

Abstract
The effects of donepezil, one of the most common cholinesterase inhibitors used for treatment of Alzheimer's disease, were studied on nicotinic receptors (nAChRs)-mediated postsynaptic currents, in dopaminergic neurons of the substantia nigra pars compacta, using the patch-clamp recording technique in slice preparations.
Donepezil (10–100 μM) selectively and reversibly depressed nicotine currents, induced by brief puffer pulses, through a glass micropipette positioned above the slice.
The peak amplitude fading of the responses generated by repeated test applications of low doses of nicotine was accelerated by donepezil, while it slowed the recovery of nicotine currents after a large, desensitising, dose of the same agonist.
Donepezil depressed even maximal responses to nicotine, revealing a noncompetitive mechanism of action; moreover, the inhibition of nAChRs was voltage and time independent.
Pretreatment with vesamicol or methamidophos did not prevent the reduction of nicotine-induced currents. The data indicated direct effect on nAChR, independent from the activity of donepezil as cholinesterase inhibitor.
Keywords: Donepezil, neuronal nicotinic receptors, dopaminergic neuron, acetylcholinesterase, allosteric modulator, Alzheimer's disease, Parkinson's disease, vesamicol, patch clamp, desensitisation
Introduction
Neuronal nicotinic acetylcholine receptors (nAChRs) belong to a family of ACh-gated cationic channels consisting of different subtypes with distinct anatomical distribution in the vertebrate central and peripheral nervous system (Paterson & Nordberg, 2000). Current interest in central nAChRs has been prompted by their involvement in a large number of neuropsychiatric disorders such as Alzheimer's disease (AD), Parkinson's disease (PD), epilepsy, addiction and schizophrenia (Perry et al., 1995; Paterson & Nordberg, 2000). Despite their different pathogeneses, these diseases share a common neurochemical deficit: a dysfunction of nAChRs, which is probably responsible for part of the clinical symptomatology (Paterson & Nordberg, 2000; Picciotto & Zoli, 2002). In particular, an impairment of cholinergic function has been associated with AD (Perry et al., 1995; Palmer, 2002) and PD (Quik & Kulak, 2002). Therefore, the most common strategy to reduce clinical symptoms and ameliorate AD is to amplify the extracellular concentration of the endogenous neurotransmitter ACh, using acetylcholinesterase inhibitors such as donepezil, physostigmine, tacrine and galanthamine (Svensson & Nordberg, 1996; Maelicke et al., 2000; Pereira et al., 2002; Prince et al., 2002; Santos et al., 2002). When considering the role of cholinergic dysfunction in PD, cumulative evidence indicates that stimulation of nicotinic receptors in the basal ganglia results in functional consequences that include the control of locomotor activity and protection against nigrostriatal degeneration. Restoring the physiological activity of the cholinergic system may thus represent an important strategy for the symptomatic treatment of Parkinson's disease, or for long-term neuroprotection (Quik & Kulak, 2002). Furthermore, it has been shown that the AChE inhibitor donepezil, currently used in the treatment of Alzheimer's disease, increases the cognitive performances of PD patients with cognitive impairment (Aarsland et al., 2002).
Nevertheless, it has been widely demonstrated that some of the drugs that are commonly used to enhance the ACh levels in the brain, namely AChE inhibitors, exert complex action on the cholinergic system. In fact, while they inhibit AChE activity in the brain, they also interact, at higher concentration, with the nicotinic receptor itself, via complex mechanisms of action. For instance, galanthamine and physostigmine have been classified as allosteric potentiating ligands of nAChRs (Pereira et al., 2002), while tacrine has been demonstrated to behave as an open-channel blocker of such receptors (Prince et al., 2002). Recent data have shown that donepezil, one of the most commonly used AChE inhibitors in the AD therapy, produces a concentration-dependent inhibition of ACh-evoked nicotinic responses in HEK-293 cells expressing the human α4β2 nAChR (Samochocki et al., 2003). However, the mechanism accounting for this inhibition and whether this behaviour is observed in the native receptor from the brain is still unknown.
Dopaminergic neurones of the substantia nigra pars compacta (SNc) express high postsynaptic levels of the typical central nicotinic α4β2 receptor (Klink et al., 2001), that can be activated and desensitised by nicotine (Calabresi et al., 1989; Pidoplichko et al., 1997; Wooltorton et al., 2003). The consideration that partial loss of central nicotinic receptors occurs in Parkinson's disease, together with reports that nicotine treatment relieves some of the symptoms of this disorder (Quik & Kulak, 2002; Paterson & Nordberg, 2000), makes dopaminergic neurones of the SNc a useful model to study the interaction of donepezil with native nAChRs.
Methods
Slice preparation for electrophysiology
Wistar rats, 4–5-weeks old, were anaesthetised with halothane and subsequently killed by decapitation. All experiments followed international guidelines on the ethical use of animals from the European Communities Council Directive of 24 November 1986 (86/609/EEC). The brain was rapidly removed from the skull and horizontal midbrain slices (240 μm) were cut in cold (8–12°C) artificial cerebrospinal fluid, using a vibratome, and left to recover at 34°C for at least 1 h. Slices were separately placed in a recording chamber, on the stage of an upright microscope (Olympus BX50WI) and submerged in a continuously flowing (2.5 ml min−1) solution at 34°C. Artificial cerebrospinal fluid (ACSF) composition was the following (in mM): NaCl, 126; KCl, 2.5; MgCl2, 1.2; CaCl2, 2.4; NaH2PO4, 1.2; NaHCO3, 19; glucose, 11; saturated with 95% O2, 5% CO2 (pH 7.4).
Patch-clamp recordings
Neurones were visualised with infrared Nomarski video microscopy. Patch-clamp recordings were obtained using glass electrodes (3–4 MΩ) filled with (in mM): 115 K-methylsulphate, 20 NaCl, 1.5 MgCl2, 5 HEPES, 0.1 EGTA, 2 ATP, 0.5 GTP (pH 7.3, with KOH). Membrane currents were recorded with a patch-clamp amplifier (Axopatch 1D; Axon Instruments, U.S.A.), filtered at 1 kHz, digitised (10 kHz) and stored on computers using the pClamp9 software (Axon Instruments). Dopaminergic neurones were identified electrophysiologically on the basis of a prominent hyperpolarisation-activated current Ih at negative voltage steps, and a typical voltage sag when negative current steps were applied in the current-clamp mode (Mercuri et al., 1995).
Drugs and application method
Donepezil, methamidophos (Fluka) and Vesamicol (Tocris) were applied to the slice via the perfusion system. In order to minimise receptor desensitisation, nicotine (Sigma) was delivered by pressure application (10–20 psi) from glass micropipette positioned over the slice in correspondence to the recorded neurone (Di Angelantonio & Nistri, 2001). Donepezil hydrochloride was a gift from Professor M.A. Sortino.
Evoked synaptic currents
Excitatory postsynaptic currents (EPSCs) were evoked in dopaminergic cells using a bipolar Ni/Cr stimulating electrode, placed 50–100 μm rostral to the recording electrode. To evoke a stable EPSC, each stimulus of 150–300 μs at 20–50 mV was delivered every 30 s. In order to block the fast GABAergic synaptic currents, picrotoxin was applied (100 μM).
Electrophysiological data analysis
Data are presented as mean±s.e.m., with statistical significance assessed using Wilcoxon test (for nonparametric data) or paired t-test (for normally distributed data). A value of P<0.05 was accepted as indicative of a statistically significant difference. Data represented in the dose–response curves are derived from repeated experiments; on each cell, all doses of nicotine or donepezil were applied. The IC50 values (concentration producing 50% reduction in nicotine current amplitude) for donepezil block were calculated using the following equation:
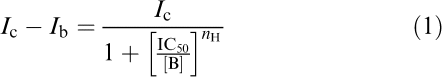 |
where Ib and Ic are amplitudes of blocked and control currents, [B] the donepezil concentration and nH the Hill coefficient (Origin 6.0, Microcal, Northampton, MA, U.S.A.); zero for the fit was set when, in the absence of agonist, the holding current was unchanged.
Results
Modulation of nicotine responses by donepezil
When nicotine was applied onto a dopaminergic neurone via a puffer pipette positioned above the slice, a rapid inward current developed, mediated by the activation of postsynaptic nAChRs. The current was indeed blocked by the nAChRs antagonist dihydro-β-erythroidine, and was left unchanged by applying tetrodotoxin 1 μM.
Figure 1a shows inward currents generated by nicotine applied on a dopaminergic neurone, via brief (100 ms) pressure pulses from a glass pipette filled with nicotine 1 mM (final dilution in ACSF), to minimise rapid desensitisation (Khiroug et al., 1997; Di Angelantonio & Nistri, 2001) (Figure 1a, left). When the same pulse was delivered in the presence of the acetylcolinesterase inhibitor donepezil (100 μM; bath applied for 5 min), the inward current was reduced (53%; Figure 1a, middle), without any direct action of donepezil on the resting membrane conductance or holding current. On average, the current was reduced to 57±3% of the control (n=16) and the depression was reversible on donepezil washout (Figure 1a, right).
Figure 1.

Depression of nicotine-induced responses by donepezil. (a) Current records obtained with 100 ms nicotine (1 mM pipette concentration; left), 3 min after starting bath application of donepezil (100 μM; middle) and 2 min after donepezil washout. Note the reversible reduction in nicotine current amplitude. (b) Time course of depression of nicotine currents (1 mM pipette concentration, 100 ms application) after application of 100 μM donepezil to six neurones. (c) Plot of the fractional reduction in current amplitude against different log concentrations of donepezil (ranging from 1 to 200 μM). The test pulse (50 ms, 1 mM) of nicotine was the same for all concentrations of donepezil (n=4, 20 cells). The calculated IC50 value for donepezil was 85±10 μM. (d) Current-clamp recording from one DA neurone on which 100 ms nicotine puff induces firing of three action potentials in control; 30 μM donepezil application reduces to one action potential the response to the same nicotine puffer pulse after 1 min, and to zero after 3 min. Partial recovery is shown after 3 min washout.
Dynamics of nicotine current reduction by donepezil
Figure 1b shows the time course of donepezil-induced depression for six neurones. It is noteworthy that, after 2.5 min of donepezil application, the extent of depression reached a steady state and that recovery was achieved 3 min after drug washout.
Figure 1c shows a plot of the fractional reduction in current amplitude against different log concentrations of donepezil. Donepezil concentrations (ranging from 1 to 200 μM) were tested on responses evoked by the same pulse duration of nicotine (50 ms, 1 mM; n=10). From these data, the calculated IC50 value for donepezil was 85±10 μM. This value is far from the inhibitory potency of donepezil towards AChE activity, that is 6.7 nM in vitro (Ogura et al., 2000). This behaviour is reminiscent of other AChE inhibitors, such as galantamine and physostigmine, that bind to and modulate nAChR at concentrations higher with respect to their potency in inhibiting AChE (Samochocki et al., 2003).
Donepezil makes dopaminergic neurones less excitable in the presence of nicotine
Additional support for a direct effect of donepezil on nAChR activity was obtained from current-clamp recordings of dopaminergic neurons of the SNc. The bath solution contained 3 μM atropine to block the effect of free ACh on muscarinic receptor when inhibiting AChE. In six of six cells tested, short (50–500 ms) puffer pulses of nicotine increased the firing frequency to 150±35% (in accordance with Mansvelder & McGehee, 2002), but they also caused strong membrane depolarisation and consequent block of firing. For this reason, cells were manually hyperpolarised to −70 mV with DC current injection. In the example given in Figure 1d, the cell hyperpolarised manually to –70 mV fired three action potentials (APs) in response to 100 ms puff of nicotine. After 1 min of donepezil application (30 μM), the same nicotine puff evoked one AP, and after 3 min of application the cell was not any more able to fire APs. After donepezil washout, the cell gradually recovered its original firing pattern (Figure 1d). This result was reproduced in six cells.
Donepezil action depended on nicotine dose, but not on membrane potential
Further tests were performed to characterise the mechanism underlying the depression by donepezil of nicotine-mediated currents. Figure 2a shows that increasing the duration (10–5000 ms) of 1 mM nicotine pulses yielded a progressively larger current with saturation at 1 s pulses. When the same protocol was repeated in the presence of 100 μM donepezil (5 min bath preapplication), currents were reduced at each tested nicotine pulse duration. Thus, the plot was downward shifted by the application of donepezil, a pattern of action which could account for a noncompetitive antagonism over nAChRs. Taking the average responses at approximately the midpoint of the curve (100 ms), 100 μM donepezil application gave a 40±10% depression from control conditions (n=12, P<0.05 for all nicotine doses). We tested the possibility that the puffer application of nicotine could wash donepezil off from the cell, especially for longer application of nicotine, by changing the puffer pipette during the experiment. The first puffer pipette, filled with 1 mM nicotine, was used for monitoring the cell in control and in donepezil. In control, condition 1 s pulse of nicotine was elicited and an inward current of 564±30 pA; when the steady state of donepezil application was reached, the same application of nicotine elicited a current of 440±40 pA. The reduction in the peak current was then 22±6% (n=3). At this time, the puffer pipette was changed with a second one filled with 1 mM nicotine plus 100 μM donepezil that was positioned nearly at the same distance from the recorded neuron. The accessibility of the cell in the slice was the limiting factor for using this protocol; however, in three cells 1 s application of nicotine plus donepezil gave a current of 396±40 pA, very similar to the one obtained with nicotine alone. This indicates that the puffer application of nicotine did not wash off donepezil from the recorded cell.
Figure 2.

Donepezil allosterically modulates nicotinic receptors. (a) Plot of nicotine current amplitude versus increasing duration of nicotine pressure pulses in control solution and in the presence of donepezil. Ordinate, current amplitude normalised with respect to the response evoked by 50 ms in control solution for each neurone. Abscissa, pulse duration of nicotine (1 mM) applications. Donepezil (100 μM) was applied for ∼5 min (n=12). Note that the data points for nicotine in donepezil solution (filled circles) differed significantly from the corresponding controls (filled squares) with P<0.01 for 10–500 ms, P<0.05 for 1, 2 and 5 s. (b) Bar chart showing an equivalent degree of peak current depression exerted by donepezil at three different holding potential (−30, −60, −90 mV, n=5). (c) Superimposed current response in control (black) and in the presence of 10 μM donepezil (grey) to repeated pulses of nicotine (0.01 Hz). Note that, in the presence of donepezil, the extent of desensitisation is more pronounced. (d) Plot of current peak amplitude, normalised with respect to the first response to nicotine, in control condition and in the presence of low donepezil doses (10 μM) for a 0.1 Hz pulse application. (e) Histograms of averaged τ values for current amplitude decay for repetitive puffer pulses in control and in the presence of 10 μM donepezil. Note that donepezil enhances the desensitisation induced by repetitive stimulation.
We then explored the possibility that the antagonism exerted by donepezil could be altered when the cell membrane potential was changed, as would be expected for an open-channel blocker. Donepezil is a tertiary amine with a pKa value of 8.82, which means that, at pH=7.4, 96% of this compound will be protonated, making possible its interaction with the strong negative charges inside the nicotinic channel.
However, histograms in Figure 2b show that donepezil elicited a similar reduction in nicotine current amplitude at −90, −60 or −30 mV holding potential. On average, the depression at −90 mV was 45±7%, a value thus not significantly different (n=5, P>0.05) from that observed at −60 mV (39±5%), and from that observed at –30 mV (43±10%). These data, therefore, suggested that the block by donepezil of nicotinic receptor-mediated responses was voltage independent, making unlikely the possibility of a channel block by donepezil.
Donepezil facilitates nAChR desensitisation
A mechanism that could account for the reduction of nicotine-induced currents exerted by donepezil is facilitation of the desensitisation process, as proposed for substance P (Clapham & Neher, 1984; Simmons et al., 1990; Valenta et al., 1993). This process could account for the depression observed also for responses induced by large doses of nicotine (33±6% for 2 s pulse), which are known to be prone to desensitisation (Valenta et al., 1993; Khiroug et al., 1997, 1998). Figure 2c shows an example of superimposed current responses to repeated nicotine pulses (50 ms, 1 mM, 10 s intervals). When this protocol was applied in the presence of low doses of donepezil (10 μM, 5 min preincubation), the extent of desensitisation was more pronounced. Namely, the Ilast/Ifirst ratio was 0.78 in control and 0.18 in the presence of donepezil, even if the peak amplitude induced by the first nicotine pulse was unchanged. The inset in Figure 2c shows the response to nicotine in control and in donepezil at the end of the desensitisation protocol, superimposed and scaled to the peak. The deactivation of the current evoked by brief puffer pulses of nicotine (50 ms) was left unchanged by donepezil application, indicating that the reduction in the peak amplitude was not due to a change in the kinetics of the channel. Figure 2d shows the averaged data, normalised with respect to the first response to nicotine (n=5), for repetitive nicotine pulse applications (0.1 Hz) in control condition and in the presence of donepezil 10 μM. For each neuron, in which this protocol was applied, taking the peak amplitude of five currents elicited by the same 50 ms pulse of nicotine at a frequency of 0.1 Hz, the time course of peak amplitude reduction in control and in the presence of donepezil were plotted versus time, and the resulting curve was fitted by a monoexponential decay function. Averaged τ values are reported in the histograms in Figure 2e. A highly significant decrease of τ values from 62±16 to 8±1 s (P<0.01, n=5) was observed in the presence of 10 μM donepezil.
Donepezil accelerates fading of desensitised responses to nicotine
In order to better characterise how donepezil could interfere with the process of nAChR desensitisation, we induced receptor desensitisation using the classical protocol of Katz & Thesleff (1957), in control and in the presence of 30 μM donepezil. The protocol consisted of repeated test applications (every 30 s) of a nondesensitising dose of nicotine (50 ms, 1 mM pipette concentration, Figure 3a left, first arrow), followed by a conditioning dose (2 s pulse, 1 mM nicotine) of the same drug, which elicits a desensitising inward current (for details, see Khiroug et al., 1998). After this conditioning pulse, the test pulse was resumed at the same rate to monitor the time course of nAChR recovery from desensitisation. When the same protocol was applied in the presence of 30 μM donepezil (Figure 3a; bottom traces), the peak amplitude of currents was depressed as expected (cf. Figure 1); in addition, the fading of the response to the conditioning pulse was faster and the recovery from desensitisation largely delayed. The fading of the 2 s nicotine-evoked currents, in control and in the presence of 30 μM donepezil, were fitted by a monoexponential function. In control condition, the τ value (τdecay−2 s pulse) was 1205±111 ms, while in the presence of donepezil (30 μM preapplied for 5 min) was significantly faster 597±180 ms (n=6, P<0.01; Figure 3b).
Figure 3.

Donepezil promotes nAChR desensitisation. (a) Current records induced by nicotine obtained with a conditioning pulse protocol consisting of repeated test applications of a nondesensitising dose of nicotine, before (first trace) and after a conditioning (desensitising; 2 s) dose, to monitor the time course of nAChR recovery from desensitisation. This protocol was tested in control conditions (top traces) and in the presence of donepezil 30 μM (bottom traces). In the presence of donepezil, the current peak amplitude is depressed and the extent and time course of recovery from desensitisation is largely delayed. (b) Bar chart showing that donepezil produces a significant acceleration of current fading, for desensitising nicotine pulses (2 s). (c) Average time course of recovery from desensitisation (evoked by 2 s nicotine application) obtained from a sample of six cells in control conditions and in the presence of 30 μM donepezil. (d) Average values of τrecovery obtained by fitting with an exponential function recovery from desensitisation for each neurone (n=6). Note that, in the presence of donepezil, the τrecovery was significantly shortened (from 132±13 to 59±9s).
Recovery from desensitisation is delayed by donepezil
Recovery from desensitisation was also significantly reduced by donepezil (Figure 3a). This was calculated by plotting the ratio I/Itest versus time, where I and Itest were the nicotinic currents evoked after and before the conditioning pulse, respectively (Figure 3c). The time course of the recovery was fitted by a monoexponential function characterised by a τ value (τrecovery). The averaged values of τrecovery in control and in the presence of donepezil (30 μM) show significant reduction from 132±13 s (control) to 59±9 s (donepezil, P<0.01, n=6). These data indicate that donepezil interacts with both onset and offset of the nAChRs desensitisation process (Figure 3d).
Action of donepezil on nAChRs is independent from the block of AChE
In order to analyse if the facilitation of nAChR desensitisation displayed by donepezil was due to an increased level of free ACh in the tissue, or to a direct allosteric modulation of nicotinic receptor, slices were depleted of ACh by treatment with vesamicol 5 μM (Zhou et al., 2001; 2002). Figure 4A, B shows that 5 μM vesamicol was effective in depleting ACh.
Figure 4.

In the presence of vesamicol, donepezil still reduces nicotine currents but not EPSCs. (A) Representative experiment showing the effect of blocking AChE with donepezil on EPSCs, recorded in control condition and in the presence of 5 μM vesamicol. (A) When donepezil was applied in control condition, the higher levels of free ACh produce a reduction of EPSCs. (B) When the same protocol was applied in the presence of 5 μM vesamicol (preapplied for 15 min), donepezil application did not affect EPSCs amplitude. (B) Averaged EPSCs amplitude in control, and in the presence of 100 μM donepezil, before and during treatment with 5 μM vesamicol (n=5). (C) Current records obtained with 50 ms pulses of 1 mM nicotine. Donepezil depresses nicotine-evoked current in control condition and in 5 μM vesamicol. (D) Average value of peak amplitude reduction obtained with donepezil in control conditions and in the presence of 5 μM vesamicol. Note that the extent of depression does not depend on the block of AChE; data are from six neurones.
It has been previously shown that AChE inhibition leads to a reduction of evoked EPSCs in dopaminergic neurons of the SNc, due to the activation of presynaptic muscarinic receptors (Grillner et al., 1999). Accordingly, donepezil 100 μM reversibly depressed EPSCs to 75±2% (n=5, P<0.05) of control. Figure 4A, B shows that, after donepezil washout (10 min) and 15 min incubation with 5 μM vesamicol (dashed line), the same application of donepezil did not affect EPSCs, indicating that no tonic ACh was released under vesamicol (97±2%, n=5, P>0.05). Conversely in the presence of 5 μM vesamicol, donepezil was still effective in reducing nicotine-induced currents (Figure 4C). Moreover, the extent of depression for all donepezil concentrations tested was very similar in control and after vesamicol application, as shown by histograms in Figure 4D (n=6). Finally, on these cells, the treatment with vesamicol did not interfere with the facilitation of desensitisation, as indicated by the significant shortening of the τdecay-2 s pulse value, that was 1053±124 ms in vesamicol, and 620±80 ms when 30 μM donepezil was added (n=5). This acceleration of the decay was very similar to the one observed with donepezil alone, suggesting that donepezil directly interacted with nAChRs, via a mechanism different from the AChE block. This action may be carried out by binding to an allosteric site of the nAChR itself, and then facilitating the desensitisation process of such receptors (Valenta et al., 1993; Lester & Dani, 1995; Khiroug et al., 1998; Dani et al., 2000; Quick & Lester, 2002). In order to address more directly this hypothetical interaction between donepezil and the desensitisation process of nAChR, we tested the ability of donepezil to affect responses elicited by carbachol, an agonist of nAChRs, which is less prone to desensitisation. In the presence of 3 μM atropine, to block muscarinic responses, charbachol-induced current (3 mM pipette concentration, 200 ms pulse) was 41±6 pA (n=4). When, on the same cells, the same pulse of carbachol was applied in the presence of donepezil 100 μM, the current was 24±4 pA. The reduction of the peak current was then 41±4% (n=4), indicating that, besides altering the kinetics of desensitisation, donepezil is also inhibiting the function of the channel in other ways.
Specificity of donepezil action
In order to address the specificity of donepezil action, we examined whether donepezil affected the postsynaptic responses to AMPA, another fast-acting receptor channel agonist (Gotz et al., 1997). When AMPA was delivered via puffer application onto four dopaminergic neurons of the SNc (10 μM; pipette concentration, 500 ms), inward currents were recorded. Bath application of 100 μM donepezil did not produce any depression of these currents (98±5%, n=4; data not shown).
Pretreatment with methamidophos does not prevent donepezil inhibition of nAChRs
It is well known that organophosphoric agents completely and irreversibly bind to the active site of AChE (Aldridge & Reiner, 1969; Aldridge, 1981). Taking advantage of this properties, we examined the action of donepezil on nAChRs under a complete inhibition of AChE by methamidophos (Camara et al., 1997). Methamidophos (200 μM) was preapplied to the slice for 15 min, and then washed before donepezil application. Figure 5a shows a representative experiment in which the application of 100 μM donepezil, after AChE block, reduced the nicotine-induced current in a reversible manner, by 51%. On a sample of six cells, the same application of 100 μM donepezil, after AChE block, caused a reduction of 45±10% (n=6, P<0.05; Figure 5b). When donepezil was applied onto dopaminergic cells in untreated slices, the reduction in nicotine current amplitude was 44±6 % (n=12), a value very similar to that obtained for treated slices (Figure 5c). These data confirm that donepezil, besides inhibiting AChE, does also exert a direct antagonism over postsynaptic nAChRs.
Figure 5.

Pretreatment with methamidophos does not prevent donepezil reduction of nicotine currents. (a) Current records evoked by 50 ms pulse of nicotine (1 mM) after pretreatment with 200 μM methamidophos, during application of 100 μM donepezil, and after washout. Note that donepezil was still able to depress nicotine-evoked current in a reversible manner even following pretreatment with methamidophos. (b) Histograms plotting the average reduction of nicotine-evoked currents when donepezil was applied after methamidophos; data are from five neurons. (c) Histograms summarising the average reduction in nicotine current amplitude in the presence of donepezil for control slices and for slices pretreated with methamidophos.
Discussion
The main finding of the present study is the demonstration of an allosteric modulation by donepezil of neuronal nAChRs in SNc dopaminergic neurones. This was evidenced by a rapid onset and agonist-insurmountable inhibition of inward currents evoked by pulse applications of nicotine. Such an effect was distinct from the inhibition of AChE exerted by donepezil, in view of its persistence even in the absence of free ACh or when AChE had previously broken down. The interaction of donepezil with central nicotinic receptors suggests that it may play an important role in the modulation of fast neuronal signalling. Donepezil action appeared to be specific for nAChRs, since AMPA receptor-mediated responses were insensitive to this drug, in the same neurones.
Characteristics of the action of donepezil on nicotine-mediated responses
Donepezil strongly depressed the inward currents induced by nicotine without changing the baseline-holding current of the dopaminergic cells. The extent of inhibition was unrelated to AChE inhibition, since depletion of ACh with vesamicol did not prevent donepezil action. This suggests that donepezil interacts directly with the nicotinic receptors.
Donepezil, a tertiary amine which is almost fully protonated at physiological pH, may block receptor channels opened by nicotine in a manner similar to that of other substances, like local and general anaesthetics (Neher & Steinbach, 1978; Mori et al., 2001), tacrine (Prince et al., 2002), or mecamylamine (Giniatullin et al., 2000). However, this mechanism seems unlikely, since donepezil effect was voltage-independent throughout a wide range of membrane potentials.
The use of nonequilibrium responses to nicotine, and the puffer-application protocol strictly precluded quantitative pharmacological analysis of donepezil antagonism. Recent work, however, has indicated that the amount of agonist delivered by 10- to 50-ms puffer (1 mM) application closely corresponds to superfusing 20–100 μM nicotine (Di Angelantonio & Nistri, 2001), thus, providing a relatively narrow range of agonist concentrations. On the other hand, an advantage in the use of puffer application of agonist is that, with short pressure applications, receptor desensitisation is minimised and the agonist can be quickly delivered, to mimic the natural course of action of the endogenous transmitters.
Even with the interpretation constraints imposed by using nonequilibrium responses to brief pulses of nicotine, it was clear that donepezil blocked all responses to nicotine and that increasing the amount of nicotine delivered to the cell did not counteract the inhibitory effect of donepezil. Indeed, the graph plotting the fractional response amplitude versus the amount of nicotine delivered by pressure pulse showed a downward shift in the presence of donepezil. This observation is consistent with a noncompetitive antagonism of donepezil on nicotinic receptors.
A process that could account for the effect of donepezil is the facilitation of desensitisation, as proposed for substance P (Clapham & Neher, 1984; Simmons et al., 1990; Valenta et al., 1993). Since nAChRs undergo profound desensitisation (for a review, see Quick & Lester, 2002), this process appears to be one potential target for the inhibitory action by donepezil. In general, nAChR desensitisation is observed as a decline in the macroscopic current response during continuous exposure to neurotransmitter, with an onset kinetics that depends on agonist exposure time and concentration. It has long been thought that neurones may utilise desensitisation to regulate receptor function, although it is not clear how extensive this phenomenon occurs under physiological conditions (Huganir & Greengard, 1990).
In agreement with this hypothesis, we found that responses induced by large doses of nicotine, which are more prone to desensitisation (Valenta et al., 1993; Lester & Dani, 1995; Khiroug et al., 1997; 1998; Quick & Lester, 2002), were inhibited by donepezil.
Moreover, in the presence of donepezil, responses induced by nicotine showed acceleration of the current decay and reversible depression to subsequent application of the same agonist. When the process of desensitisation was induced using the classical protocol of Katz & Thesleff (1957), not only was the peak current depressed in the presence of donepezil, but also recovery from desensitisation was largely delayed.
From these observations, the emerging pattern of donepezil action is binding to an allosteric site on the nAChR, to generate transient downregulation of nicotinic receptor activity.
Physiological and clinical implication
When trying to predict the clinical efficacy by extrapolating from in vitro results, it is important to consider the concentration of the drug that can be achieved in in vivo conditions. Pharmacokinetic study suggests that the clinically achievable concentration of donepezil are similar to the concentrations of the drug used in this study; in fact, the steady-state plasma concentration (Cmax) of donepezil in patients repeatedly treated with this drug at the oral dose of 10 mg kg−1 for 28 days was 1127.8 ng h ml−1) (Tiseo et al., 1998). Taking into account that, in rodents, the concentration of donepezil in the brain may be up to 10 times higher than in the plasma (Kosasa et al., 2000), it is conceivable to hypothesise a brain concentration in humans between 1 and 10 μM.
Our experimental evidence of a direct interaction of donepezil with the nicotinic receptors adds new insights into the mechanism of action of this drug. This has to be taken into account, especially when considering the large clinical use of this cholinesterase inhibitor in the treatment of the early phases of AD (Palmer, 2002), and of the cognitive impairment in PD (Aarsland et al., 2002). While donepezil should indeed raise the levels of free ACh (pathologically low in AD patients) by acting as a cholinesterase inhibitor, it might also induce a parallel nicotinic receptor desensitisation, thus promoting adaptive brain processes.
Since all nAChRs are Ca2+ permeable (McGehee & Role, 1995), a donepezil-induced receptor inhibition could prevent neuronal toxicity. In line with this hypothesis, Akasofu et al. (2003) found that 10 μM donepezil exerted a neuroprotective effect against oxygen glucose deprivation in rat cortical neurones. Other AChE inhibitors did not share this effect.
More interestingly, donepezil might have a therapeutic use in heavy smokers, by diminishing the excitatory effects of nicotine on dopaminergic neurones in the ventral midbrain, hence decreasing the rewarding effects of nicotine (Mansvelder & McGehee, 2002). In fact, current-clamp recordings, showed that APs firing evoked by nicotine was suppressed by donepezil, independently from muscarinic receptor activity, indicating that donepezil makes dopaminergic neurones less excitable by nicotine.
Acknowledgments
We thank Dr Nicola Berretta for carefully reading this manuscript. This work was supported by grants from the MIUR (FIRB proneuro) and Minustero della Salute (Ra00.86).
Abbreviations
- Ach
acetylcholine
- AChE
acetylcholinesterase
- AD
Alzheimer's disease
- AP
action potential
- EPSCs
excitatory postsynaptic currents
- nAChRs
neuronal nicotinic acetylcholine receptors
- PD
Parkinson's disease
- SNc
substantia nigra pars compacta
References
- AARSLAND D., LAAKE K., LARSEN J.P., JANVIN C. Donepezil for cognitive impairment in Parkinson's disease: a randomised controlled study. J. Neurol. Neurosurg. Psychiatry. 2002;72:708–712. doi: 10.1136/jnnp.72.6.708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AKASOFU S., KOSASA T., KIMURA M., KUBOTA A. Protective effect of donepezil in a primary culture of rat cortical neurons exposed to oxygen-glucose deprivation. Eur. J. Pharmacol. 2003;472:57–63. doi: 10.1016/s0014-2999(03)01865-x. [DOI] [PubMed] [Google Scholar]
- ALDRIDGE W.N. Organophosphorus compounds: molecular basis for their biological properties. Sci. Prog. 1981;67:131–147. [PubMed] [Google Scholar]
- ALDRIDGE W.N., REINER E. Acetylcholinesterase. Two types of inhibition by an organophosphorus compound: one the formation of phosphorylated enzyme and the other analogous to inhibition by substrate. Biochem. J. 1969;115:147–162. doi: 10.1042/bj1150147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CALABRESI P., LACEY M.G., NORTH R.A. Nicotinic excitation of rat ventral tegmental neurones in vitro studied by intracellular recording. Br. J. Pharmacol. 1989;98:135–140. doi: 10.1111/j.1476-5381.1989.tb16873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMARA A.L., BRAGA M.F., ROCHA E.S., SANTOS M.D., CORTES W.S., CINTRA W.M., ARACAVA Y., MAELICKE A., ALBUGUERGUE E.X. Methamidophos: an anticholinesterase without significant effects on postsynaptic receptors or transmitter release. Neurotoxicology. 1997;18:589–602. [PubMed] [Google Scholar]
- CLAPHAM D.E., NEHER E. Substance P reduces acetylcholine-induced currents in isolated bovine chromaffin cells. J.Physiol. 1984;347:255–277. doi: 10.1113/jphysiol.1984.sp015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DANI J.A., RADCLIFFE K.A., PIDOPLICHKO V.I. Variations in desensitization of nicotinic acetylcholine receptors from hippocampus and midbrain dopamine areas. Eur. J. Pharmacol. 2000;393:31–38. doi: 10.1016/s0014-2999(00)00003-0. [DOI] [PubMed] [Google Scholar]
- DI ANGELANTONIO S., NISTRI A. Calibration of agonist concentrations applied by pressure pulses or via rapid solution exchanger. J. Neurosci. Methods. 2001;110:155–161. doi: 10.1016/s0165-0270(01)00437-x. [DOI] [PubMed] [Google Scholar]
- GINIATULLIN R.A., SOKOLOVA E.M., DI ANGELANTONIO S., SKORINKIN A., TALANTOVA M.V., NISTRI A. Rapid relief of block by mecamylamine of neuronal nicotinic acetylcholine receptors of rat chromaffin cells in vitro: an electrophysiological and modeling study. Mol. Pharmacol. 2000;58:778–787. doi: 10.1124/mol.58.4.778. [DOI] [PubMed] [Google Scholar]
- GOTZ T., KRAUSHAAR U., GEIGER J., LUBKE J., BERGER T., JONAS P. Functional properties of AMPA and NMDA receptors expressed in identified types of basal ganglia neurons. J. Neurosci. 1997;17:204–215. doi: 10.1523/JNEUROSCI.17-01-00204.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRILLNER P., BONCI A., SVENSSON T.H., BERNARDI G., MERCURI N.B. Presynaptic muscarinic (M3) receptors reduce excitatory transmission in dopamine neurons of the rat mesencephalon. Neuroscience. 1999;91:557–565. doi: 10.1016/s0306-4522(98)00619-8. [DOI] [PubMed] [Google Scholar]
- HUGANIR R.L., GREENGARD P. Regulation of neurotransmitter receptor desensitization by protein phosphorylation. Neuron. 1990;5:555–567. doi: 10.1016/0896-6273(90)90211-w. [DOI] [PubMed] [Google Scholar]
- KATZ B., THESLEFF S. A study of the ‘desensitization' produced by acetylcholine at the motor end-plate. J. Physiol. (Lond.) 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHIROUG L., GINIATULLIN R., SOKOLOVA E., TALANTOVA M., NISTRI A. Imaging of intracellular calcium during desensitization of nicotinic acetylcholine receptors of rat chromaffin cells. Br. J. Pharmacol. 1997;122:1323–1332. doi: 10.1038/sj.bjp.0701518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KHIROUG L., SOKOLOVA E., GINIATULLIN R., AFZALOV R., NISTRI A. Recovery from desensitization of neuronal nicotinic acetylcholine receptors of rat chromaffin cells is modulated by intracellular calcium through distinct second messengers. J. Neurosci. 1998;18:2458–2466. doi: 10.1523/JNEUROSCI.18-07-02458.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KLINK R., DE KERCHOVE D.A., ZOLI M., CHANGEUX J.P. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J. Neurosci. 2001;21:1452–1463. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOSASA T., KURIYA Y., MATSUI K., YAMANISHI Y. Inhibitory effect of orauy administrated donepezil hydrocloride (E2020), a novel treatment for Alzheimer's disease, on cholinesterase activity in rats. Eur. J. Pharmacol. 2000;389:173–179. doi: 10.1016/s0014-2999(99)00876-6. [DOI] [PubMed] [Google Scholar]
- LESTER R.A., DANI J.A. Acetylcholine receptor desensitization induced by nicotine in rat medial habenula neurons. J. Neurophysiol. 1995;74:195–206. doi: 10.1152/jn.1995.74.1.195. [DOI] [PubMed] [Google Scholar]
- MAELICKE A., SCHRATTENHOLZ A., SAMOCHOCKI M., RADINA M., ALBUQUERQUE E.X. Allosterically potentiating ligands of nicotinic receptors as a treatment strategy for Alzheimer's disease. Behav. Brain Res. 2000;113:199–206. doi: 10.1016/s0166-4328(00)00214-x. [DOI] [PubMed] [Google Scholar]
- MANSVELDER H.D., MCGEHEE D.S. Cellular and synaptic mechanisms of nicotine addiction. J. Neurobiol. 2002;53:606–617. doi: 10.1002/neu.10148. [DOI] [PubMed] [Google Scholar]
- MCGEHEE D.S., ROLE L.W. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu. Rev. Physiol. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- MERCURI N.B., BONCI A., CALABRESI P., STEFANI A., BERNARDI G. Properties of the hyperpolarization-activated cation current Ih in rat midbrain dopaminergic neurons. Eur. J. Neurosci. 1995;7:462–469. doi: 10.1111/j.1460-9568.1995.tb00342.x. [DOI] [PubMed] [Google Scholar]
- MORI T., ZHAO X., ZUO Y., AISTRUP G.L., NISHIKAWA K., MARSZALEC W., YEH J.Z., NARAHASHI T. Modulation of neuronal nicotinic acetylcholine receptors by halothane in rat cortical neurons. Mol. Pharmacol. 2001;59:732–743. doi: 10.1124/mol.59.4.732. [DOI] [PubMed] [Google Scholar]
- NEHER E., STEINBACH J.H. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J. Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OGURA H., KOSASA T., KURIYA Y., YAMANISHI Y. Comparison of inhibitory activities of donepezil and other cholinesterase inhibitors on acetylcholinesterase and butyrylcholinesterase in vitro. Methods Find. Exp. Clin. Pharmacol. 2000;22:609–613. doi: 10.1358/mf.2000.22.8.701373. [DOI] [PubMed] [Google Scholar]
- PALMER A.M. Pharmacotherapy for Alzheimer's disease: progress and prospects. Trends Pharmacol. Sci. 2002;23:426–433. doi: 10.1016/s0165-6147(02)02056-4. [DOI] [PubMed] [Google Scholar]
- PATERSON D., NORDBERG A. Neuronal nicotinic receptors in the human brain. Prog. Neurobiol. 2000;61:75–111. doi: 10.1016/s0301-0082(99)00045-3. [DOI] [PubMed] [Google Scholar]
- PEREIRA E.F., HILMAS C., SANTOS M.D., ALKONDON M., MAELICKE A., ALBUQUERQUE E.X. Unconventional ligands and modulators of nicotinic receptors. J. Neurobiol. 2002;53:479–500. doi: 10.1002/neu.10146. [DOI] [PubMed] [Google Scholar]
- PERRY E.K., MORRIS C.M., COURT J.A., CHENG A., FAIRBAIRN A.F., MCKEITH I.G., IRVING D., BROWN A., PERRY R.H. Alteration in nicotine binding sites in Parkinson's disease, Lewy body dementia and Alzheimer's disease: possible index of early neuropathology. Neuroscience. 1995;64:385–395. doi: 10.1016/0306-4522(94)00410-7. [DOI] [PubMed] [Google Scholar]
- PICCIOTTO M.R., ZOLI M. Nicotinic receptors in aging and dementia. J. Neurobiol. 2002;53:641–655. doi: 10.1002/neu.10102. [DOI] [PubMed] [Google Scholar]
- PIDOPLICHKO V.I., DEBIASI M., WILLIAMS J.T., DANI J.A. Nicotine activates and desensitizes midbrain dopamine neurons. Nature. 1997;390:401–404. doi: 10.1038/37120. [DOI] [PubMed] [Google Scholar]
- PRINCE R.J., PENNINGTON R.A., SINE S.M. Mechanism of tacrine block at adult human muscle nicotinic acetylcholine receptors. J. Gen. Physiol. 2002;120:369–393. doi: 10.1085/jgp.20028583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- QUICK M.W., LESTER R.A. Desensitization of neuronal nicotinic receptors. J. Neurobiol. 2002;53:457–478. doi: 10.1002/neu.10109. [DOI] [PubMed] [Google Scholar]
- QUIK M., KULAK J.M. Nicotine and nicotinic receptors; relevance to Parkinson's disease. Neurotoxicology. 2002;23:581–594. doi: 10.1016/s0161-813x(02)00036-0. [DOI] [PubMed] [Google Scholar]
- SAMOCHOCKI M., HOFFLE A., FEHRENBACHER A., JOSTOCK R., LUDWIG J., CHRISTNER C., RADINA M., ZERLIN M., ULLMER C., PEREIRA E.F., LUBBERT H., ALBUQUERQUE E.X., MAELICKE A. Galantamine is an allosterically potentiating ligand of neuronal nicotinic but not of muscarinic acetylcholine receptors. J. Pharmacol. Exp. Ther. 2003;305:1024–1036. doi: 10.1124/jpet.102.045773. [DOI] [PubMed] [Google Scholar]
- SANTOS M.D., ALKONDON M., PEREIRA E.F., ARACAVA Y., EISENBERG H.M., MAELICKE A., ALBUQUERQUE E.X. The nicotinic allosteric potentiating ligand galantamine facilitates synaptic transmission in the mammalian central nervous system. Mol. Pharmacol. 2002;61:1222–1234. doi: 10.1124/mol.61.5.1222. [DOI] [PubMed] [Google Scholar]
- SIMMONS L.K., SCHUETZE S.M., ROLE L.W. Substance P modulates single-channel properties of neuronal nicotinic acetylcholine receptors. Neuron. 1990;4:393–403. doi: 10.1016/0896-6273(90)90051-g. [DOI] [PubMed] [Google Scholar]
- SVENSSON A.L., NORDBERG A. Tacrine interacts with an allosteric activator site on alpha 4 beta 2 nAChRs in M10 cells. Neuroreport. 1996;7:2201–2205. doi: 10.1097/00001756-199609020-00029. [DOI] [PubMed] [Google Scholar]
- TISEO P.J., ROGERS S.L., FRIEDHOFF L.T. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following evening administration. Br. J. Clin. Pharmacol. 1998;46:13–18. doi: 10.1046/j.1365-2125.1998.0460s1013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VALENTA D.C., DOWNING J.E., ROLE L.W. Peptide modulation of ACh receptor desensitization controls neurotransmitter release from chicken sympathetic neurons. J. Neurophysiol. 1993;69:928–942. doi: 10.1152/jn.1993.69.3.928. [DOI] [PubMed] [Google Scholar]
- WOOLTORTON J.R., PIDOPLICHKO V.I., BROIDE R.S., DANI J.A. Differential desensitization and distribution of nicotinic acetylcholine receptor subtypes in midbrain dopamine areas. J. Neurosci. 2003;23:3176–3185. doi: 10.1523/JNEUROSCI.23-08-03176.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHOU F.M., LIANG Y., DANI J.A. Endogenous nicotinic cholinergic activity regulates dopamine release in the striatum. Nat. Neurosci. 2001;4:1224–1229. doi: 10.1038/nn769. [DOI] [PubMed] [Google Scholar]
- ZHOU F.M., WILSON C.J., DANI J.A. Cholinergic interneuron characteristics and nicotinic properties in the striatum. J. Neurobiol. 2002;53:590–605. doi: 10.1002/neu.10150. [DOI] [PubMed] [Google Scholar]