

Abstract
Maturity-onset diabetes of the young, a monogenic form of Type II diabetes mellitus, is most commonly caused by mutations in hepatic nuclear factor 1α (HNF-1α). Here, the dimerization motif of HNF-1α is shown to form an intermolecular four-helix bundle. One face contains an antiparallel coiled coil whereas the other contains splayed α-helices. The “mini-zipper” is complementary in structure and symmetry to the top surface of a transcriptional coactivator (dimerization cofactor of homeodomains). The bundle is destabilized by a subset of mutations associated with maturity-onset diabetes of the young. Impaired dimerization of a β-cell transcription factor thus provides a molecular mechanism of metabolic deregulation in diabetes mellitus.
Keywords: diabetes mellitus, gene regulation, protein structure, NMR spectroscopy, four-helix bundle
Diabetes mellitus (DM) is a heterogeneous group of diseases characterized by hyperglycemia caused by impaired insulin secretion or action. A general feature is pancreatic β-cell failure resulting from either autoimmune destruction (Type I) (1) or inadequate compensation for insulin demand (Type II) (2). Monogenic forms of Type II DM [autosomal dominant syndromes designated maturity-onset diabetes of the young (MODY)] provide an opportunity to study mechanisms of β-cell dysfunction (3). Genetic analyses (4, 5) have highlighted the importance of a transcriptional cascade involving hepatic nuclear factors (HNFs) 1α, 1β, and 4. Despite the latter nomenclature, the MODY phenotype is restricted to the β cell, a site of HNF expression. The β-cell HNF cascade regulates genes required for glucose-stimulated insulin secretion (6, 7). The genetic association between DM and transcriptional deregulation had not been anticipated.
The most common form of MODY (subtype 3) is caused by mutations in HNF-1α (4, 8–13). Such mutations—otherwise rare in human populations—also occur in a subset of adults with classic Type II DM (14) and in children carrying a clinical diagnosis of Type I DM (15). HNF-1α is a modular protein containing at least four functional regions: an N-terminal dimerization domain, bipartite DNA-binding domain, and C-terminal transcriptional activation region (16–19). The N-terminal domain, an autonomous module flexibly linked to the DNA-binding domain (18, 19), also functions as a target of transcriptional coactivator dimerization cofactor of homeodomains (DCoH) (20–28). MODY-associated mutations occur in each domain (4, 8–13). Representative mutations have been shown in cell culture to attenuate HNF-1α-mediated transcriptional activation (13).
The present study focuses on the dimerization domain of HNF-1α (29, 30). This domain, like dimerization motifs in other transcription factors, coordinates recognition of an extended DNA site (29) and is required in culture for the protein's gene-regulatory activity (13). An homologous motif occurs in HNF-1β and mediates combinatorial homo- and heterodimerization (31). Like the leucine zipper (LZ) (32, 33), the HNF-1 motif contains multiple leucines, forms a stable dimer, and exhibits a two-state unfolding transition between folded dimer and unfolded monomer (34). Unlike the LZ, however, the peptide lacks a regular heptad repeat and does not form a continuous coiled-coil (35). Qualitative characterization by NMR, performed under acidic conditions, revealed an incompletely ordered N-terminal strand followed by an α-helix-turn-helix (35, 36). We present here the domain's three-dimensional structure and characterize thermodynamic effects of DM-associated mutations. Our results define an antiparallel four-helix bundle (4HB) and suggest that impaired dimerization of a human β-cell transcription factor can cause DM.
Materials and Methods
Peptide Synthesis.
Peptides were prepared by solid-phase synthesis with continuous-flow 9-fluorenylmethoxycarbony1 chemistry. Syntheses were performed on the 0.1 mM scale; the resin was split at intermediate steps to allow analogs to be synthesized. Peptide resins were cleaved by trifluoroacetic acid in the presence of scavengers. After filtration, peptides were precipitated with diethyl ether and were purified by reversed-phase high performance liquid chromatography (RP-HPLC). The C terminus was in each case amidated; a C-terminal tryptophan (Table 1) was added to facilitate measurement of peptide concentration by ultraviolet (UV) absorbance. Norleucine (“X” in Table 1) was used instead of methionine. Fidelity of synthesis was assessed by matrix-assisted laser-desorption ionization time-of-flight mass spectrometry (MS); purity (>96%) was estimated by analytical RP-HPLC. Dimerization was verified by analytical ultracentrifugation. To facilitate crystallographic analysis, an analog was prepared containing selenomethionine at position one.
Table 1.
Synthetic HNF-1α peptide sequences
| Wild type (wt) | XVSKL SQLQT ELLAA LLESG LSKEA LIQAL GEW |
| L12H | XVSKL SQLQT EHLAA LLESG LSKEA LIQAL GEW |
| wt-KEK | KEK XVSKL SQLQT ELLAA LLESG LSKEA LIQAL GEW |
| G20R-KEK | KEK XVSKL SQLQT ELLAA LLESR LSKEA LIQAL GEW |
X, norleucine. Inclusion of W33 (underlined in top sequence) does not alter the domain's CD helix content. Sites of substitution in analogs are underlined. “KEK” designates charged N-terminal extension.
1H-NMR Studies.
Spectra were obtained at 25°C in 10 mM potassium phosphate (pH 7.0 or pD 6.6, direct meter reading) and 50 mM KCl. The peptide concentration was 2 mM. Spectra in water (10% D2O) were obtained in the absence of solvent presaturation through the use of a WATERGATE-type pulse sequence (37, 38). Sequential assignment (see supplemental material on the PNAS web site, www.pnas.org) was obtained based on two-dimensional NMR methods (39). Resonance line widths are consistent with a dimeric molecular mass of 7 kDa; chemical shifts are similar in the peptide concentration range 0.2–2.0 mM.
NMR Structure Determination.
Distance geometry/simulated annealing (DG/SA) calculations (see supplemental material; ref. 40) were performed by using Insight II and x-plor (http://xplor.csb.yale.edu) (Biosym Technologies, San Diego). Initial calculations employed short- and medium-range helix-related restraints and a subset of interprotomeric nuclear Overhauser enhancements (NOEs). The latter were defined as contacts otherwise inconsistent with the secondary structure of a protomer. Preliminary models enabled additional long-range NOEs to be classified as intraprotomeric, dimer-related, or ambiguous. NOEs between residues 9 and 19 (spanning helix 1) indicated a dimer-related antiparallel contact (i.e., 9–19′), as an intraprotomeric 9–19 NOE would require a distorted α-helix inconsistent with the restraints. Control models were calculated with this or other assumptions, such as imposition of parallel or antiparallel covalent disulfide-bridged tethers. The final model employs 230 NOEs, 56 φ restraints, and 24 hydrogen-bond restraints per dimer. Root-mean-square deviations (rmsd) in the dimer (residues 3–31 and 3′–31′) are 0.77 Å (main chain) and 1.28 Å (side chain) relative to the average coordinates.
X-Ray Crystallography.
Crystals, grown by hanging drop/vapor diffusion in 2 weeks, were flash-frozen at 100 K by using glycerol as cryoprotectant. X-ray diffraction data to 1.9 Å resolution were collected by using a Rigaku (Tokyo) x-ray generator and R-axis IIC area-detector. Data are 97% complete with an Rsym of 6.5%. The crystals belong to space group P4222 with unit-cell dimensions a = b = 42.787, c = 29.128 Å. The volume of the unit cell implies one molecule per asymmetric unit with 27% solvent content. Multiple anomalous dispersion data were obtained at Argonne National Laboratories (Advanced Photon Source beam line 14D) using crystals of the selenomethionine peptide analog.
Circular Dichroism.
Spectra were obtained by using an Aviv Associates (Lakewood, NJ) spectropolarimeter. Peptide concentration was 5–50 μM in NMR buffer (above) at pH 7.4 and 4°C. Guanidine denaturation curves were obtained at 4°C by using an automated titration unit; the peptide concentration was 50 μM in the stock solution and 5 μM in the cuvette. Data were analyzed by nonlinear least-squares curve fitting (41). Thermal melting curves were obtained at 222 nm by using a thermister-controlled sample chamber.
Mass Spectrometry.
Matrix-assisted laser-desorption ionization time-of-flight MS was carried out on a Voyager-DE instrument (PerSeptive Biosystems, Framingham, MA) as described by the vendor. Samples were air-dried on the plate and were run in linear mode by using a 20-kV accelerating voltage. Insulin was used as an external standard.
Modeling of DCoH-HNF-1α Complex.
Modeling employed the insightii package. The Cα atoms of HNF-1α residues 10–14 and 14′–10′ were respectively aligned with residues 53–49 and 49′–53′ of subunits C and D of the crystallographic DCoH tetramer (22–24). In this alignment, the two-fold symmetry axes of DCoH and HNF-1α are superposed. No steric clash occurs between DCoH (subunits A and B) and HNF-1α.
Results
Structure at Neutral pH.
Studies focus on a 33-residue peptide at pH 7.0 (Table 1). 1H-NMR spectra at pH 2.7 have been described (35, 36). Acidic pH, which minimizes base-catalyzed exchange of amide protons, was presumably chosen to maximize the intensity of amide resonances after solvent presaturation. Reinvestigation at pH 7.0 yields 1H-NMR spectra similar in chemical shift but richer in density of interresidue NOEs. Resonance assignment is consistent with previous findings (35). A single spin system is observed for each residue, indicating that symmetry-related environments in the dimer are maintained on the NMR time scale. Secondary structural elements at pH 7.0 are similar to those at pH 2.7 (35, 36): the domain contains a well defined α-helix (residues 8–18), β-turn (residues 19–22), and second α-helix (residues 23–29). The N-terminal segment (residues 1–7) is not well ordered. Protected amide protons are observed in each helix.
The structure was obtained by iterative DG/SA (see Materials and Methods). In brief, the protocol begins with elements of secondary structure and then addresses their interrelation. The overall symmetry follows from initial classification of selected long-range NOEs spanning helix 1 (across the mini-zipper; see below). The essential observation utilizes contacts between side chains at opposite ends of this helix (e.g., between residues 9 and 19′). Inconsistent with the pattern of contacts predicted between ends of an α-helix, such NOEs are instead compatible with adjoining antiparallel α-helices. Preliminary crystallographic analysis of multiple anomalous dispersion data obtained from crystals of a selenomethionine analog likewise suggests an antiparallel dimer, which utilizes the crystallographic two-fold axis. In successive DG/SA models, essentially all interresidue NOEs could be rationalized as either intraprotomeric, dimer-related, or both. The latter were not included in the calculations. Dimer-related NOEs occur between helix 1 and helix 1′ and elsewhere (residues 5–21′, 9–19′, 9–21′, and 17–33′). Contacts between α-helices within a protomer include those between residues 12–30, 14–23, 16–23, 17–22, 17–23, and 17–24. Representative NOE spectroscopy spectra, diagonal NOE plot, restraint information, and statistical parameters are provided as supplemental material.
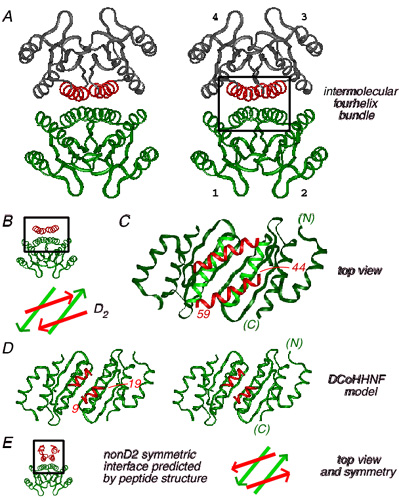
Structures are shown in Fig. 1 A (ensemble) and B (ribbon); for clarity, disordered N- and C-terminal residues are omitted. An individual protomer, as extracted from the dimer, consists of an acute helix-turn-helix (Fig. 1C). The angle between α-helices is near 31 ± 4°; the mean distance of closest approach between helical axes is 7.3 Å. These values are typical of 4HBs containing splayed α-helices (ref. 42; see Discussion). One face of the dimer is dominated by the central α-helices (residues 8–18; Fig. 1D). This interface contains four leucine side chains (L13, L17, and symmetry-related residues; Fig. 1D) and may be considered as a “mini-leucine zipper” antiparallel in orientation. The opposite face of the dimer is formed by skewed C-terminal α-helices (not shown).
Figure 1.

Structure of HNF-1α dimerization domain (stereo panels). (A) Ensemble of 14 main-chain structures. One protomer is shown in red and the other in blue. Ensemble was aligned according to main-chain atoms of residues 8–18 and 23–30. The dimer-related ensemble was positioned according to the mean orientation of the two protomers as obtained in dimeric DG/SA models. (B) Ribbon model of one dimer oriented as in A. (C) Ensemble of protomers showing selected side chains. MODY-associated sites of mutation L12 and G20 are highlighted in red. Ensemble was aligned according to main-chain atoms of residues 8–18 and 22–29. (D) Structure of dimer interface (“mini-zipper”) comprising residues 9–18 and 18′-9′ (α-helix 1 and 1′). L12 is highlighted in red. Ensemble was aligned according to the main-chain atoms of residues 9–18.
The structure of an “extracted monomer” is not physical as its folding is coupled to dimerization (35). This model nonetheless allows stepwise analysis of side-chain accessibilities in the protomer and dimer (see Fig. 7 in the supplemental material). A subset of side chains is buried in the protomer's helix–helix interface (L12, L13, K23, I27, and L30). Integral to this interface is the side chain of L12, a site of MODY mutation (highlighted in red in Figs. 1 and 2). The least accessible side chain in the protomer (fractional exposure 15 ± 4 percent), L12 projects into a pocket bounded by the side chains of L8, L13, I27, and L30. The other site of MODY mutation (G20, also highlighted in red in Figs. 1 and 2) is exposed in the central turn. The protomer's helix–helix interface (Fig. 2A) contains two distinct surfaces (Fig. 2B). A side view of these surfaces is shown in Fig. 2B relative to the symmetry-related protomer (gray ribbon). One surface (L8, E11, L16, E18, L26, and L30; green in Fig. 2) is exposed to solvent whereas the other (comprising the side chains of L5, Q9, T10, L13, L17, L21, K23, E24 and Q28; blue) constitutes the dimer interface. The flatness of the protomer is striking. Dimerization allows 658 Å2 of mean surface area per protomer to be buried. The dimer interface is nonpolar. The methylene portions of the side chains of T10, K23, E24, and Q28 pack in or adjoin the interface whereas their polar or charged functional groups are solvent exposed. These side chains exhibit significant dimer-specific reductions in fractional solvent accessibility. The side chain of L12, inaccessible within a protomer and projecting away from the internal interface, is not buried further in the dimer. The central β-turn is exposed on the surface of the dimer. The side chain of I27 (a site of neutral polymorphism I27L; magenta in Fig. 2) is shielded in part within the protomer and in part within the dimer. The exposed surface of the dimer contains a putative DCoH-binding site (Fig. 2B; see Discussion).
Figure 2.

(A) Upper and lower surfaces of helix-turn-helix protomer. Shown is a stereo pair showing side chains in the helix–helix interface (arrow): upper surface (residues 8, 11, 16, 26, and 30; green) and lower surface (residues 5, 9, 13, 17, 21, 23, 24; blue). The side chain of L12 (red) is buried in this interface. I27 is shown in magenta. The position of G20 Cα is shown as a red sphere. The main chain is shown in gray; carbonyl oxygens are omitted. (B) Stereo pair showing structure of one protomer relative to the other (gray ribbon). The coloring scheme is as in A. One surface of the protomer forms an internal dimeric interface whereas the other is predicted to bind to DCoH. Residues 7 and 29 (gray) belong to neither vertical surface. The view is rotated by 90° from that in A.
MODY Mutations Destabilize the 4HB.
To test whether MODY mutations impair dimerization, analogs were prepared containing substitutions L12H or G20R (Table 1; ref. 14). The solubility of the L12H analog was indistinguishable from that of the native dimer whereas G20R impairs solubility. Solubility of the G20R analog was restored by addition of a charged N-terminal extension to the disordered N terminus (Table 1). This extension has no significant effect on the native CD spectrum (Fig. 3 A and B) or stability (Table 2).
Figure 3.

CD spectra of analogs probing effects of MODY variants. (A) Comparison of far-UV CD spectra of wild-type and L12H peptides. (B) Comparison of far-UV CD spectra of KEK-extended native and G20R peptides. (C) Comparison of guanidine unfolding curves. (D) Thermal melting curves as monitored at 222 nm. Spectra in each panel were obtained at 4°C. For clarity, KEK native controls are omitted in C and D; their unfolding curves are essentially identical to those of the native peptide.
Table 2.
Synthetic HNF-1α peptides' thermodynamic stabilities
| Peptide | ΔGu (H2O) | Peptide | ΔGu (H2O) |
|---|---|---|---|
| Wild type (wt) | 12.0 ± 0.1 | L12H | 8.3 ± 0.2 |
| wt-KEK | 11.8 ± 0.1 | G20R-KEK | 7.2 ± 0.3 |
ΔGu values (kcal/mol) were extrapolated to zero denaturant concentration at a 1 M standard-state peptide concentration. “KEK” designates N-terminal extension (see Table 1).
CD spectra of the homodimeric analogs at 4°C are similar to those of the native dimer (Fig. 3 A and B), demonstrating that neither substitution precludes formation of α-helices. The variant structures nonetheless exhibit decreased thermal and thermodynamic stabilities (Fig. 3 C and D). Thermal unfolding studies (monitored at a helix-sensitive wavelength) demonstrate that the variant domains exhibit decreased thermal stability. Guanidine unfolding transitions, in each case consistent with a two-state process (34), demonstrate that the variant peptides exhibit significantly reduced thermodynamic stabilities (Table 2): ΔΔG values are 3.7 ± 0.3 kcal/mol (L12H) and 4.6 ± 0.4 kcal/mol (G20R). Because dimerization and peptide folding are coupled, these observations imply that dimerization is in each case weakened by at least 300-fold. These data do not address the relative stabilities of heterodimers comprised of native and variant subunits.
Discussion
The goal of the present study was to determine the structure of the HNF-1α dimerization domain and to test whether MODY mutations destabilize this structure. Our results define an antiparallel “mini-zipper” within a 4HB and characterize structural sites of mutation. In addition, the symmetry of the domain immediately suggests how it may bind transcriptional coactivator DCoH. These implications are discussed in turn.
Structure Reconciles Cross-Linking Paradox.
A previous study of the HNF-1α domain employed C-terminal GGC extensions to probe the symmetry of the dimer. This approach was motivated by studies of the LZ (33). Because the LZ consists of a parallel coiled coil, C-terminal cysteines adjoin and readily oxidize to form an intermolecular disulfide (33). Similar results were obtained with N-terminal CGG extensions. The LZ's parallel orientation precludes formation of antiparallel covalent dimers.
An analogous C-terminal-GGC-extended HNF-1α peptide has likewise been shown to form a covalent dimer (36). Because its 1H-NMR spectrum was similar to that of the native (noncovalent) dimer (36), such cross-linking suggested—by analogy to the LZ—that the orientation of the HNF-1α dimer is parallel. This conclusion is inconsistent with the present structure. The discrepancy is reconciled by inspection of distances between peptide termini. Respective C termini are positioned on the same face of the structure. Their proximity (the 33-Cα/33′-Cα distance is 16 ± 1 Å) enables interposition of a covalent GGC-disulfide-CGG tether (maximal length < 19 Å). Such accommodation is unrelated to the antiparallel orientation of the mini-zipper. Because the logic of LZ cross linking (33) is rigorous only for an extended structure (and does not generalize to globular domains), the previous conclusion (36) is not compelling. We note in passing that the distance between N termini of the HNF-1α dimer, although not well defined in the ensemble, is usually is too large (1-Cα/1′-Cα 40 ± 5 Å) to allow bridging by N-terminal CGG extensions. As predicted, the yield of such covalent dimers is negligible under mildly oxidizing conditions (M.Z. and M.A.W., unpublished results).
The HNF-1α Domain Differs from Classic Bundles.
4HBs are ubiquitous among protein structures (42, 43) and have been extensively investigated by mutagenesis and design (44–48). Two classes of 4HBs are recognized (see Fig. 8 in the supplemental material). The first, based on the coiled coil, is illustrated by repressor of primer (Rop). Rop consists of a dimer of coiled coils (49). Adjoining helices in the protomer and dimer are antiparallel and remain in contiguity by means of superhelical twisting. This requires a repeat of 3.5 residues per helical turn (rather than 3.6 as ordinarily occurs in an α-helix) and is associated with a heptad sequence repeat. The angle between helices is close to 20°, as originally proposed by Crick (50).
The second class of 4HBs is exemplified by cytochrome b562 (51). Its helices do not bend or supercoil and hence diverge. The helical repeat is 3.6 residues per turn, incommensurate with a heptad repeat (42). Splaying of helices can allow binding of ligands in a pocket adjoining the hydrophobic core. The up-down-up-down topology of cytochrome b562 is characterized by antiparallel packing of helices adjacent in the sequence. Because loops in other proteins may be of variable length, antiparallel interactions can occur between helices not contiguous in sequence. An example is provided by the up-up-down-down topology of the cytokine family (52).
The HNF-1α dimerization domain exhibits features of both classes of 4HBs. On the one hand, helix 1 forms an antiparallel “mini-zipper” in which dimer-related helices remain in contiguity throughout their length. On the other hand, the C-terminal helices are splayed, reminiscent of cytochrome b562 and analogous Class II structures. Thus, the structure in its entirety does not conform to either class. Whether the helices in the HNF-1α domain exhibit 3.5 or 3.6 residues per turn awaits determination of a high-resolution crystal structure.
A parallel 4HB occurs in the bHLH family of transcription factors (53–55). This structure contains large interhelical angles, which are more typically associated with non-4HB globular proteins. Long loops permit displacement of the second helix within a parallel dimer. Divergent N-terminal helices extend to form basic arms whose folding is coupled to DNA binding. By orienting the arms, the symmetry of this motif is integral to the mechanism of bHLH-DNA recognition. By contrast, the symmetry of the HNF-1α dimer is unrelated to DNA binding: “domain swap” experiments established that the HNF-1α domain may be replaced by either a parallel LZ or antiparallel 4HB (Rop) without change in the protein's DNA-binding properties (19). The lack of relationship between the symmetry of the HNF-1α dimer and DNA binding reflects the flexibility of the linker connecting the dimerization domain (residues 1–32) to the DNA-binding domain (residues 97–280).
The Symmetry of the HNF-1α 4HB Matches that of the DCoH Saddle.
The dimerization domain of HNF-1α mediates binding of DCoH (20, 21, 26). Although not known to be a site of mutation in DM, DCoH functions as an HNF-1-specific transcriptional coactivator: a bridge between HNF-1α and a preinitiation complex (25). [DCoH is also a pterin-4α-carbinolamine dehydratase, but this activity is not required for either HNF-1α binding or transcriptional activation (27, 28).] The crystal structure of DCoH has been determined as a dimer of dimers (Fig. 4A; refs. 22–24). The tetramer does not bind HNF-1α, presumably because its binding surface is occluded. The functional dimer is saddle-shaped (green in Fig. 4A) but, unlike the TATA-binding protein, does not bind DNA. A seeming paradox was posed by the incongruity between the symmetry of the DCoH dimer (antiparallel) (22–24) and the parallel model of the HNF-1α dimerization domain previously proposed (36). Such incongruity, precluding alignment of respective symmetry axes, implied an asymmetric mechanism of protein–protein recognition (24).
Figure 4.

Structure of transcriptional coactivator DCoH (PDB ID code 1dch; refs. 22–24) (A) and model of the DCoH-HNF-1α complex (B). (A) The DCoH homotetramer is formed by an antiparallel dimer of saddles (upper and lower dimers). The lower dimer is shown in green relative to binding helices (red) of upper dimer (gray). (B Upper) The tetramer interface of DCoH contains an antiparallel 4HB (box) with dihedral (D2) symmetry. (Lower) Side view of proposed model of HNF-1α (residues 5–31; red in box) atop the DCoH dimer (green). The predicted interface's symmetry differs from that of the DCoH-DCoH tetramer.
The symmetry of the present structure resolves this paradox and immediately suggests a model of the HNF-1α/DCoH complex (Fig. 4). Just as the symmetry of DCoH is exploited in the crystallographic tetramer [in which α-helices pack atop the saddle with dihedral symmetry to form an intermolecular 4HB (Fig. 4B Upper)], we suggest an analogous mode of DCoH/HNF-1α recognition. In particular, alignment of respective symmetry axes predicts that HNF-1α's mini-zipper sits atop the saddle to form an analogous intermolecular 4HB (Fig. 4B Lower; supplemental material).
MODY Mutations Highlight Specific Features of the Motif.
Residues 12 and 20 occupy unique positions in the structure of the HNF-1α domain. The side chain of L12 projects into a well ordered pocket within the protomer. The size and shape of this pocket are commensurate with that of leucine. Modeling suggests that the side chain of histidine can be accommodated with only local adjustments. Because of the flatness of the ring, however, the variant structure is predicted to exhibit packing defects. In addition, insertion of histidine in the nonpolar pocket may be less favorable because of the polar character of the imidazole ring. Invariance of leucine at position 12 is likely to be enjoined by a combination of shape, size, and electrostatic selectivity within this pocket. It is of future interest to investigate a variety of substitutions in the pocket as probes of the motif's architectural requirements. Whether such substitutions can alter tertiary structure will be addressed by comparative NMR or crystallographic studies.
Understanding the instability of the G20R domain will require a high-resolution analysis of analogs. Modeling suggests that an R20 side chain would project from the surface of the protomer, solvating the charged guanidinium group. Why this would be destabilizing is not clear. Among 4HB proteins, the sequences of turns are not well conserved and can accommodate diverse substitutions (56). Although destabilizing mutations in turns occur (57, 58), the predominant role of helical residues in determining structure has been demonstrated in cytochrome b562 (56) and in model peptides (59, 60). Because glycine is invariant at position 20, it is possible that its substitution leads to a global change in tertiary structure. Such a perturbation can in principle reflect an aberrant interaction by the R20 side chain; i.e., the absence of the variant side chain disallows an otherwise competing fold. A second possibility is unrelated to arginine: the helix-turn-helix structure may be unable to accommodate main-chain (φ, ψ) dihedral angles in the L region of the Ramachandran plot, independent of the identity of the L-amino acid. The configuration of G20 is not well defined in the present ensemble.
In summary, the HNF-1α dimerization domain has been shown to be a member of the 4HB superfamily, containing features of both Class I and Class II motifs. Its symmetry is antiparallel and thus matches that of transcriptional coactivator DCoH. MODY mutations in HNF-1α significantly weaken dimerization, a finding that rationalizes loss of HNF-1α-dependent transcriptional activation in cell culture (13). Impaired dimerization of a β-cell transcription factor provides a mechanism of metabolic deregulation in a monogenic form of DM.
Supplementary Material
Acknowledgments
We thank G. I. Bell, K. Polonsky, and D. F. Steiner for discussions; A. Khatri and G. Reddy for assistance with peptide synthesis; and T. Sosnick for advice regarding thermodynamic analyses. The work was supported in part by the Diabetes Research & Training Center at The University of Chicago.
Abbreviations
- DCoH
dimerization cofactor of homeodomains
- DG
distance geometry
- DM
diabetes mellitus
- 4HB
four-helix bundle
- HNF
hepatic nuclear factor
- LZ
leucine zipper
- MODY
maturity-onset diabetes mellitus of the young
- NOE
nuclear Overhauser enhancement
- rmsd
rms deviation
- SA
simulated annealing
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The structural coordinates have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1DT8).
References
- 1.Schranz D B, Lernmark A. Diabetes Metab Rev. 1998;14:3–29. doi: 10.1002/(sici)1099-0895(199803)14:1<3::aid-dmr206>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 2.Efendic S, Khan A, Ostenson C G. Diabetes Metab. 1994;20:81–86. [PubMed] [Google Scholar]
- 3.Velho G, Froguel P. Diabetes Metab. 1997;23:7–17. [PubMed] [Google Scholar]
- 4.Yamagata K, Oda K, Kaisaki P J, Menzel S, Furuta H, Vaxillaire M, Southam L, Cox R D, Lathrop M, Boriraj V V, et al. Nature (London) 1996;384:455–458. doi: 10.1038/384455a0. [DOI] [PubMed] [Google Scholar]
- 5.Yamagata K, Futura F, Oda N, Kaisaki P J, Menzel S, Cox N J, Fajans S S, Signorini S, Stoffel M, Bell G I. Nature (London) 1996;384:458–460. doi: 10.1038/384458a0. [DOI] [PubMed] [Google Scholar]
- 6.Byrne M M, Sturis J, Menzel S, Yamagata K, Fajans S S, Dronsfield M J, Bain S C, Hattersley A T, Valho G, Froguel P, et al. Diabetes. 1996;45:1503–1510. doi: 10.2337/diab.45.11.1503. [DOI] [PubMed] [Google Scholar]
- 7.Lehto M, Tuomi T, Mahtani M M, Widen E, Forsblom C, Sarelin L, Gullstrom M, Isomma B. J Clin Invest. 1997;99:582–591. doi: 10.1172/JCI119199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Glucksmann A, Lehto M, Tayber O, Scotti S, Berkemeier L, Pulido J, Wu Y, Nir W, Fang L, Markel P, et al. Diabetes. 1997;46:1081–1086. doi: 10.2337/diab.46.6.1081. [DOI] [PubMed] [Google Scholar]
- 9.Hansen T, Eiberg H, Rouard M, Vaxillaire M, Moller A M, Rasmussen S K, Fridberg M, Urhanmmer S A, Holst J J, Almind K, et al. Diabetes. 1997;46:726–730. doi: 10.2337/diab.46.4.726. [DOI] [PubMed] [Google Scholar]
- 10.Vaxillaire M, Rouard M, Yamagata K, Oda N, Kaisaki P, Boriraj V, Chévre J, Boccio V, Cox R, Lathrop G, et al. Hum Mol Genet. 1997;6:583–586. doi: 10.1093/hmg/6.4.583. [DOI] [PubMed] [Google Scholar]
- 11.Elbein S C, Teng K, Yount P, Scroggin E. J Clin Endocrinol Metab. 1998;83:2059–2065. doi: 10.1210/jcem.83.6.4874. [DOI] [PubMed] [Google Scholar]
- 12.Yamagata K, Yang Q, Yamamoto K, Iwahashi H, Miyagawa J, Okita K, Yoshiuchi I, Miyazaki J, Noguchi T, Nakajima H, et al. Diabetes. 1998;47:1231–1235. doi: 10.2337/diab.47.8.1231. [DOI] [PubMed] [Google Scholar]
- 13.Yamada S, Tomura H, Nishigori H, Sho K, Mabe H, Iwatani N, Takumi T, Kito Y, Moriya N, Muroya K, et al. Diabetes. 1999;48:645–648. doi: 10.2337/diabetes.48.3.645. [DOI] [PubMed] [Google Scholar]
- 14.Iwasaki N, Oda N, Ogata M, Hara M, Hinokio Y, Oda Y, Yamagata K, Kanematsu S, Ohgawara H, Omori Y, Bell G I. Diabetes. 1997;46:1504–1508. doi: 10.2337/diab.46.9.1504. [DOI] [PubMed] [Google Scholar]
- 15.Yamada S, Nishigori H, Onda H, Utsugi T, Yanagawa T, Maruyama T, Onigata K, Nagashima K, Nagai R, Morikawa A, et al. Diabetes. 1997;46:1643–1647. doi: 10.2337/diacare.46.10.1643. [DOI] [PubMed] [Google Scholar]
- 16.Mendel D B, Crabtree G R. J Biol Chem. 1991;266:677–680. [PubMed] [Google Scholar]
- 17.Tronche F, Yaniv M. BioEssays. 1992;14:579–587. doi: 10.1002/bies.950140902. [DOI] [PubMed] [Google Scholar]
- 18.Frain M, Swart G, Monaci P, Nicosia A, Stampfli S, Frank R, Cortese R. Cell. 1989;59:145–157. doi: 10.1016/0092-8674(89)90877-5. [DOI] [PubMed] [Google Scholar]
- 19.Tomei L, Cortese R, Francesco R D. EMBO J. 1992;11:4119–4129. doi: 10.1002/j.1460-2075.1992.tb05505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mendel D B, Khavari P A, Conley P B, Graves M K, Hansen L P, Admon A, Crabtree G R. Science. 1991;254:1762–1767. doi: 10.1126/science.1763325. [DOI] [PubMed] [Google Scholar]
- 21.Hansen L P, Crabtree G R. Curr Opin Genet Dev. 1993;3:246–253. doi: 10.1016/0959-437x(93)90030-s. [DOI] [PubMed] [Google Scholar]
- 22.Endrizzi J A, Cronk J D, Wang W, Crabtree G R, Alber T. Science. 1995;268:556–559. doi: 10.1126/science.7725101. [DOI] [PubMed] [Google Scholar]
- 23.Ficner R, Sauer U H, Stier G, Suck D. EMBO J. 1995;14:2034–2042. doi: 10.1002/j.1460-2075.1995.tb07195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cronk J D, Endrizzi J A, Alber T. Protein Sci. 1996;5:1963–1972. doi: 10.1002/pro.5560051002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suck D, Ficner R. FEBS Lett. 1996;389:35–39. doi: 10.1016/0014-5793(96)00573-x. [DOI] [PubMed] [Google Scholar]
- 26.Rhee K H, Stier G, Becker P B, Suck D, Sandaltzopoulos R. J Mol Biol. 1997;265:20–29. doi: 10.1006/jmbi.1996.0708. [DOI] [PubMed] [Google Scholar]
- 27.Sourdive D J, Transy C, Garbay S, Yaniv M. Nucleic Acids Res. 1997;25:1476–1484. doi: 10.1093/nar/25.8.1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Johnen G, Kaufman S. Proc Natl Acad Sci USA. 1997;94:13469–13474. doi: 10.1073/pnas.94.25.13469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Nicosia A, Monaci P, Tomei L, De Francesco R, Nuzzo M, Stunnenberg H, Cortese R. Cell. 1990;61:1225–1236. doi: 10.1016/0092-8674(90)90687-a. [DOI] [PubMed] [Google Scholar]
- 30.Chouard T, Blumenfeld M, Bach I, Vandekerckhove J, Cereghini S, Yaniv M. Nucleic Acids Res. 1990;18:5853–5863. doi: 10.1093/nar/18.19.5853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bach I, Yaniv M. EMBO J. 1993;12:4229–4242. doi: 10.1002/j.1460-2075.1993.tb06107.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Landschulz W H, Johnson P F, McKnight S L. Science. 1988;240:1759–1764. doi: 10.1126/science.3289117. [DOI] [PubMed] [Google Scholar]
- 33.O'Shea E K, Rutkowski R, Kim P S. Science. 1989;243:538–542. doi: 10.1126/science.2911757. [DOI] [PubMed] [Google Scholar]
- 34.De Francesco R, Pastore A, Vecchio G, Cortese R. Biochemistry. 1991;30:143–147. doi: 10.1021/bi00215a021. [DOI] [PubMed] [Google Scholar]
- 35.Pastore A, De Francesco R, Barbato G, Castiglione-Morelli M A, Motta A, Cortese R. Biochemistry. 1991;30:148–153. doi: 10.1021/bi00215a022. [DOI] [PubMed] [Google Scholar]
- 36.Pastore A, De Francesco R, Castiglione-Morelli M A, Nalis D, Cortese R. Protein Eng. 1992;5:749–757. doi: 10.1093/protein/5.8.749. [DOI] [PubMed] [Google Scholar]
- 37.Smallcombe S H. J Am Chem Soc. 1993;115:4776–4785. [Google Scholar]
- 38.Jahnke W, Kessler H. J Biomol NMR. 1994;4:735–740. doi: 10.1007/BF00404281. [DOI] [PubMed] [Google Scholar]
- 39.Wuthrich K. NMR of Proteins and Nucleic Acids. New York: Wiley; 1986. [Google Scholar]
- 40.Havel T. Prog Biophys Mol Biol. 1991;56:43–78. doi: 10.1016/0079-6107(91)90007-f. [DOI] [PubMed] [Google Scholar]
- 41.Bhattacharyya R P, Sosnick T R. Biochemistry. 1999;38:2601–2609. doi: 10.1021/bi982209j. [DOI] [PubMed] [Google Scholar]
- 42.Kamtekar S, Hecht M H. FASEB J. 1995;9:1013–1022. doi: 10.1096/fasebj.9.11.7649401. [DOI] [PubMed] [Google Scholar]
- 43.Weber P C, Salemme F R. Nature (London) 1980;287:82–84. doi: 10.1038/287082a0. [DOI] [PubMed] [Google Scholar]
- 44.Robinson C R, Sligar S G. Protein Sci. 1993;2:826–837. doi: 10.1002/pro.5560020512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vlassi M, Steif C, Weber P, Tsernoglou D, Wilson K S, Hinz H, Kokkinidis M. Struct Biol. 1994;1:706–716. doi: 10.1038/nsb1094-706. [DOI] [PubMed] [Google Scholar]
- 46.Hecht M H, Richardson J S, Richardson D C, Ogden R C. Science. 1990;249:884–891. doi: 10.1126/science.2392678. [DOI] [PubMed] [Google Scholar]
- 47.Choma C T, Lear J D, Nelson M J, Dutton P L, Robertson D E, Degrado W F. J Am Chem Soc. 1994;116:856–865. [Google Scholar]
- 48.Munson M, O'Brien R, Sturtevant J M, Regan L. Protein Sci. 1994;3:2015–2022. doi: 10.1002/pro.5560031114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Banner D W, Kokkinidis M, Tsernoglou D. J Mol Biol. 1987;196:657–675. doi: 10.1016/0022-2836(87)90039-8. [DOI] [PubMed] [Google Scholar]
- 50.Crick F H C. Acta Crystallogr. 1953;6:689–697. [Google Scholar]
- 51.Lederer F, Glatigny A, Bethge P H, Bellamy H D, Matthews F S. J Mol Biol. 1981;148:427–448. doi: 10.1016/0022-2836(81)90185-6. [DOI] [PubMed] [Google Scholar]
- 52.De Vos A M, Ultsch M, Kossiakoff A. Science. 1992;255:306–312. doi: 10.1126/science.1549776. [DOI] [PubMed] [Google Scholar]
- 53.Ellenberger T, Fass D, Arnaud M, Harrison S C. Genes Dev. 1994;8:970–980. doi: 10.1101/gad.8.8.970. [DOI] [PubMed] [Google Scholar]
- 54.Ma P C, Rould M A, Weintraub H, Pabo C O. Cell. 1994;77:451–459. doi: 10.1016/0092-8674(94)90159-7. [DOI] [PubMed] [Google Scholar]
- 55.Ferre-D'Amare A R, Prendergast G C, Ziff E B, Burley S K. Nature (London) 1993;363:38–45. doi: 10.1038/363038a0. [DOI] [PubMed] [Google Scholar]
- 56.Brunet A P, Huang E S, Huffine M E, Loeb J E, Weltman R J, Hecht M H. Nature (London) 1993;364:355–358. doi: 10.1038/364355a0. [DOI] [PubMed] [Google Scholar]
- 57.Castagnoli L, Vetriani C, Cesarini C. J Mol Biol. 1994;237:378–387. doi: 10.1006/jmbi.1994.1241. [DOI] [PubMed] [Google Scholar]
- 58.Zhou H X, Hoess R H, DeGrado W F. Nat Struct Biol. 1996;3:446–451. doi: 10.1038/nsb0596-446. [DOI] [PubMed] [Google Scholar]
- 59.Degrado W F, Wasserman Z R, Lear J D. Science. 1989;243:622–628. doi: 10.1126/science.2464850. [DOI] [PubMed] [Google Scholar]
- 60.Harbury P B, Zhang T, Kim P S, Alber T. Science. 1993;262:1401–1407. doi: 10.1126/science.8248779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}