

Antiepileptic drugs (AED) protect against seizures through interactions with a variety of cellular targets. The actions on these targets are often categorized into three broad groups: (1) modulation of voltage-gated ion channels (mainly sodium and also calcium channels); (2) effects on GABA systems, including enhancement of synaptic inhibition mediated by GABAA receptors; and (3) inhibition of synaptic excitation mediated by ionotropic glutamate receptors (Rogawski and Löscher, 2004). The critical downstream action of AEDs that act on voltage-dependent sodium channels—such as phenytoin, carbamazepine and lamotrigine—may be to reduce action potential-dependent glutamate release, particularly that dependent upon prolonged high frequency firing as occurs during epileptic discharges (Lingamaneni and Hemmings, 1999; Rogawski, 2002). Voltage-dependent sodium channel block also may reduce the propagation of action potentials from the soma into dendrites (Jung et al., 1997; Colbert et al., 1997) and may reduce the dendritic amplification of synaptic potentials (Schwindt and Crill, 1995). Together, these actions inhibit the spread of epileptiform activity. Electrophysiological recordings of synaptic responses demonstrate that sodium and calcium channel blocking anticonvulsants inhibit action potential-dependent synaptic events without affecting action potential-independent (‘miniature’) synaptic events. This is expected since these drugs do not block postsynaptic glutamate receptors and do not interfere directly with the synaptic release machinery. Interestingly, such drugs seem to have a preferential action on glutamate release and only weakly affect GABA release, possibly as a result of differences in excitation-contraction coupling in glutamatergic and GABAergic neurons (Prakriya and Mennerick, 2000; Westphalen and Hemmings, 2003).
In the past year, two entirely new AED targets have emerged. The targets were identified through similar pathways. The marketed AEDs gabapentin and levetiracetam—which were originally identified by virtue of their activity in empirical, mechanism-neutral screening models—were each found to bind with high affinity to unique recognition sites in brain. Two distinct proteins representing these recognition sites were identified and various lines of converging evidence supported the view that the binding proteins were the actual drug targets responsible for therapeutic activity. The two binding proteins have distinct subcellular localizations and undoubtedly have very different biological roles, although in both cases these roles are still largely obscure. Remarkably, both binding proteins may have roles in regulating vesicular neurotransmitter release. In the case of gabapentin (and the related AED pregabalin), drug binding has been shown to reduce neurotransmitter release. Because the levetiracetam target is localized to synaptic vesicles, it too may also influence neurotransmitter release. Recognizing that much remains to be learned, it is now appropriate to add a fourth category to the classification of AED mechanisms, as shown in Table 1 (see also illustration in Fig. 1). Here we describe evidence that one of these proteins—the calcium channel α2-δ subunit—is an AED target; the other target, SV2A, is considered elsewhere (Rogawski, 2006).
Table 1.
Revised classification of AED mechanisms
|
Figure 1.
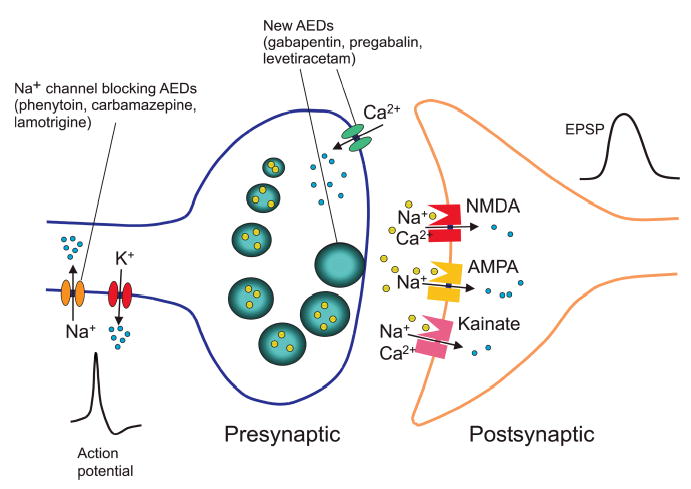
Model synapse illustrating interaction of Na+ channel blocking AEDs with voltage-activated Na+ channels and putative sites of action of newer AEDs that may more directly interact with release machinery. Gabapentin and pregabalin bind to α2-δ, which may inhibit voltage-activated Ca2+ entry through high voltage-activated Ca2+ channels or affect the way in which Ca2+ channels interact with vesicular release. Levetiracetam may also affect release by binding to synaptic vesicles protein SV2A. Action potentials are mediated by voltage-activated Na+ and K+ channels; Na+ channel blocking AEDs suppress epileptiform action potential firing, which leads to inhibited release. Small blue circles indicate ions; larger yellow circles represent glutamate within synaptic vesicles and free in the synaptic cleft. Glutamate acts on ionotropic receptors (of the NMDA, AMPA and kainate types) to generate an excitatory postsynaptic potential (EPSP) in the postsynaptic neuron.
The putative AED target α2-δ actually represents a family of four related proteins (MW, ~125 kD) encoded by separate genes (Klugbauer et al., 2003; Arikkath and Campbell, 2003; Qin et al., 2002). The first member to be identified, α2-δ type 1, is expressed ubiquitously. The related proteins α2-δ type 2, α2-δ type 3 and α2-δ type 4 are 54%, 26% and 25% identical, respectively, to α2-δ-1 at the amino acid level and share common structural motifs. α2-δ-2 is found in several tissues including brain and heart, α2-δ-3 is brain-specific, and α2-δ-4 is found in endocrine tissues and at very low levels in brain. The α2-δ subunits are post-translationally cleaved into α2- and δ-peptides, which are then covalently linked by a disulfide bridge. α2-δ-1 and α2-δ-2 are believed to form complexes with many calcium channel types (represented by different a1 isoforms), allosterically enhancing current amplitude and also promoting channel trafficking to the membrane (Canti et al., 2003). The mouse mutant ducky, which is associated with mutations in the α2-δ-2 gene, exhibits spontaneous spike-wave seizures (Barclay et al., 2001). Similarly, targeted deletion of the α2-δ-2 gene results in enhanced seizure susceptibility (Ivanov et al., 2004). These mouse models confirm a role for α2-δ-2 in the regulation of seizure susceptibility.
Gabapentin has been known to bind α2-δ-1 (encoded by the CACNA2D1 gene) for nearly a decade (Gee et al., 1996). However, it is only recently that evidence has been developed that strongly links binding to the anticonvulsant activity of gabapentin and pregabalin [S-(+)-3-isobutyl GABA]. Indeed, several alternative theories have been proposed to explain the mechanism of action of these two drugs (Taylor, 2002). While gabapentin and pregabalin are GABA analogs and were originally synthesized with the intention of targeting GABA systems, they do not interact with GABAA or GABAB receptors or GABA transporters (Sills, 2006). Moreover, neither compound is active at other target sites commonly associated with anticonvulsant effects, including voltage-activated Na+ channels or glutamate receptors (NMDA, strychnine-insensitive glycine, AMPA, KA or mGluR1, mGluR5). Both compounds, in contrast, are high-affinity ligands for the α2-δ-1 and α2-δ-2 proteins (but not α2-δ-3 or α2-δ-4) labeled with either [3H]gabapentin (Suman-Chauhan et al., 1993; Marais et al., 2001; Qin et al., 2002) or [3H]pregabalin (Piechan et al., 2004). Mutagenesis experiments have shown that the anticonvulsant binding site is probably confined to the α2 protein and/or the external portion of the associated δ component (Brown and Gee, 1998; Wang et al., 1999). Recent structure-activity studies with a variety of compounds related to gabapentin and pregabalin suggest that high affinity binding to α2-δ is required for anticonvulsant activity with compounds structurally similar to these drugs (Bryans et al., 1998; Belliotti et al., 2005).
Additional strong evidence linking the pharmacological actions of gabapentin and pregabalin to α2-δ-1 binding has come from studies with mice bearing a site-directed mutation in α2-δ-1. With in vitro single amino acid mutagenesis experiments of the recombinant α2-δ protein, substitution of arginine 217 with alanine was found to greatly reduce [3H]gabapentin-binding (Wang et al., 1999). Mice were then generated with this mutation (Bramwell et al., 2004; Taylor, 2004; Bian et al., 2004). Brains from these animals showed reduced [3H]pregabalin binding in the neocortex, amygdala, entorhinal cortex and hippocampus. The mutant mice exhibited pain responses and seizures that were indistinguishable from those of wild-type controls derived from crossing the same inbred lines. In addition, morphine was analgesic and phenytoin protected against seizures in the R217A animals. However, pregabalin (30 mg/kg) had no analgesic activity although it was analgesic in wild type controls. R217A mutant mice still show anticonvulsant responses to pregabalin in the maximal electroshock test, although the ED50 value is shifted from 7.8 to 20 mg/kg (M. Vartanian and S. Baron, unpublished). These results indicate that α2-δ-1 binding is required for the analgesic activity of pregabalin and contributes to its anticonvulsant activity. It seems plausible that the additional anticonvulsant activity could result from binding to α2-δ-2, but this will require experimental verification. In addition, further experiments with R217A mice that are genetically uniform (in contrast to the hybrid mice used for these early experiments) will clarify the present results.
How does binding to the α2-δ proteins protect against seizures? Numerous studies have examined the effects of gabapentin or pregabalin on voltage-gated calcium channel function. There are several reports that the drugs reduce calcium current in neuronal cell body membranes (Stefani et al., 1998; Martin et al., 2003; McClelland et al., 2004). However, in other studies with neurons (van Hooft et al., 2002) or recombinant systems (Canti et al., 2003), gabapentin was inactive. Despite these conflicting data, studies of calcium influx measured with fluorescent probes in synaptosomes from rat neocortex (van Hooft et al., 2002) or human brain tissue obtained at epilepsy surgery (Fink et al., 2002) indicate that both gabapentin and pregabalin reduce calcium influx into presynaptic terminals. In addition, gabapentin and pregabalin have been shown to produce subtle but reproducible reductions in the calcium-dependent release of glutamate and other neurotransmitters, including norepinephrine, serotonin and dopamine, from neocortical tissues (Dooley et al., 2000a,b, 2002), and also to inhibit the release of glutamate stimulated by the peptide neurotransmitters substance P and calcitonin gene related peptide (CGRP) from sensory neurons (Maneuf et al., 2001). In some systems, pretreatment with agents that induce inflammation or with neuropeptides or protein kinase activators are required before clear drug-induced reductions in neurotransmitter release are seen (Fehrenbacher et al., 2003).
Recently, several studies have indicated that not only calcium-dependent release of neurotransmitters, but also asynchronous (spontaneous), calcium-independent release of individual transmitter vesicles from glutamate synapses in spinal cord slices (Shimoyama et al., 2000; Patel et al., 2000), entorhinal cortex slices (Cunningham et al., 2004) and cultured rat hippocampal neurons (K. Micheva and S. Smith, personal communication) is reduced by treatment with gabapentin or pregabalin. These results suggest that the actions of α2-δ ligands to reduce neurotransmitter release may not require inhibition of calcium influx, and therefore may be mediated by an interaction of α2-δ (or the calcium-channel complex containing α2-δ) with synaptic proteins that are involved in the release or trafficking of synaptic vesicles.
Could sites of action other than α2-δ contribute to the pharmacological activity of gabapentin and pregabalin? It is, of course, impossible to rule out contributions from effects on other, as yet unidentified targets. However, the evidence accumulated to date suggests that other targets where the drugs are known to act are unlikely to contribute in a significant way to anticonvulsant activity. For example, 3-substituted GABA analogs like gabapentin and pregabalin have the ability to activate the GABA synthethetic enzyme L-glutamic acid decarboxylase (GAD) (Andruszkiewicz and Silverman, 1989). However, the required drug concentrations are very high, and there is no correlation between enzyme activation and anticonvulsant activity. In structure-activity studies, pregabalin had the most potent anticonvulsant activity but was a weak activator of GAD, whereas other more potent activators of GAD had very weak anticonvulsant activity (Silverman et al., 1991). Gabapentin also inhibits GABA-transaminase (GABA-T), the enzyme responsible for degradation of GABA, although this effect is very weak and is not shared by pregabalin (Goldlust et al., 1995). If GABA-T inhibition were relevant to the anticonvulsant activity of gabapentin or pregabalin, the drugs should increase brain GABA levels, as is the case for the anticonvulsant drug vigabatrin. In the rat, neither gabapentin nor pregabalin increases GABA levels (Sills et al., 2003; Errante and Petroff, 2003), although small increases have been reported with gabapentin in human brain tissue slices (Errante et al., 2002). [Increases in GABA also were seen in human neocortex with topiramate (Petroff et al., 2001) and lamotrigine (Kuzniecky et al., 2002) which are not known to alter the synthesis or metabolism of GABA.] Since both gabapentin and pregabalin are effective anticonvulsants in the rat, it is clear that GABA-T inhibition is not necessary for anticonvulsant activity in the model systems where it has been tested. Whether inhibition of GABA-T or elevated brain GABA concentrations contribute to the therapeutic activity in humans remains to be demonstrated.
Another target of gabapentin is the cytosolic isoform of branched-chain aminotransferase (Hutson et al., 1998). However, this potential target is not inhibited by pregabalin (T.-Z. Su, personal communication).
Finally, it is well recognized that gabapentin and pregabalin are substrates for the large neutral amino acid system L (leucine) transporter (Thurlow et al., 1993; Su et al., 2005). In fact, to be absorbed after oral administration and for active transport into the brain, 3-substituted GABA analogs also must be system L substrates (Fig. 2). The structural requirements for α2-δ binding differ from those that confer system L substrate activity (Belliotti et al., 2005). Both S(+)- and R(−)-isobutyl GABA are system L substrates (IC50 values for inhibition of [3H]L-leucine uptake are 158 and 355 μM, respectively), but the S-enantiomer (pregabalin) binds to α2-δ with 10-fold greater affinity than the R-enantiomer. Consistent with the role of α2-δ binding in mediating the anticonvulsant activity of pregabalin, only the S-enantiomer confers seizure protection in the maximal electroshock test (Taylor et al., 1993). Moreover, it is notable that analogs that bind to α2-δ but are not system L substrates exhibit anticonvulsant activity but only when administered intracerebroventricularly (Schwarz et al., 2005). Clearly, the system L transporter is not a target for the pharmacodynamic activity of gabapentin and pregabalin, although substrate activity is essential for efficacy when the drugs are administered systemically. In sum, it does not appear that any of the previously described targets for 3-substituted GABA analogs other than α2-δ are relevant to the pharmacodynamic actions of this class of drugs.
Figure 2.
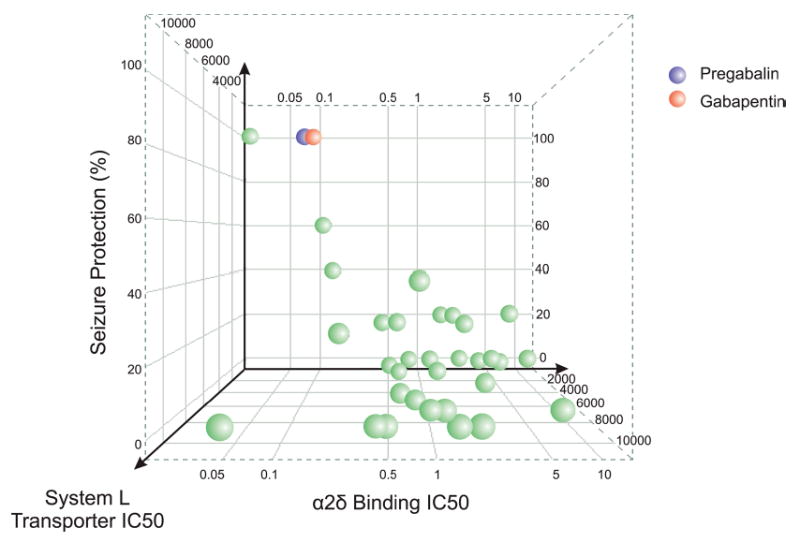
Relationship between α2-δ binding affinity, system L transporter affinity, and potency for protection against audiogenic seizures in DBA/2 mice for gabapentin, pregabalin and structural analogs. The Y-axis represents the percent protection (out of 5 animals) in the seizure model at a dose of 30 mg/kg, p.o. The Z-axis represents the concentration (μM) producing half-maximal inhibition of [3H]L-leucine uptake by the system L transporter in CHO-K1 cells. The X-axis represents the concentration (μM) producing half-maximal inhibition of specific [3H]gabapentin binding to pig brain membranes. Seizure protection correlates with α2-δ binding only for those compounds that are substrates for the system L transporter, which appears to be required for absorption by the gut and delivery to the brain (D.J. Wustrow and C.P. Taylor, unpublished).
Since the discovery of gabapentin in the mid 1970s and pregabalin in 1990, various theories have been proposed to explain the anticonvulsant properties of these molecules (Taylor, 2002; Sills, 2006). Gabapentin and pregabalin show a similar spectrum of activity in animal models that is distinct from that of other AEDs, suggesting that they share a common mechanism of action (Vartanian et al., 2006). Recently, converging lines of evidence have pointed to the α2-δ proteins as critical targets for the anticonvulsant effects of both drugs. However, since pregabalin protects against seizures in α2-δ-1 R217A mutant mice (albeit with reduced potency), an important question is whether binding to α2-δ-2 is responsible for the residual anticonvulsant activity or whether there are additional targets.
While it is now clear that α2-δ-1 represents a site of action for gabapentin and pregabalin, it is still not fully defined how binding results in therapeutic effects. There is evidence that presynaptic inhibition of neurotransmitter release (presumably of glutamate) is the critical effect that emerges from drug binding. However, to what extent this occurs through modulation of nerve terminal calcium channel activity or alternatively by direct effects on the release of synaptic vesicles remains to be determined. Finally, much needs to be learned about the functional roles of α2-δ proteins: how they modulate calcium channel behavior or whether they have other roles in the cell. Only then will it be possible to have a complete picture of the pharmacology of anticonvulsant drugs that act at this novel target.
Footnotes
Published in final edited form within: Loscher W, Schmidt D “New horizons in the development of antiepileptic drugs: innovative strategies” Epilepsy Res. 2006 June; 69(3): 183–272. doi: 10.1016/j.eplepsyres.2006.03.014.
References
- Andruszkiewicz, R.S., Silverman, R.B., 1989. A convenient synthesis of 3-alkyl-4-aminobutanoic acids. Synthesis 953–955.
- Arikkath J, Campbell KP. Auxiliary subunits: essential components of the voltage-gated calcium channel complex. Curr Opin Neurobiol. 2003;13:298–307. doi: 10.1016/s0959-4388(03)00066-7. [DOI] [PubMed] [Google Scholar]
- Barclay J, Balaguero N, Mione M, Ackerman SL, Letts VA, Brodbeck J, Canti C, Meir A, Page KM, Kusumi K, Perez-Reyes E, Lander ES, Frankel WN, Gardiner RM, Dolphin AC, Rees M. Ducky mouse phenotype of epilepsy and ataxia is associated with mutations in the Cacna2d2 gene and decreased calcium channel current in cerebellar Purkinje cells. J Neurosci. 2001;21:6095–6104. doi: 10.1523/JNEUROSCI.21-16-06095.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belliotti TR, Capiris T, Ekhato IV, Kinsora JJ, Field MJ, Heffner TG, Meltzer LT, Schwarz JB, Taylor CP, Thorpe AJ, Vartanian MG, Wise LD, Zhi-Su T, Weber ML, Wustrow DJ. Structure-activity relationships of pregabalin and analogues that target the α2-δ protein. J Med Chem. 2005;48:2294–2307. doi: 10.1021/jm049762l. [DOI] [PubMed] [Google Scholar]
- Bian, F., Davis, M.D., McCormick, J., Taylor, C.P., Walker, J.C., 2004. Calcium channel alpha2–delta (α2δ) type 1 subunit is the major binding protein for pregabalin in neocortex, hippocampus, amygdala, and spinal cord: an ex vivo autoradiographic study in genetically modified mice. Program No. 906.5. Abstract Viewer/Itinerary Planner. Society for Neuroscience, Washington, D.C. [DOI] [PubMed]
- Bramwell, S.R., Cox, P.J., Melrose, H., Stott, E., Wain, L., Corradini, L., Rees, H., Offord, J., Ti-Zhi, S., Williams, D., Field, M.J., 2004. The analgesic actions of pregabalin are mediated through its binding to the α2δ-1 subunit of voltage gated calcium channels. Program No. 523.19. Abstract Viewer/Itinerary Planner. Society for Neuroscience, Washington, D.C.
- Brown JP, Gee NS. Cloning and deletion mutagenesis of the α2δ calcium channel subunit from porcine cerebral cortex. J Biol Chem. 1998;273:25458–25465. doi: 10.1074/jbc.273.39.25458. [DOI] [PubMed] [Google Scholar]
- Bryans JS, Davies N, Gee NS, Dissanayake VU, Ratcliffe GS, Horwell DC, Kneen CO, Morrell AI, Oles RJ, O’Toole JC, Perkins GM, Singh L, Suman Chauhan N, O’Neil JA. Identification of novel ligands for the gabapentin binding site on the α2δ subunit of a calcium channel and their evaluation as anticonvulsant agents. J Med Chem. 1998;41:1838–1845. doi: 10.1021/jm970649n. [DOI] [PubMed] [Google Scholar]
- Canti C, Davies A, Dolphin AC. Calcium channel α2δ subunits: Structure, functions and target site for drugs. Curr Neuropharmacol. 2003;1:209–217. [Google Scholar]
- Colbert CM, Magee JC, Hoffman DA, Johnston D. Slow recovery from inactivation of Na+ channels underlies the activity-dependent attenuation of dendritic action potentials in hippocampal CA1 pyramidal neurons. J Neurosci. 1997;17:6512–6521. doi: 10.1523/JNEUROSCI.17-17-06512.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham MO, Woodhall GL, Thompson SE, Dooley DJ, Jones RS. Dual effects of gabapentin and pregabalin on glutamate release at rat entorhinal synapses in vitro. Eur J Neurosci. 2004;20:1566–1576. doi: 10.1111/j.1460-9568.2004.03625.x. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Donovan CM, Meder WP, Whetzel SZ. Preferential action of gabapentin and pregabalin at P/Q-type voltage-sensitive calcium channels: inhibition of K+-evoked [3H]-norepinephrine release from rat neocortical slices. Synapse. 2002;45:171–190. doi: 10.1002/syn.10094. [DOI] [PubMed] [Google Scholar]
- Dooley DJ, Donovan CM, Pugsley TA. Stimulus-dependent modulation of [3H]norepinephrine release from rat neocortical slices by gabapentin and pregabalin. J Pharmacol Exp Ther. 2000a;295:1086–1093. [PubMed] [Google Scholar]
- Dooley DJ, Mieske CA, Borosky SA. Inhibition of K+-evoked glutamate release from rat neocortical and hippocampal slices by gabapentin. Neurosci Lett. 2000b;280:107–110. doi: 10.1016/s0304-3940(00)00769-2. [DOI] [PubMed] [Google Scholar]
- Errante LD, Petroff OA. Acute effects of gabapentin and pregabalin on rat forebrain cellular GABA, glutamate, and glutamine concentrations. Seizure. 2003;12:300–306. doi: 10.1016/s1059-1311(02)00295-9. [DOI] [PubMed] [Google Scholar]
- Errante LD, Williamson A, Spencer DD, Petroff OA. Gabapentin and vigabatrin increase GABA in the human neocortical slice. Epilepsy Res. 2002;49:203–210. doi: 10.1016/s0920-1211(02)00034-7. [DOI] [PubMed] [Google Scholar]
- Fehrenbacher JC, Taylor CP, Vasko MR. Pregabalin and gabapentin reduce release of substance P and CGRP from rat spinal tissues only after inflammation or activation of protein kinase C. Pain. 2003;105:133–141. doi: 10.1016/s0304-3959(03)00173-8. [DOI] [PubMed] [Google Scholar]
- Fink K, Dooley DJ, Meder WP, Suman-Chauhan N, Duffy S, Clusmann H, Gothert M. Inhibition of neuronal Ca2+ influx by gabapentin and pregabalin in the human neocortex. Neuropharmacology. 2002;42:229–236. doi: 10.1016/s0028-3908(01)00172-1. [DOI] [PubMed] [Google Scholar]
- Gee NS, Brown JP, Dissanayake VU, Offord J, Thurlow R, Woodruff GN. The novel anticonvulsant drug, gabapentin (Neurontin), binds to the α2δ subunit of a calcium channel. J Biol Chem. 1996;271:5768–5776. doi: 10.1074/jbc.271.10.5768. [DOI] [PubMed] [Google Scholar]
- Goldlust A, Su TZ, Welty DF, Taylor CP, Oxender DL. Effects of anticonvulsant drug gabapentin on the enzymes in metabolic pathways of glutamate and GABA. Epilepsy Res. 1995;22:1–11. doi: 10.1016/0920-1211(95)00028-9. [DOI] [PubMed] [Google Scholar]
- Hutson SM, Berkich D, Drown P, Xu B, Aschner M, LaNoue KF. Role of branched-chain aminotransferase isoenzymes and gabapentin in neurotransmitter metabolism. J Neurochem. 1998;71:863–874. doi: 10.1046/j.1471-4159.1998.71020863.x. [DOI] [PubMed] [Google Scholar]
- Ivanov SV, Ward JM, Tessarollo L, McAreavey D, Sachdev V, Fananapazir L, Banks MK, Morris N, Djurickovic D, Devor-Henneman DE, Wei MH, Alvord GW, Gao B, Richardson JA, Minna JD, Rogawski MA, Lerman MI. Cerebellar ataxia, seizures, premature death, and cardiac abnormalities in mice with targeted disruption of the Cacna2d2 gene. Am J Pathol. 2004;165:1007–1018. doi: 10.1016/S0002-9440(10)63362-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung HY, Mickus T, Spruston N. Prolonged sodium channel inactivation contributes to dendritic action potential attenuation in hippocampal pyramidal neurons. J Neurosci. 1997;17:6639–6646. doi: 10.1523/JNEUROSCI.17-17-06639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klugbauer N, Marais E, Hofmann F. Calcium channel α2δ subunits: differential expression, function, and drug binding. J Bioenerg Biomembr. 2003;35:639–647. doi: 10.1023/b:jobb.0000008028.41056.58. [DOI] [PubMed] [Google Scholar]
- Klugbauer N, Lacinová L, Marais E, Hobom M, Hofmann F. Molecular diversity of the calcium channel α2δ subunit. J Neurosci. 1999;19:684–691. doi: 10.1523/JNEUROSCI.19-02-00684.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuzniecky R, Ho S, Pan J, Martin R, Gilliam F, Faught E, Hetherington H. Modulation of cerebral GABA by topiramate, lamotrigine, and gabapentin in healthy adults. Neurology. 2002;58:368–372. doi: 10.1212/wnl.58.3.368. [DOI] [PubMed] [Google Scholar]
- Lingamaneni R, Hemmings HC., Jr Effects of anticonvulsants on veratridine-and KCl-evoked glutamate release from rat cortical synaptosomes. Neurosci Lett. 1999;276:127–130. doi: 10.1016/s0304-3940(99)00810-1. [DOI] [PubMed] [Google Scholar]
- Maneuf YP, Hughes J, McKnight AT. Gabapentin inhibits the substance P-facilitated K+-evoked release of [3H]glutamate from rat caudial trigeminal nucleus slices. Pain. 2001;93:191–196. [Google Scholar]
- Marais E, Klugbauer N, Hofmann F. Calcium channel α2δ subunits — structure and gabapentin binding. Mol Pharmacol. 2001;59:1243–1248. doi: 10.1124/mol.59.5.1243. [DOI] [PubMed] [Google Scholar]
- Martin DJ, McClelland D, Herd MB, Sutton KG, Hall MD, Lee K, Pinnock RD, Scott RH. Gabapentin-mediated inhibition of voltage-activated Ca2+ channel currents in cultured sensory neurones is dependent on culture conditions and channel sub-unit expression. Neuropharmacology. 2003;42:353–366. doi: 10.1016/s0028-3908(01)00181-2. [DOI] [PubMed] [Google Scholar]
- McClelland D, Evans RM, Barkworth L, Martin DJ, Scott RH. A study comparing the actions of gabapentin and pregabalin on the electrophysiological properties of cultured DRG neurones from neonatal rats. BMC Pharmacol. 2004;4:14. doi: 10.1186/1471-2210-4-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel MK, Gonzalez MI, Bramwell S, Pinnock RD, Lee K. Gabapentin inhibits excitatory synaptic transmission in the hyperalgesic spinal cord. Br J Pharmacol. 2000;130:1731–1734. doi: 10.1038/sj.bjp.0703530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroff OA, Hyder F, Rothman DL, Mattson RH. Topiramate rapidly raises brain GABA in epilepsy patients. Epilepsia. 2001;42:543–548. doi: 10.1046/j.1528-1157.2001.18800.x. [DOI] [PubMed] [Google Scholar]
- Piechan, J. L., Donevan, S. D., Taylor, C. P., Dickerson, M. R., Li, Z., 2004. Pregabalin, a novel anticonvulsant, analgesic, and anxiolytic drug, exhibits class-specific alpha2-delta-1 and alpha2-delta-2 calcium channel subunit binding. Soc. Neurosci. Abstr., Program No. 115.11.
- Prakriya M, Mennerick S. Selective depression of low-release probability excitatory synapses by sodium channel blockers. Neuron. 2000;26:671–682. doi: 10.1016/s0896-6273(00)81203-9. [DOI] [PubMed] [Google Scholar]
- Qin N, Yagel S, Momplaisir M-L, Codd EE, D’Andrea MR. Molecular cloning and characterization of the human voltage-gated calcium channel α2–δ-4 subunit. Mol Pharmacol. 2002;62:485–496. doi: 10.1124/mol.62.3.485. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Diverse mechanisms of antiepileptic drugs in the development pipeline. Epilepsy Res. 2006;69:273–294. doi: 10.1016/j.eplepsyres.2006.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogawski MA, Löscher W. The neurobiology of antiepileptic drugs. Nat Rev Neurosci. 2004;5:553–564. doi: 10.1038/nrn1430. [DOI] [PubMed] [Google Scholar]
- Rogawski MA. Principles of antiepileptic drug action. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. Fifth. Lippincott Williams & Wilkins; Philadelphia: 2002. pp. 3–22. [Google Scholar]
- Schwarz JB, Gibbons SE, Graham SR, Colbry NL, Guzzo PR, Le VD, Vartanian MG, Kinsora JJ, Lotarski SM, Li Z, Dickerson MR, Su TZ, Weber ML, El-Kattan A, Thorpe AJ, Donevan SD, Taylor CP, Wustrow DJ. Novel cyclopropyl β-amino acid analogues of pregabalin and gabapentin that target the α2–δ protein. J Med Chem. 2005;48:3026–3035. doi: 10.1021/jm0491086. [DOI] [PubMed] [Google Scholar]
- Schwindt PC, Crill WE. Amplification of synaptic current by persistent sodium conductance in apical dendrite of neocortical neurons. J Neurophysiol. 1995;74:2220–2224. doi: 10.1152/jn.1995.74.5.2220. [DOI] [PubMed] [Google Scholar]
- Shimoyama M, Shimoyama N, Hori Y. Gabapentin affects glutamatergic excitatory neurotransmission in the rat dorsal horn. Pain. 2000;85:405–414. doi: 10.1016/S0304-3959(99)00283-3. [DOI] [PubMed] [Google Scholar]
- Sills GJ, Butler E, Forrest G, Ratnaraj N, Patsalos PN, Brodie MJ. Vigabatrin, but not gabapentin or topiramate, produces concentration-related effects on enzymes and intermediates of the GABA shunt in rat brain and retina. Epilepsia. 2003;44:886–892. doi: 10.1046/j.1528-1157.2003.04203.x. [DOI] [PubMed] [Google Scholar]
- Sills, G.J., 2006. The mechanisms of action of gabapentin and pregabalin. Curr. Opin. Pharmacol. Epub ahead of print [DOI] [PubMed]
- Silverman RB, Andruszkiewicz R, Nanavati SM, Taylor CP, Vartanian MG. 3-Alkyl-4-aminobutyric acids: the first class of anticonvulsant agents that activates L-glutamic acid decarboxylase. J Med Chem. 1991;34:2295–2198. doi: 10.1021/jm00111a053. [DOI] [PubMed] [Google Scholar]
- Stefani A, Spadoni F, Bernardi G. Gabapentin inhibits calcium currents in isolated rat brain neurons. Neuropharmacol. 1998;37:83–91. doi: 10.1016/s0028-3908(97)00189-5. [DOI] [PubMed] [Google Scholar]
- Su TZ, Feng MR, Weber ML. Mediation of highly concentrative uptake of pregabalin by L-type amino acid transport in chinese hamster ovary and Caco-2 cells. J Pharmacol Exp Ther. 2005;313:1–10. doi: 10.1124/jpet.104.082255. [DOI] [PubMed] [Google Scholar]
- Suman-Chauhan N, Webdale L, Hill DR, Woodruff GN. Characterisation of [3H]gabapentin binding to a novel site in rat brain: homogenate binding studies. Eur J Pharmacol. 1993;244:293–301. doi: 10.1016/0922-4106(93)90155-3. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Gabapentin mechanisms of action. In: Levy RH, Mattson RH, Meldrum BS, Perucca E, editors. Antiepileptic Drugs. Fifth. Lippincott Williams & Wilkins; Philadelphia: 2002. pp. 321–334. [Google Scholar]
- Taylor CP. Meeting report. The biology and pharmacology of calcium channel α2–δ proteins Pfizer Satellite Symposium to the 2003 Society for Neuroscience Meeting, Sheraton New Orleans Hotel, New Orleans, LA November 10, 2003. CNS Drug Rev. 2004;10:183–188. doi: 10.1111/j.1527-3458.2004.tb00012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor CP, Vartanian MG, Yuen PW, Bigge C, Suman-Chauhan N, Hill DR. Potent and stereospecific anticonvulsant activity of 3-isobutyl GABA relates to in vitro binding at a novel site labeled by tritiated gabapentin. Epilepsy Res. 1993;14:11–15. doi: 10.1016/0920-1211(93)90070-n. [DOI] [PubMed] [Google Scholar]
- Thurlow RJ, Brown JP, Gee NS, Hill DR, Woodruff GN. [3H]gabapentin may label a system-L-like neutral amino acid carrier in brain. Eur J Pharmacol. 1993;247:341–345. doi: 10.1016/0922-4106(93)90204-m. [DOI] [PubMed] [Google Scholar]
- van Hooft JA, Dougherty JJ, Endeman D, Nichols RA, Wadman WJ. Gabapentin inhibits presynaptic Ca2+ influx and synaptic transmission in rat hippocampus and neocortex. Eur J Pharmacol. 2002;449:221–228. doi: 10.1016/s0014-2999(02)02044-7. [DOI] [PubMed] [Google Scholar]
- Vartanian MG, Radulovic LL, Kinsora JJ, Serpa KA, Vergnes M, Bertram E, Taylor CP. Activity profile of pregabalin in rodent models of epilepsy and ataxia. Epilepsy Res. 2006;68:189–205. doi: 10.1016/j.eplepsyres.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Wang M, Offord J, Oxender DL, Su TZ. Structural requirement of the calcium channel subunit α2δ for gabapentin binding. Biochem J. 1999;342:313–320. [PMC free article] [PubMed] [Google Scholar]
- Westphalen RI, Hemmings HC., Jr Selective depression by general anesthetics of glutamate versus GABA release from isolated cortical nerve terminals. J Pharmacol Exp Ther. 2003;304:1188–1196. doi: 10.1124/jpet.102.044685. [DOI] [PubMed] [Google Scholar]