

Abstract
In this study, we have used Kir6.1/Kir6.2 chimeric proteins and current recordings to investigate the molecular basis of PNU-37883A inhibition of cloned KATP channels.
Rat Kir6.1, Kir6.2 and Kir6.1/Kir6.2 chimeras were co-expressed with either SUR2B or SUR1, following RNA injection into Xenopus oocytes, and fractional inhibition of KATP currents by 10 μM PNU-37883A reported.
Channels containing Kir6.1/SUR2B were more sensitive to inhibition by PNU-37883A than those containing Kir6.2/SUR2B (mean fractional inhibition: 0.70, cf. 0.07).
On expression with SUR2B, a chimeric channel with the Kir6.1 pore and the Kir6.2 amino- and carboxy-terminal domains was PNU-37883A insensitive (0.06). A chimera with the Kir6.1 carboxy-terminus and Kir6.2 amino-terminus and pore was inhibited (0.48). These results, and those obtained with other chimeras, suggest that the C-terminus is an important determinant of PNU-37883A inhibition of Kir6.1. Similar results were seen when constructs were co-expressed with SUR1. Further chimeric constructs localised PNU-37883A sensitivity to an 81 amino-acid residue section in the Kir6.1 carboxy-terminus.
Our data show that structural differences between Kir6.1 and Kir6.2 are important in determining sensitivity to PNU-37883A. This compound may prove useful in probing the structural and functional differences between the two channel subtypes.
Keywords: ATP-sensitive potassium channel, Kir6.1, Kir6.2, sulphonylurea receptor, SUR2B, SUR1, PNU-37883A, inwardly rectifying potassium channel
Introduction
ATP-sensitive potassium channels (KATP) couple the metabolic state of the cell to membrane excitability. KATP channels play key roles in several vital physiological functions, including insulin secretion by the pancreas, protection of cardiac muscle during ischaemia and hypoxic vasodilatation of arterial smooth muscle (reviewed by Seino & Miki, 2003). They are composed of hetero-octamers of four pore-forming Kir6.0 and four sulphonylurea receptor (SUR) subunits (Clement et al., 1997; Inagaki et al., 1997; Shyng & Nichols, 1997). The SUR subunit, which is a member of the ATP-binding cassette (ABC) protein superfamily, does not contribute to the potassium-conducting part of the channel, but is required for correct trafficking of Kir6.0 subunits to the plasma membrane and confers sensitivity to drugs of the sulphonylurea class (Aguilar-Bryan et al., 1995; Zerangue et al., 1999; Schwappach et al., 2000). There are two isoforms of the pore-forming subunit Kir6.1 (Inagaki et al., 1995a) and Kir6.2 (Inagaki et al., 1995b; Sakura et al., 1995), and three main isoforms of the sulphonylurea receptor SUR1 (Aguilar-Bryan et al., 1995), SUR2A (Chutkow et al., 1996) and SUR2B (Isomoto et al., 1996). The pharmacological differences between KATP channels recorded in different tissues have been attributed to differing combinations of Kir6.0 and SUR subunits. For example, the pancreatic KATP channel is Kir6.2/SUR1 (Inagaki et al., 1995b), the cardiac channel is Kir6.2/SUR2A (Inagaki et al., 1996) and the vascular channel is attributed to Kir6.1/SUR2B (Yamada et al., 1997). In addition to ATP, channel opening can be inhibited pharmacologically by the application of sulphonylureas such as glibenclamide (Sturgess et al., 1985). Channel opening can be stimulated by ADP (Dunne & Petersen, 1986; Kakei et al., 1986) and pharmacologically by potassium channel openers (KCOs) such as pinacidil (Robertson & Steinberg, 1990). Both sulphonylureas and KCOs have been shown to interact with specific regions of the SUR subunit of the channel (Ashfield et al., 1999; Uhde et al., 1999; Babenko et al., 2000). In contrast, although sulphonylurea receptors contain two nucleotide-binding domains, the primary site of inhibitory ATP binding has been mapped to the pore-forming Kir6.0 subunit (Tucker et al., 1997).
Previous studies in native tissue have shown the morpholinoguanidine drug PNU-37883A to be selective for the vascular form of KATP channel (Meisheri et al., 1993; Guillemare et al., 1994; Humphrey, 1999; Wellman et al., 1999). We and others have shown that, in recombinant channels, this compound shows selectivity for channels containing Kir6.1 (Surah-Narwal et al., 1999; Kovalev et al., 2001). In this study, we have used chimeras of Kir6.1 and Kir6.2 expressed in Xenopus oocytes to identify a region of the Kir6.1 subunit which confers sensitivity to PNU-37883A.
Methods
Molecular biology
Rat Kir6.1 (D42145), Kir6.2 (D86039) and SUR2B (AF087838) (kind gifts from F. Ashcroft, Y. Kurachi and S. Seino, respectively) were subcloned into pBFT, a modified version of pBF (a kind gift from B. Fakler), carrying both bacteriophage SP6 and T7 RNA polymerase promoters. Rat SUR1 (L40624) cloned in pBF was a kind gift from F. Ashcroft. Inserts in pBF/pBFT are flanked by the 5′ and 3′ untranslated regions of the Xenopus laevis β-globin gene and followed by a synthetic poly-A tract.
Chimeras of Kir6.1 and Kir6.2 with junctions after Kir6.1/6.2 residues 64/63 and 196/186 were produced by site-directed mutagenesis and subcloning. A Sal1 site was introduced into both subunit cDNAs (just upstream of TM1), and an Sph1 site (corresponding to that in Kir6.2) was introduced into Kir6.1 by silent mutagenesis using the GeneEditor™ in vitro Site-Directed Mutagenesis System (Promega, Southampton, U.K.). The carboxy-terminal domain was subdivided by the introduction of an EcoRV site (corresponding to that in Kir6.1) into the Kir6.2 cDNA by mutagenesis. This allowed creation of chimeras with junctions after residues 280/270 (Kir6.1/Kir6.2). In order to maximise the functionality of the resulting channels, all chimeras were based on an amino-acid sequence alignment of the two isoforms, and did not result in any insertions or duplications of residues. All constructs were verified by restriction digestion and sequencing.
Capped mRNA transcripts for microinjection were generated from plasmid DNA, linearised with Mlu1, using the mMESSAGE mMACHINE™ in vitro transcription kit (Ambion, Austin, TX, U.S.A.).
Electrophysiology
Xenopus laevis were killed by immersion in 0.35% 3-amino benzoic acid ethyl ester methane sulphonate salt (Sigma, Poole, U.K.), followed by destruction of the brain and spinal cord in accordance with Schedule 1 of the Animals (Scientific Procedures) Act of 1986. Egg sacs were removed and rinsed in ND962+ (containing, in mM, 96 NaCl, 2 KCl, 5 HEPES, 1 MgCl2, 2 CaCl2, 5 Na pyruvate, pH 7.5). Sacs were then cut into clumps of around 50 oocytes and transferred to OR2−, a low Ca2+-containing solution (in mM; 96 NaCl, 2 KCl, 1 MgCl2, 5 HEPES, pH 7.5). Oocytes were then enzyme treated for 50 min in type 1 collagenase (1 mg ml−1 in OR2−) (Sigma, Poole, U.K.), washed several times in OR2−, and transferred to ND962+. Single oocytes were manually defolliculated using forceps, and stored in ND962+ at 18°C. Oocytes were injected the following day with 1 ng of Kir6.x and 25 ng SURx by an intracellular microinjector (Inject+Matic, Geneva). They were then returned to the 18°C incubator, and membrane currents recorded 3–6 days post injection.
Two-microelectrode voltage clamp was used to record whole-cell currents from oocytes (Axon Instrument Geneclamp 500, Burlingame, CA, U.S.A.). KATP current was measured at a membrane potential of −60 mV. Oocytes were initially bathed in an extracellular solution containing 2 mM K+ (2 K solution (mM): 2 KCl, 96 NaCl, 5 HEPES, 1 MgCl2, 2 CaCl2, pH 7.4). After electrode impalement, the extracellular solution was changed to one containing 98 mM K+ (98 K solution (mM): 98 KCl, 5 HEPES, 1 MgCl2, 2 CaCl2, pH 7.4). KATP current was induced by 100 μM pinacidil (SUR2B containing combinations) or 100 μM diazoxide and 10 μM carbonyl cyanide m-chlorophenyl-hydrazone (CCCP, and SUR1-containing combinations). Pinacidil and diazoxide are synthetic KATP channel openers (Quayle et al., 1997). Pinacidil is selective for the SUR2B-containing channel, while diazoxide activates both SUR1- and SUR2B-containing channels. CCCP is a protonophore that dissipates the mitochondrial membrane potential, causing the ATP synthase to enter the reverse mode, and so depleting cellular ATP. Solutions were delivered by perfusion through the experimental chamber (flow rate 2 ml min−1; chamber volume 200 μl). All experiments were conducted at room temperature.
Data analysis
Concentration–effect relationships were fitted with the Hill equation.
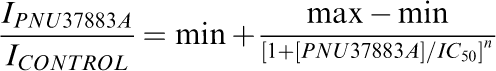 |
min is the initial Y-axis value, max the final Y-axis value, IPNU37883A is the current in the presence of PNU-37883A, ICONTROL is the current in the absence of PNU-37883A, [PNU-37883A] is the PNU-37883A concentration, IC50 the concentration at which current is inhibited by half and n is the Hill slope factor. IPNU37883A and ICONTROL were measured relative to the current in the presence of 10 μM glibenclamide. Fits to the data used the nonlinear curve-fitting routines provided in SigmaPlot8 (SPSS Inc., Chicago, IL, U.S.A.).
Data are presented as mean±s.e.m., and n indicates the number of cells. Statistical significance was assessed by one-way analysis of variance (ANOVA) with Tukey's test for multiple comparisons used as a post hoc analysis. P<0.05 indicates significance.
Solutions and drugs
Chemicals and drugs were obtained from Sigma (Poole, U.K.) or Merck (Poole, U.K.), except 4-morpholinecarboxamidine-N-1-adamantyl-N′-cyclohexyl-hydrochloride (PNU-37883A), which was a gift from Pharmacia Upjohn. Pinacidil, glibenclamide and CCCP were dissolved in DMSO to give 10 mM stock solutions. Diazoxide was prepared as a 200 mM stock solution in DMSO. PNU-37883A was dissolved in water as a 10 mM stock solution. Drugs were diluted in 98 K solution to give the final desired concentration.
Results
Xenopus oocytes injected with Kir6.1/SUR1, Kir6.1/SUR2B, Kir6.2/SUR1 or Kir6.2/SUR2B all expressed KATP currents (Figure 1). Expression of the Kir6.1/SUR2B combination was relatively poor, but the properties of all combinations were typical of those previously reported for cloned KATP channels (Seino & Miki, 2003). For instance, currents were activated by the potassium channel openers diazoxide and pinacidil, they were activated by metabolic inhibition by CCCP, they were inhibited by 10 μM glibenclamide, they were K+ selective (not shown), and they showed little intrinsic voltage dependence (not shown). No KATP currents were seen in response to 100 μM pinacidil in uninjected oocytes, water injected oocytes, or oocytes injected with either Kir6.1 or Kir6.2 in the absence of SUR (n=10 cells, data not shown).
Figure 1.

Recordings of KATP current in Xenopus oocytes injected with Kir6.1/SUR2B, Kir6.2/SUR2B, Kir 6.1/SUR1 and Kir6.2/SUR1 mRNAs, as indicated. 98 K and 2 K solutions were perfused, as indicated by the arrows. For Kir6.1/SUR2B and Kir6.2/SUR2B, 100 μM pinacidil was added to the extracellular solution to activate currents. For Kir 6.1/SUR1 and Kir6.2/SUR1, 100 μM diazoxide and 10 μM CCCP were added to activate currents. PNU-37883A and glibenclamide were added in the presence of pinacidil or diazoxide/CCCP. Membrane current was measured at a potential of −60 mV, and the dashed line indicates the zero current level.
PNU-37883A inhibition of cloned KATP channels
Cloned KATP channels formed by Kir6.1/SUR are more sensitive to inhibition by PNU-37883A than those formed by Kir6.2/SUR (Surah-Narwal et al., 1999; Kovalev et al., 2001). Figure 1 (upper two panels) illustrates the effect of PNU-37883A on KATP current in oocytes injected with Kir6.1 or Kir6.2 in combination with SUR2B. Initially, current was measured in 2 K solution at a membrane potential of −60 mV. The extracellular solution was then exchanged for one containing 98 K, changing the driving force on K+ movement to an inward direction and resulting in an inward basal current. Further, KATP current was then induced by 100 μM pinacidil, a synthetic KATP channel opener that is effective on SUR2B-containing combinations. For Kir 6.1/SUR2B (upper left panel), 3 and 10 μM PNU-37883A inhibited current when added to the extracellular solution in the continued presence of pinacidil. The remaining current was blocked by 10 μM glibenclamide. In comparison, 10 μM PNU-37883A had little effect on the Kir6.2/SUR2B combination (upper right panel). These results support previous observations that KATP channels that include Kir6.1 are more sensitive to PNU-37883A inhibition than those containing Kir6.2. PNU-37883A (10 μM) inhibited Kir6.1/SUR2B currents by 0.70±0.05 (n=10 cells), while Kir6.2/SUR2B currents were inhibited by 0.07±0.03 (n=5 cells). A concentration–effect relationship for PNU-37883A inhibition of Kir6.1/SUR2B is shown in Figure 2. Data were fitted with the Hill equation with half inhibition occurring at a concentration of 5 μM.
Figure 2.

Concentration–response relationship for PNU-37883A inhibition of Kir6.1/SUR2B. Inhibition is expressed as a fraction of the 10 μM glibenclamide-sensitive current at −60 mV. The curve was fitted to the data with the Hill equation with a slope factor of 1.67 and an IC50 of 4.88 μM. Data for Kir6.1/SUR1, Kir6.2/SUR2B and Kir6.2/SUR1 are also shown. All data are mean±s.e.m., n=2–13.
PNU-37883A inhibition of cloned KATP channels expressed in mammalian cells is also influenced by the SUR subunit (Cui et al., 2003). In Xenopus oocytes, currents encoded by Kir6.1/SUR1 were inhibited by PNU-37883A less effectively than those encoded by Kir6.1/SUR2B (compare the upper and lower left panels, Figure 1; see also Figure 2). As when co-expressed with SUR2B, PNU-37883A was selective for Kir6.1 over Kir6.2 (compare the upper and lower panels, Figure 1). PNU-37883A (100 μM) inhibited Kir6.1/SUR1 currents by 0.74±0.04 (n=5 cells) and Kir6.2/SUR1 currents by 0.05±0.02 (n=5 cells). Our data therefore show that PNU-37883A is a Kir6.1-selective KATP channel inhibitor and that sensitivity to this compound is influenced by the SUR subunit.
Effect of PNU-37883A on Kir6.1/Kir6.2 channel chimeras
Kir6.1/Kir6.2 chimeric proteins were made to probe the molecular basis for the observed difference in sensitivity of Kir6.1 and Kir6.2 to PNU-37883A. Chimeras of Kir6.1/6.2 were made at junctions after residues 64/63 and 196/186 (see Methods and Takano et al., 1998). This effectively divides the channel into three domains: an intracellular amino-terminal domain, a region containing the pore signature sequence and the two trans-membrane segments, and an intracellular carboxy-terminal domain. Domains can then be exchanged between Kir6.1 and Kir6.2 to study their role in drug sensitivity. In this scheme, Kir6.1 is represented as 1-1-1, Kir6.2 as 2-2-2 and chimeras as 1-2-1, etc. Figure 3 summarises the effect of 10 μM PNU-37883A on 100 μM pinacidil-induced currents through several Kir6.1/6.2 chimeras when co-expressed with SUR2B. It is apparent from the data that the C-terminal domain of Kir6.1 confers high sensitivity to the inhibitor, while the C-terminal domain of Kir6.2 confers low inhibitor sensitivity. For instance, 10 μM PNU-37883A inhibits 1-1-2 by 0.10±0.04 (n=6 cells), while 2-2-1 is inhibited by 0.48±0.04 (n=7 cells).
Figure 3.

PNU-37883A inhibition of the current through Kir6.1/Kir6.2 chimeric channels co-expressed with SUR2B. The figure shows fractional inhibition by 10 μM PNU-37883A of the 100 μM pinacidil-induced current. Inhibition is expressed relative to the 10 μM glibenclamide-sensitive current, except in the case of Kir6.2Δ26C, which is relative to 1 mM Ba2+. *P<0.05, ***P<0.001 when compared to Kir6.2.
The truncation mutant of Kir6.2 Kir6.2Δ26C, which expresses in the absence of SUR, is blocked by PNU-37883A when expressed in HEK 293 cells (IC50=4.6 μM, Cui et al., 2003). These data suggested that PNU-37883A may interact with the Kir6.2 subunit of the KATP channel. However, in Xenopus oocytes, when the Kir6.2Δ26C construct was expressed and activated by metabolic inhibition (1 μM CCCP), the resulting currents were insensitive to 10 μM PNU-37883A (Figure 3). Kir6.2Δ26C currents were blocked by 1 μM Ba2+, a KATP channel inhibitor (Quayle et al., 1997) (not shown).
Some chimeras showed rather small currents when co-expressed with SUR2B. Results were, therefore, confirmed and extended by co-expression with SUR1, which generally gave robust currents. Original current recordings (Figure 4) and summary data (Figure 5) for these SUR1 experiments support the conclusion that the C-terminal domain is important in determining PNU-37883A sensitivity. For instance, 100 μM PNU-37883A inhibited currents induced by 1-1-2 by 0.06±0.01 (n=5 cells), and currents induced by 2-2-1 by 0.58±0.04 (n=5 cells). The larger currents seen on co-expression with SUR1 also enabled the effect of PNU-37883A to be tested on basal KATP current (i.e. in the absence of activators). Basal Kir6.1 current was inhibited by 100 μM PNU-37883A to a similar extent to the current that was activated by diazoxide/CCCP (0.74±0.04 (n=5 cells) cf. 0.65±0.03 (n=5 cells); see Figure 5). Basal Kir6.2 current was not inhibited by 100 μM PNU-37883A (n=3 cells). These results suggest that activation of current by potassium channel openers and metabolic inhibition does not alter the sensitivity to PNU-37883A.
Figure 4.

Effect of 100 μM PNU-37883A on chimeric Kir6.1/Kir6.2 channels co-expressed with SUR1. Constructs are 1-2-1, 1-1-2, 2-2-1 and 1-1(1-2), as indicated. 98 K and 2 K solutions were perfused, as indicated by the arrows. All currents were activated by adding 100 μM diazoxide and 10 μM CCCP to the 98 K extracellular solution. PNU-37883A and glibenclamide were added in the presence of diazoxide/CCCP. Membrane current was measured at a potential of −60 mV, and the dashed line indicates the zero current level.
Figure 5.

PNU-37883A inhibition of the current through Kir6.1/Kir6.2 chimeric channels co-expressed with SUR1. The figure shows fractional inhibition by 100 μM PNU-37883A of the 100 μM diazoxide/10 μM CCCP-induced current. Inhibition is expressed relative to the 10 μM glibenclamide-sensitive current. ***P<0.001 when compared to Kir6.2.
The data described above suggest that the C-terminal domain of Kir6.1 is important in determining the sensitivity of cloned KATP channels to PNU-37883A. To further localise important regions within the C-terminus, additional Kir6.1/Kir6.2 chimeras were prepared. To do this, the C-terminus was split at a junction 248 bp downstream of the Sph1 site, making use of an EcoRV site at Kir6.1/Kir6.2 residues 281/271. By doing this, the C-terminus can be subdivided into two further segments, that between the Sph1 site and the EcoRV site, and that between the EcoRV site and the C-terminus. These constructs are named according to the convention that Kir6.1 is 1-1-(1-1), and Kir6.2 is 2-2-(2-2). The following constructs were prepared: 1-1-(1-2), 1-1-(2-1), 2-2-(2-1) and 2-2-(1-2).
The C-terminal ends, from the EcoRV restriction site to the C-terminus, were swapped between Kir6.1 and Kir6.2, forming 1-1-(1-2) and 2-2-(2-1), and these were co-expressed with SUR1. Currents were activated by 100 μM diazoxide/10 μM CCCP, and inhibition by 10 μM PNU-37883A was tested. 1-1-(1-2) was inhibited by 0.35±0.01 (n=8 cells), while 2-2-(2-1) was inhibited by 0.06±0.01 (n=9 cells). These results suggest that the 81 amino-acid section encoded between the Sph1 and EcoRV sites is important for PNU-37883A inhibition. The construct 1-1-(2-1) was also expressed with SUR1, and this was insensitive to 10 μM PNU-37883A (fractional inhibition of 0.001, n=8 cells), further supporting the important role of the 81-residue section. However, 2-2-(1-2), when co-expressed with SUR2B and activated by 100 μM pinacidil, was not sensitive to PNU-37883A 10 μM, inhibiting this construct by 0.044 (n=5 cells). This final swap shows that, although the 81-residue section is important in PNU-37883A inhibition, transfer of this region of Kir6.1 into a background of Kir6.2 is insufficient to confer PNU-37883A sensitivity on the channel, suggesting that other residues or structural constraints in the C-terminus are also important.
Discussion and conclusion
Previous evidence suggests that PNU-37883A, in contrast to other KATP active drugs, does not bind to the SUR subunit. PNU-37883A and glyburide work synergistically to inhibit KCO-mediated vasorelaxation of isolated rabbit mesenteric artery (Ohrnberger et al., 1993). PNU-37883A does not affect pinacidil binding to rat aorta and only blocks glyburide binding nonspecifically at high concentration (Löffler-Walz & Quast, 1998). There is also a body of evidence, both pharmacological and physiological, indicating that PNU-37883A is selective for the vascular form of KATP channel. 3H-PNU-37883A shows specific, displaceable, binding to rabbit mesenteric artery (Kd=65 nM) (Oleynek & Meisheri, 1992), but not to pancreatic RINm5F cell membranes (Guillemare et al., 1994). PNU-37883A blocked dog coronary artery KATP channels (IC50=0.72 μM), antagonised lemakalim (IC50=1 μM) in rat mesenteric artery cells and antagonised pinacidil-mediated vasorelaxation in norepinephrinecontracted rabbit mesenteric artery (IC50=0.78 μM) (Meisheri et al., 1993), but failed to depolarise RINm5F cells (Guillemare et al., 1994) or guinea-pig ventricular myocytes (Higden et al., 1995). In a more comprehensive electrophysiological analysis, Wellman et al. (1999) showed that PNU-37883A inhibited pinacidil-activated KATP currents in single rat mesenteric artery smooth muscle cells, but had little effect on those elicited in skeletal or cardiac myocytes. The vascular KATP channel is widely held to be comprised of Kir6.1 and SUR2B, suggesting Kir6.1 as the site of action of PNU-37883A. Consistent with these observations, we and others have previously reported selectivity of PNU-37883A for recombinant KATP channels containing Kir6.1 expressed in Xenopus oocytes (Surah-Narwal et al., 1999; Kovalev et al., 2001). In this study, we have confirmed and extended these observations by examining the sensitivity of Kir6.1/Kir6.2 chimeric channels to PNU-37883A. We have been able to identify a region of the carboxy-terminal domain of the pore-forming subunit, which determines the sensitivity to this compound.
It is clear that, while the pore-forming subunit of the KATP channel is the major determinant of action for this compound, the choice of SUR subunit can have a significant effect on the potency of inhibition, with IC50 ranging from around 5 μM with SUR2B (this study) to 32 μM with SUR1 (Surah-Narwal et al., 1999). This dependence on SUR is seen, too, for the inhibitory potency of ATP on KATP channels. Although the Kir6.0 subunit has clearly been identified as the site of inhibitory ATP action (Tucker et al., 1997), the Ki for ATP is 10-fold higher when Kir6.2 is expressed with SUR2A than when it is expressed with SUR1 (Inagaki et al., 1995b; 1996). When Kir6.2 is released from dependence on co-expression with SUR for insertion into the plasma membrane, by truncation of the final 26 amino acids, it remains ATP-sensitive, but the Ki for ATP is similar to that observed when SUR2A is present (Tucker et al., 1997).
In this study, we have assigned the property of PNU-37883A sensitivity to a carboxy-terminal region of Kir6.1 (amino acids 200–424). Only chimeras including this carboxy-terminal portion of Kir6.1 show high sensitivity to this compound. We have also demonstrated that the distal portion of this region (amino acids 281–424) contributes little to the interaction; swapping the-terminal amino acids for those of Kir6.2 only causes a small reduction in fractional inhibition; thus, the region between residues 200 and 280 plays a key role in determining sensitivity to PNU-37883A. Replacement of this region of Kir6.1 with the corresponding region of Kir6.2 abolishes sensitivity, but the reciprocal swap is not sufficient to confer sensitivity on Kir6.2, suggesting that residues or structural determinants outside this core region must also make a contribution. The region we have identified contains 81 amino-acid residues. Of these, 57 residues are shared between Kir6.1 and Kir6.2, and 12 are conservative substitutions. Analysis of predicted secondary structure for the region using the PIX program (www.hgmp.mrc.ac.uk/Registered/Webapp/pix/) shows no major differences (data not shown).
Recently, it has been reported that PNU-37883A selectivity for Kir6.1 over Kir6.2 is much less marked in HEK293 expressed recombinant KATP channels (Cui et al., 2003). Cui et al. report an IC50 for inhibition of Kir6.1/SUR2B channels of 6 μM, which is similar to that reported here (5 μM) but, in contrast to our findings, they also found Kir6.2-containing channels to show some sensitivity to PNU37883A (Kir6.2/SUR2B IC50=15 μM). Why this should be the case is not clear. However, in this study, we have shown that PNU-37883A sensitivity is not affected by the use of potassium channel openers or metabolic inhibition. Given the evidence from other studies (discussed above), it seems unlikely that selectivity is a peculiarity of the oocyte system.
The observation by Cui et al. (2003) that Kir6.2ΔC26 shows considerably greater sensitivity to PNU-37883A when expressed alone than when expressed with SUR2B raises several interesting possibilities. It may be that the presence of SUR in some way hinders access to or modifies the conformation of a drug-binding site on the pore-forming subunit. In this context, it is interesting to note that the region 208–279 of Kir6.2 (corresponding to residues 218–288 in Kir6.1) has been identified as a putative SUR interaction domain (Giblin et al., 1999). It could be that PNU-37883A binds to a site which is conserved between Kir6.1 and Kir6.2, but that the region we have identified influences access or binding to this site in some way differentially. A model where PNU-37883A binds equally to the two isoforms but only Kir6.1 has the residues necessary to allow inhibition of channel opening seems unlikely though, given previous drug-binding data which shows binding to rabbit mesenteric artery (Oleynek & Meisheri, 1992), but not to pancreatic RINm5F cell membranes (Oleynek & Meisheri, 1992; Guillemare et al., 1994). It is possible that the differences in sensitivity to PNU37883A shown by Kir6.1 and Kir6.2 may reflect differences in the intrinsic gating of the channels. Channels containing Kir6.2 are activated by depletion of ATP, whereas those containing Kir6.1 are not (Yamada et al., 1997). Previous analysis of chimeric Kir6.1/Kir6.2 channels mapped the residues responsible for spontaneous opening, in the absence of ATP, to two short regions of the Kir6.0 subunit; nine residues in the amino-terminal region and six in the proximal carboxy-terminal region (Kondo et al., 1998). It is perhaps interesting to note that these latter six residues fall within the region responsible for sensitivity to PNU37883A identified in this study.
Our failure to transfer sensitivity to PNU-37883A from Kir6.1 to Kir6.2 with the 81 amino-acid region may reflect some subtle influence of the overall conformation of the recombinant channel. It is worth noting that, even with closely related channel isoforms such as the Kir6.0s, certain chimeras express quite poorly in the oocyte system, suggesting that, despite its undoubted usefulness in determining biochemical interactions, the technique of using chimeric channels may lose some of the subtleties of native channel structure, function and/or regulation.
In this study, we have demonstrated that sensitivity to the vascular-selective morpholinoguanidine PNU-37883A maps to the proximal part of the carboxy-terminal domain of the Kir6.1 subunit. Determination of the precise residues responsible will require the analysis of specific point mutants. This compound may prove useful in probing structural and functional differences between the two channel subtypes.
Acknowledgments
We thank Professors A.R. North and A.-M. Surprenant for advice on setting up the Xenopus expression system. We thank the following for their kind gifts: Professor F. Ashcroft for Kir6.1 and SUR1, Professor Y. Kurachi for Kir6.2 and Professor S. Seino for SUR2B, Professor B. Fakler for pBF and Dr S.J. Humphrey for PNU-37883A. This work was supported by the British Heart Foundation.
Abbreviations
- CCCP
carbonyl cyanide m-chlorophenyl-hydrazone
- DMSO
dimethylsulphoxide
- KATP
ATP-sensitive potassium channels
- KCO
potassium channel opener
- Kir
inwardly rectifying potassium channel
- PNU-37883A
4-morpholinecarboxamidine-N-1-adamantyl-N′-cyclohexyl-hydrochloride
- SUR
sulphonylurea receptor
References
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT J.P., IV, BOYD A.E., GONZALEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- ASHFIELD R., GRIBBLE F.M., ASHCROFT S.J., ASHCROFT F.M. Identification of the high-affinity tolbutamide site on the SUR1subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- BABENKO A.P., GONZALEZ G., BRYAN J. Pharmaco-topology of sulfonylurea receptors. Separate domains of the regulatory subunits of KATP channel isoforms are required for selective interaction with K+ channel openers. J. Biol. Chem. 2000;275:717–720. doi: 10.1074/jbc.275.2.717. [DOI] [PubMed] [Google Scholar]
- CHUTKOW W.A., SIMON M.C., LE BEAU M.M., BURANT C.F. Cloning, tissue expression, and chromosomal localization of SUR2, the putative drug-binding subunit of cardiac, skeletal muscle, and vascular KATP channels. Diabetes. 1996;45:1439–1445. doi: 10.2337/diab.45.10.1439. [DOI] [PubMed] [Google Scholar]
- CLEMENT IV J.P., KUNJILWAR K GONZALEZ G., SCHWANSTESCHERM M., PANTEN U., AGUILAR-BRYAN L., BRYAN J. Association and stoichiometry of KATP channel subunits. Neuron. 1997;18:827–838. doi: 10.1016/s0896-6273(00)80321-9. [DOI] [PubMed] [Google Scholar]
- CUI Y., TINKER A., CLAPP L.H. Different molecular sites of action for the KATP channel inhibitors, PNU-99963 and PNU-37883A. Br. J. Pharmacol. 2003;139:122–128. doi: 10.1038/sj.bjp.0705228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUNNE M.J., PETERSEN O.H. Intracellular ADP activates K+ channels that are inhibited by ATP in an insulin-secreting cell line. FEBS Lett. 1986;208:58–62. doi: 10.1016/0014-5793(86)81532-0. [DOI] [PubMed] [Google Scholar]
- GIBLIN J.P., LEANEY J.L., TINKER A. The molecular assembly of ATP-sensitive potassium channels: determinants on the pore forming subunit. J. Biol. Chem. 1999;274:22652–22659. doi: 10.1074/jbc.274.32.22652. [DOI] [PubMed] [Google Scholar]
- GUILLEMARE E., HONORE E., DE WEILLE J., FOSSET M., LAZDUNSKI M., MEISHERI K. Functional receptors in Xenopus oocytes for U-37883A, a novel ATP-sensitive K+ channel blocker: comparison with rat insulinoma cells. Mol. Pharmacol. 1994;46:139–145. [PubMed] [Google Scholar]
- HIGDEN N.R., XU X., KHAN S.A., WANG J., LEE K., MEISHERI K.D. Selective vascular KATP channel blockade by U-37883A: comparison with cardiac KATP channel. FASEB J. 1995;9:A614. [Google Scholar]
- HUMPHREY S.J. Pharmacology of the KATP channel blocking morpholinoguanidine PNU-37883A. Cardiovasc. Drug Rev. 1999;17:295–328. [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulphonylurea receptor. Science. 1995b;270:1166–1169. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT IV J.P., WANG C.Z., AGUILAR-BRYAN L., BRYAN J., SEINO S. A family of sulfonylurea receptors determines the properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., SEINO S. Subunit stoichiometry of the pancreatic beta-cell ATP-sensitive K+ channel. FEBS Lett. 1997;409:232–236. doi: 10.1016/s0014-5793(97)00488-2. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., TSUURA Y., NAMBA N., MASUDA K., GONOI T., HORIE M., SEINO Y., MIZUTA M., SEINO S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J. Biol. Chem. 1995a;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- ISOMOTO S., KONDO C., YAMADA M., MATSUMOTO S., HIGASHIGUCHI O., HORIO Y., MATSUZAWA Y., KURACHI Y. A novel sulfonylurea receptor forms with BIR (Kir6.2) a smooth muscle type ATP-sensitive K+ channel. J. Biol. Chem. 1996;271:24321–24324. doi: 10.1074/jbc.271.40.24321. [DOI] [PubMed] [Google Scholar]
- KAKEI M., KELLY R.P., ASHCROFT S.J.H., ASHCROFT F.M. The ATP-sensitivity of K+ channels in rat pancreatic B-cells is modulated by ADP. FEBS Lett. 1986;208:63–66. doi: 10.1016/0014-5793(86)81533-2. [DOI] [PubMed] [Google Scholar]
- KONDO C., REPUNTE V.P., SATOH E., YAMADA M., HORIO Y., MATSUZAWA Y., POTT L., KURACHI Y. Chimeras of Kir6.1 and Kir6.2 reveal structural elements involved in spontaneous opening and unitary conductance of the ATP-sensitive K+ channels. Recept. Channel. 1998;6:129–140. [PubMed] [Google Scholar]
- KOVALEV H., LODWICK D., QUAYLE J.M. Inhibition of cloned KATP channels by the morpholinoguanidine PNU-37883A. J. Physiol. (Lond.) 2001;531P:S173. [Google Scholar]
- LÖFFLER-WALZ C., QUAST U. Interaction of the diuretics torasemide and U-37883A with the KATP channel in rat isolated aorta. Naunyn-Schmiedeberg's Arch. Pharmacol. 1998;358:230–237. doi: 10.1007/pl00005247. [DOI] [PubMed] [Google Scholar]
- MEISHERI K.D., HUMPHREY S.J., KHAN S.A., CIPKUS-DUBRAY L.A., SMITH M.P., JONES A.W. 4-morpholinecarboximidine-N-1-adamantyl-N′-cyclohexylhydrochloride (U-37883A): pharmacological characterization of a novel antagonist of vascular ATP-sensitive K+ channel openers. J. Pharmacol. Exp. Ther. 1993;266:655–665. [PubMed] [Google Scholar]
- OHRNBERGER C.E., KHAN S.A., MEISHERI K.D. Synergistic effects of glyburide and U-37883A, two structurally different vascular ATP-sensitive potassium channel antagonists. J. Pharmacol. Exp. Ther. 1993;267:25–30. [PubMed] [Google Scholar]
- OLEYNEK J., MEISHERI K. Intact tissue binding characteristics of 3H-U-37883A, a vascular KATP antagonist. FASEB J. 1992;6:A1778. [Google Scholar]
- QUAYLE J.M., NELSON M.T., STANDEN N.B. Inward rectifier and ATP-sensitive potassium channels in arterial smooth muscle. Physiol. Rev. 1997;77:1165–1232. doi: 10.1152/physrev.1997.77.4.1165. [DOI] [PubMed] [Google Scholar]
- ROBERTSON D.W., STEINBERG M.L. Potassium channel modulators: scientific applications and therapeutic promise. J. Med. Chem. 1990;33:1529–1541. doi: 10.1021/jm00168a001. [DOI] [PubMed] [Google Scholar]
- SAKURA H., AMMALA C., SMITH P.A., GRIBBLE F.M., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- SCHWAPPACH B., ZERANGUE N., JAN Y.N., JAN L.Y. Molecular basis for KATP assembly: transmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26:155–167. doi: 10.1016/s0896-6273(00)81146-0. [DOI] [PubMed] [Google Scholar]
- SEINO S., MIKI T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Prog. Biophys. Mol. Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- SHYNG S.-L., NICHOLS C.G. Octameric stoichiometry of the KATP channel complex. J. Gen. Physiol. 1997;110:655–664. doi: 10.1085/jgp.110.6.655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STURGESS N.C., ASHFORD M.L., COOK D.L., HALES C.N. The sulphonylurea receptor may be an ATP-sensitive potassium channel. Lancet. 1985;2:474–475. doi: 10.1016/s0140-6736(85)90403-9. [DOI] [PubMed] [Google Scholar]
- SURAH-NARWAL S., XU S.Z., MCHUGH D., MCDONALD R.L., HOUGH E., CHEONG A., PARTRIDGE C., SIVAPRASADARAO A., BEECH D.J. Block of human aorta Kir6.1 by the vascular KATP channel inhibitor U37883A. Br. J. Pharmacol. 1999;128:667–672. doi: 10.1038/sj.bjp.0702862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TAKANO M., XIE L.-H., OTANI H., HORIE M. Cytoplasmic terminus domains of Kir6.x confer different nucleotide-dependent gating on the ATP-sensitive K+ channel. J. Physiol. (Lond.) 1998;512:395–406. doi: 10.1111/j.1469-7793.1998.395be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- UHDE I., TOMAN A., GROSS I., SCWANSTECHER M. Identification of the potassium channel opener site on sulfonylurea receptors. J. Biol. Chem. 1999;274:28079–28082. doi: 10.1074/jbc.274.40.28079. [DOI] [PubMed] [Google Scholar]
- WELLMAN G.C., BARRETT-JOLLEY R., KÖPPEL H., EVERITT D., QUAYLE J.M. Inhibition of vascular KATP channels by U-37883A: a comparison with cardiac and skeletal muscle. Br. J. Pharmacol. 1999;128:909–916. doi: 10.1038/sj.bjp.0702868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- YAMADA M., ISOMOTO S., MATSUMOTO S., KONDO C., SHINDO T., HORIO Y., KURACHI Y. Sulphonylurea receptor 2B and Kir6.1 form a sulphonylurea-sensitive but ATP-insensitive K+ channel. J. Physiol. (Lond.) 1997;499:715–720. doi: 10.1113/jphysiol.1997.sp021963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZERANGUE N., SCHWAPPACH B., JAN Y.N., YAN L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]