophy and heart failure after myocardial infarction in rats. Am. J. Physiol. 2001;280:H738–H745. doi: 10.1152/ajpheart.2001.280.2.H738. [DOI] [PubMed] [Google Scholar]
- MARANO G., PALAZZESI S., FADDA A., VERGARI A., FERRARI A.U. Attenuation of aortic banding-induced cardiac hypertrophy by propranolol is independent of β-adrenoceptor blockade. J. Hypertens. 2002;20:763–769. doi: 10.1097/00004872-200204000-00036. [DOI] [PubMed] [Google Scholar]
- MEGURO T., HONG C., ASAI K., TAKAGI G., MCKINSEY T.A., OLSON E.N., VATNER S.F. Cyclosporine attenuates pressure-overload hypertrophy in mice while enhancing susceptibility to decompensation and heart failure. Circ. Res. 1999;84:735–740. doi: 10.1161/01.res.84.6.735. [DOI] [PubMed] [Google Scholar]
- OIE E., BJORNERHEIM R., CLAUSEN O.P., ATTRAMADAL H. Cyclosporin A inhibits cardiac hypertrophy and enhances cardiac dysfunction during postinfarction failure in rats. Am. J. Physiol. 2000;278:H2115–H2130. doi: 10.1152/ajpheart.2000.278.6.H2115. [DOI] [PubMed] [Google Scholar]
- PENG M., HUANG L., XIE Z., HUANG W.H., ASKARI A. Partial inhibition of Na+/K+-ATPase by ouabain induces the Ca2+-dependent expressions of early-response genes in cardiac myocytes. J. Biol. Chem. 1996;271:10372–10378. doi: 10.1074/jbc.271.17.10372. [DOI] [PubMed] [Google Scholar]
- POGWIZD S.M., SIPIDO K.R., VERDONCK F., BERS D.M. Intracellular Na in animal models of hypertrophy and heart failure: contractile function and arrhythmogenesis. Cardiovasc. Res. 2003;57:887–896. doi: 10.1016/s0008-6363(02)00735-6. [DOI] [PubMed] [Google Scholar]
- SCHOLZ W., ALBUS U., COUNILLON L., GOGELEIN H., LANG H.J., LINZ W., WEICHERT A., SCHOLKENS B.A. Protective effects of HOE642, a selective sodium–hydrogen exchange subtype 1 inhibitor, on cardiac ischaemia and reperfusion. Cardiovasc. Res. 1995;29:260–268. [PubMed] [Google Scholar]
- TAKEWAKI S., KURO-O M., HIROI Y., YAMAZAKI T., NOGUCHI T., MIYAGISHI A., NAKAHARA K., AIKAWA M., MANABE I., YAZAKI Y., NAGAI R. Activation of Na+–H+ antiporter (NHE-1) gene expression during growth, hypertrophy and proliferation of the rabbit cardiovascular system. J. Mol. Cell. Cardiol. 1995;27:729–742. doi: 10.1016/s0022-2828(08)80063-6. [DOI] [PubMed] [Google Scholar]
- VERDONCK F., VOLDERS P.G., VOS M.A., SIPIDO K.R. Increased Na+ concentration and altered Na/K pump activity in hypertrophied canine ventricular cells. Cardiovasc. Res. 2003;57:1035–1043. doi: 10.1016/s0008-6363(02)00734-4. [DOI] [PubMed] [Google Scholar]
- YAMAZAKI T., KOMURO I., KUDOH S., ZOU Y., NAGAI R., AIKAWA R., UOZUMI H., YAZAKI Y. Role of ion channels and exchangers in mechanical stretch-induced cardiomyocyte hypertrophy. Circ. Res. 1998;82:430–437. doi: 10.1161/01.res.82.4.430. [DOI] [PubMed] [Google Scholar]
- YOKOYAMA H., YASUTAKE M., AVKIRAN M. α1-adrenergic stimulation of sarcolemmal Na+–H+ exchanger activity in rat ventricular myocytes: evidence for selective mediation by the α1A-adrenoceptor subtype. Circ. Res. 1998;82:1078–1085. doi: 10.1161/01.res.82.10.1078. [DOI] [PubMed] [Google Scholar]