

Abstract
The growing interest in the chemistry of C-nitroso compounds (RN=O; R = alkyl or aryl group) is due in part to the recognition of their participation in various metabolic processes of nitrogen-containing compounds. C-Nitroso compounds have a rich organic chemistry in their own right, displaying interesting intra- and intermolecular dimerization processes and addition reactions with unsaturated compounds. In addition, they have a fascinating coordination chemistry. While most of the attention has been directed towards C-nitroso compounds containing a single –NO moiety, there is an emerging area of research dealing with dinitroso and polynitroso compounds. In this critical review, we present and discuss the synthetic routes and properties of these relatively unexplored dinitroso and polynitroso compounds, and suggest areas of further development involving these compounds. (126 references.)
1 Introduction
The family of C-nitroso compounds (RN=O; R = alkyl or aryl group) is an important class of compounds that has relevance to organic chemistry and biochemistry. Preparative routes to C-nitroso compounds have been reviewed recently.1 Nitroso groups may be constituents of various metabolites in vivo, and their coordination chemistry drives many of their interactions with iron-containing biomolecules such as cytochrome P450, nitric oxide synthase, and myoglobin.2 The earlier reviews have focused on mononitroso compounds and their reactions,3–10 and to the best of our knowledge, the corresponding area of dinitroso and polynitroso compounds has not been previously reviewed. In this article, we link together the various categories of dinitroso and polynitroso compounds and indicate the potential for further study in a variety of areas.
2 Dinitroso compounds
A distinct family of molecules is provided by the relatively small number of compounds in which two nitroso groups are to be found within the same molecule. It is possible to subdivide these molecules into three major classes, namely
dinitroso compounds in which the two nitroso groups do not interact with each other,
dinitroso compounds in which there is intramolecular interaction between the two nitroso groups leading to the formation of either a cis-azodioxy group or a furoxan group (Fig. 1),
dinitroso compounds in which two or more molecules interact intermolecularly to form trans- or cis-azodioxy groups, the resulting products being dimers, oligomers or polymers.
Fig. 1.

Structures of cis-azodioxy and furoxan groups.
There is a further series of dinitroso compounds where these carry a negative charge.
2.1 Non-interacting dinitroso compounds
These compounds exhibit the characteristic physical and chemical properties of the monomeric –N=O group; they are blue/green with a molar extinction coefficient for the n → π* transition approximately twice that for the corresponding structurally related RNO monomer. An example is provided by trans-1,4-dichloro-1,4-dinitrosocyclohexane in 1,1,1-tri-fluoroethanol11 for which λmax is 640 nm and ɛmax is 42 dm3mol−1cm−1. This can be compared with 1-chloro-1-nitrosocyclohexane in cyclohexane12 for which λmax is 655 nm and ɛmax is 22 dm3mol−1cm−1. Such dinitroso compounds are unaltered structurally on change of state or on dissolution. A further example is provided by 4,4′-dinitroso-N,N-diphenylpiperazine (1) which displays similar structural features to those of the monomeric 4-nitroso-N,N-dimethylaniline (2). It is to be expected that the joining ‘end-to-end’ of other substituted nitrosoanilines will result in similar dinitroso compounds and eight further examples (3–10) have been synthesized.13

A 1,7-dinitroso-m-carborane (11) has been prepared14 in ~30% yield from reaction of nitrosyl chloride with 1,7-dilithio-m-carborane in ether–hexane. It is unfortunate that the only physical property reported is the melting point (145–146 °C), but it is most probable that this sole reported example of a dinitroso carborane contains two entirely separate nitroso groups. All known nitroso carboranes (with NO directly attached to the carboranyl cage) are monomeric. The distance between the two nitroso groups in (11) is such that intramolecular interaction between the two NO groups is very unlikely; intermolecular interactions in nitrosocarboranes are possible, although they have not yet been reported.

Compounds containing two gem-nitrosoacetates are proposed to form in low yield from the reaction of 1,3-cyclohexanedionedioxime and 1-methyl-2,6-cyclohexanedione-dioxime with lead tetraacetate; in some cases, rearrangements to the azodioxy forms occur.15
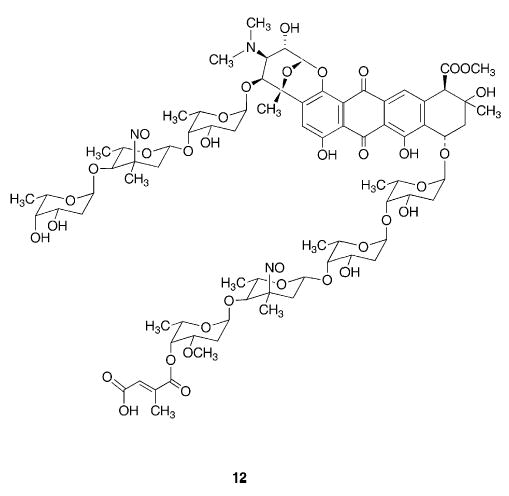
Another example of a dinitroso compound in which the two nitroso groups act independently of each other is provided by the anthracycline antibiotic viriplanin A16–19 an antibiotic with activity against Herpes simplex viruses. In this large molecule (12) the two nitroso groups are each bonded to tertiary carbon atoms in β-pyranoside rings in different sections of the molecule. Viriplanin A from Ampullariella regularis (strain SE47) is light sensitive, and is best isolated in the dark.16 The 13C NMR signal for the C-NO group appears at 101.5 ppm18 which is 12.6 ppm higher than that for the corresponding C-NO2 group and consistent with the trend observed for related aliphatic or acyclic C-NO/C-NO2 pairs (the signal due to any C-N2O2-dimer present would be at significantly lower ppm values). The characteristic visible absorption spectrum of the compound in methanol shows λmax at 710 nm with ɛmax of 34 dm3mol−1cm−1, in keeping with the range of values expected for two nitroso groups.18 A further example of a large molecule containing two independent nitroso groups is provided by 5,10-bis(4-nitrosophenyl)-15,20-bis(4-pyridyl)por-phyrin,20 prepared by electroreduction of the precursor nitro compound.
There are relatively few examples of vicinal dinitrosoalkanes or dinitrosocycloalkanes, although examples exist of metal-stabilized vicinal dinitroso compounds in which both nitroso groups are σN-coordinated to the same metal atom (e.g., 13 and 14).21–24 In these cases, the formation of the vicinal dinitrosoalkanes were assisted by the metal. The formation of such “free” organic compounds (e.g., 15) has been invoked in the reaction of nitric oxide with methyl methacrylate during the formation of the nitro nitroso final product,25 and a patent26 claims the preparation of 1,2-dinitrosoperfluoroethane and 1,2-dinitrosotrifluoromonochloroethane from the reaction of the appropriate fluoroalkene with nitric oxide in the presence of infrared radiation. The preparation of difluorodinitrosomethane (ONCF2NO)27 from the photolysis of a mixture of CF2I2 and NO is the first report of a geminal dinitroso compound, but it lacks thermal stability and is light-sensitive. The same authors report the formation of hexafluoro-1,3-dinitrosopropane from this reaction. This suggests that other geminal and terminal dinitrosoperfluoroalkanes could be prepared via similar routes.

The preparation of a dinitroso derivative of a vinylogous tetrathiafulvalene has been reported.28 The compound (16) has vicinal –N=O groups, and the proton NMR spectrum suggests a symmetrical structure which can be rationalized by a stabilization of the two NO groups by S+---O− interactions as shown in (17). The compound is brown in colour unlike other dinitroso compounds, and does not exhibit the characteristic absorption band in the red.29 The formation of a 4,9-dinitroso compound (18) is claimed when nitrous acid reacts with 3,5,8,10-tetraoxo-3,4,5,8,9,10-hexahydropyrene,30 but as the colour of the solid is a light yellowish gray, the material is either polymeric or the isomeric dioxime.

The reaction intermediate 1,4-di(nitrosomethyl)benzene is proposed to account for the linear telomeric reaction products of nitric oxide with the reactive diradical para-xylylene.31 There are also some short lived dinitroso compounds which result in the formation of other, more stable, structures at room temperature. To these we now turn.
2.2 Dinitroso compounds with intramolecular interactions
The first of these types of interaction between two –N=O groups results in the formation of a cis-azodioxy group (due to an N…N interaction) and the formation of a heterocyclic ring. It is apparent that the formation of an intramolecular trans-azodioxy linkage is virtually impossible on steric grounds, and no claims for such compounds have appeared in the literature. A wide range of ‘cis-fixed’ azodioxy compounds is known,2 and in a small number of cases these can undergo thermal dissociation to dinitroso compounds (eqn. (1) and Fig. 2).32–36
Fig. 2.

cis-Fixed azodioxy compounds that undergo thermal conversion to dinitroso species.
 |
The formation of an internal cis-dimer is usually a consequence of nitroso N…N interactions and is frequently associated with the initial formation of a blue colour due presumably to the dinitroso compound. Thermal dissociation of the internal cis-fixed compound is confined to the six-membered and larger heterocyclic rings although not all of these can dissociate to give the monomers. The smaller C3N2 and C2N2 cis-azodioxy systems are resistant to thermal dissociation.34 The report in 189837 of a colourless dinitroso compound 2,6-dimethyl-2,6-dinitrosoheptan-4-one (19) (prepared from HgO oxidation of the bis-hydroxylamine precursor) which gave a blue melt and blue solutions in organic solvents, is an early example of a cis-fixed compound.

The second form of interaction results in the formation of a furoxan ring structure due to the dinitroso –(O)N…O(N)– interaction (eqn. (2)).
 |
The first examples of this were obtained when attempts to synthesize o-dinitrosobenzenes were made. The resultant furoxan can dissociate to the short-lived o-dinitrosobenzene on heating (eqn. (3)), and this clearly permits the interconversion of substituted furoxans to occur on heating (eqn. (4)).38
 |
 |
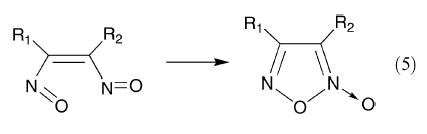
It is possible to synthesize benzodifuroxan39 (20) and benzotrifuroxan40 (21) (formerly known as hexanitrosobenzene); the latter forms charge-transfer complexes with aromatic π-donors. Similarly, the presumed dinitrosoalkenes form a furoxan ring (eqn. (5)), although this would not appear to occur were the dinitrosoalkene to be formed in the trans-configuration unless a mechanism such as that shown in eqn. (6) involving formation of a C–C single bond could participate as suggested by Grundmann et al.41 for dimesitylfuroxan.

 |
 |
The reverse of eqn. (5) was invoked by Mallory and Cammarata42 to explain the thermal interconversions of the two methylphenylfuroxans and the two ethylmethyl furoxans via the corresponding dinitrosoalkenes. Furoxan ring formation can only occur when the carbon–carbon bond to which the two NO groups are attached has either aromatic or double bond character. It is of interest to note a suggestion by Hwang et al.,43 based on mass spectrometric data, that the dinitroso compounds formed from furoxans may decompose to release two molecules of nitric oxide under appropriate conditions. In a related earlier study, Feelisch et al. reported that the vasodilator action of furoxans could be attributed to their reactions with thiols resulting in the release of NO.44 The NO-releasing abilities of furoxans has been reviewed recently.45
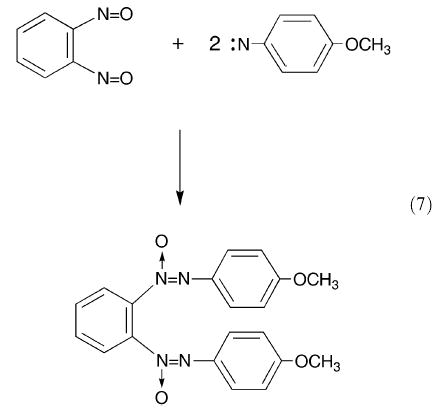
The chemistry of the furoxan ring system has been studied extensively, and there are a number of detailed review articles in which the historical development of the structural evidence is presented,38,46–48 and spectroscopic and crystallographic data are discussed.48 The ring opening eqn. (3) is of theoretical importance and was included in a computational study by Hoffmann et al.49 of the dissociation mechanism of dimeric nitroso compounds. More recent studies of the molecular rearrangements of some substituted benzofuroxans have been made using different families of quantum chemical methods, in particular semi-empirical methods, density functional methods and ab initio methods.50–52 These higher level calculations are applied to the ring opening reaction in benzofuroxans (see also Section 4). Interestingly, crystalline-state photolysis of 1-azido-2-nitrobenzene results in heterocycle formation to give the benzofuroxan which was identified by X-ray crystallography;53 this result is in agreement with computational and pyrolysis studies.54 Because nitroso compounds such as some nitroso-carbonyl compounds and nitrosoalkenes are highly reactive, their presence has often been demonstrated by Diels–Alder reactions.55,56 Similarly, there is an example of the use of a trapping reaction to demonstrate the formation of the o-dinitrosobenzene intermediate57 in eqn. (3); benzofuroxan is heated with p-anisyl azide in bromobenzene at 155 °C, and one of the three products obtained (in 40% yield) could be the result of addition of two molecules of the resulting nitrene to the o-dinitrosobenzene intermediate (eqn. (7)).
 |
Other examples of the use of a trapping reaction are provided by the reaction of furazano[3,4-b]quinoxaline 1-oxide with cyclopentadiene in which each of the NO groups of the 2,3-dinitroso form undergoes a Diels–Alder reaction with the diene,58 and by the trapping by means of cyclohexadiene of each of the two –N=O groups resulting from the thermal dissociation of 6-nitro[1,2,5]oxadiazolo[3,4-b]pyridine 1-oxide (eqn. (8)).
 |
59 Conclusive direct evidence of the production of the dinitroso intermediate is available from spectroscopic studies in which IR and UV spectroscopy are employed to detect the presence of o-dinitrosobenzene produced by the photolysis of either benzofuroxan or o-nitrophenyl azide in Ar matrices at 12–14 K.60–62 15N labeling of benzofuroxan showed60 that the singly labelled dinitroso species had two νN=O bands at 1516 and 1501 cm−1 whereas the unlabelled species had only one at 1516 cm−1 suggesting that coupling between the two N=O vibrations giving rise to an asymmetric and a symmetric stretch is very small.
2.3 Dinitroso compounds with intermolecular interactions
Intermolecular interaction of dinitroso compounds is exemplified by p-dinitrosobenzene. This compound exists at room temperature as an amorphous yellow/brown polymeric solid. This colour is in marked contrast to the blue/green colour of a monomeric nitroso compound or the colourless/creamy colour of a dimeric nitrosoarene. It is inherent in the chemistry of a dinitroso compound (22) that it has the potential to undergo polymerization by the formation of –N2O2– groups, either trans or cis or both, provided that there are no outweighing electronic, geometric or steric counter effects. The properties of the resultant polymers (23 and 24) will be dependent upon the structure of the R group. From 1887 onwards, when the first polymeric dinitrosobenzene was reported by Nietzki and Kehrmann,63 there have been only a small number of reported syntheses. For p-dinitrosobenzene itself, there is a considerable patent literature mainly relating to its use as a cross-linking agent in the manufacture of synthetic rubbers. Brydson64 has suggested a reaction sequence (eqn. (9)) to account for this behaviour.
 |

The polymeric p-dinitrosobenzenes identified are based upon the monomer unit (25) where X = Y = Z = H,36,63,65–71 X = CH3 and Y = Z = H,66,72–74 X = OCH3 and Y = Z = H,75 X = Y = CH3 and Z = H,71,76 X = Z = CH3 and Y = H,71 X = CH3 and Y = Cl and Z = H,77 X = CH3 and Y = i-Pr and Z = H,66,71,74,78 X = Y = t-Bu and Z = H,71 and X = Y = i-Pr and Z = H.71 In the case where X = Y = i-Pr, Z = H the spectroscopic properties show that this compound has a green/blue colour (λmax 800 nm) in solution consistent with the presence of N=O end groups in the monomer, but exists as a mixture of monomer and dimer in the solid state.71 In contrast, when X = Y = t-Bu and Z = H, polymerization does not appear to occur even in the solid state. Other polymeric aromatic dinitroso compounds are m-dinitrosobenzene6,70,79,80 and 1,4-dinitrosonaphthalene.73

All of the papers published before 1957 are primarily synthetic and the preparative route for the p-dinitrosoarenes required the preparation of the appropriate p-benzoquinone dioxime followed by oxidation using potassium ferricyanide, the yields being ~80%. The later use of chlorine as the oxidant67,69 increased the product yields to >90%. Catalytic autoxidation of the p-benzoquinone dioximes with nitrogen dioxide under an oxygen atmosphere in methylene chloride resulted in yields of 88–95% of the p-dinitrosoarenes.71 Electrochemical oxidation of p-benzoquinone dioxime is a simple two electron process producing p-dinitrosobenzene;81 this electrosynthetic route has been exploited for preparative purposes.71 Another possible route to a dinitroso compound could be from a trapping reaction of a diradical with two molecules of nitric oxide. A successful preparation of p-dinitrosobenzene has been reported82 from the pyrolysis of cis-3-hexen-1,5-diyne in the presence of nitric oxide at temperatures exceeding 150 °C (eqn. (10)).
 |
The preparation of m-dinitrosobenzene is from reduction of m-dinitrobenzene to the dihydroxylamine followed by oxidation.79 It may be noted that there have been no fully substantiated reports of the synthesis of any substituted m-dinitrosobenzene; the claimed synthesis83 of 2,4-dimethyl-3,5-dinitrosophenylglyoxylic acid and 2,4-dimethyl-3,5-dinitrosobenzoic acid lacks detailed evidence. A related claim for the preparation of 4-nitro-2,6-dinitroso-3-acetoxy-benzylacetate needs further investigation.84 The suggested6 formation of 1,3-dinitroso-4,6-di-monomethylaminobenzene85 and of 1,3-dinitroso-4,6-dinitrobenzene86 is not substantiated by the original papers. Kinetic measurements have been reported for reactions of some other substituted p-dinitrosobenzenes (X = C5H11, c-C6H11, C7H15, OC2H5, OCH(CH3)2, OC5H11, OC8H17, Y = Z = H)87 all of which were liberated in situ as monomers and not isolated.
In the early papers, attention was frequently directed to the solubility of the polymeric materials in a range of solvents and to the colours of the resultant solutions and also of the solid products. The very low solubility in non-polar solvents such as benzene led to attempts to recrystallize from nitrobenzene; yellow powders resulted from the dark green solutions. It was noted that when solution of some of the pale yellow p-dinitrosobenzene took place that dark brown residues remained. Ruggli and coworkers65,66 developed a purification strategy by sublimation to a water-cooled tube at a pressure of 12 mm Hg and bath temperatures of 120–160 °C. Up to 90% of the material would sublime under these conditions giving first a yellow coating and then small emerald-green crystals, the residue remaining being a dark brown amorphous powder. Bartusch88 observed that at the highest bath temperatures graphite-black colours appeared within the sublimate, and that opening the sublimate to the atmosphere always produced a yellow powdery sublimate. In our experience,70,74 sublimation at 10−3 mm Hg to a thimble at −78 °C from much lower bath temperatures (50–80 °C) gives a green sublimate which produces a yellow solid on warming; the polymerization of the green monomer to the yellow polymer probably occuring below room temperature.
It is important to know whether polymeric dinitrosobenzenes contain either or both of the azodioxy structures. The IR data of Boyer36 et al. and of Lüttke80 suggested that the trans-azodioxy group was present for the p- and m-dinitrosobenzenes, and this was confirmed later70 using both IR and Raman spectroscopy. The most direct evidence for the polymers containing only the trans-azodioxy group comes from solid state CP MAS 13C NMR spectroscopy. It is known11,89 that the chemical shifts of C-cis-N2O2 differ from those of C-trans-N2O2 by up to 1 ppm, and thus the participation of both isomeric forms in the polymer would be apparent from the solid state 13C NMR spectrum. For the polymers of p- and m-dinitrosobenzene, the solid state spectra show one quaternary carbon (both p- and m-) and one (p-) and three (m-) non-quaternary carbons, confirming that only the trans-azodioxy group is present in the solid polymer.11 The absence of any signal in the 165 ppm region characteristic of the C-NO group implies that these dinitrosobenzenes exhibit a very high degree of polymerization. Although the early literature reports an ‘ageing’ effect on the appearance and solubility of p-dinitrosobenzene, the solid state 13C NMR spectrum is unchanged over long periods of time.
The high degree of polymerization for the p- and m-dinitrosobenzenes is in marked contrast to the values established by Donaruma and coworkers90,91 for oligomeric 1,4-dinitrosocyclohexanes for which the synthetic route involves reduction of 1,4-dinitrocyclohexane to the corresponding dihydroxylamine, followed by mild oxidation to the dinitroso compound. It was shown that n = 2 or 3 in the oligomeric product (26).90 The later paper reports the synthesis of two further oligomers (27); for the first of these, n = 87 and for the second n = 10–11. These are the only reported syntheses of polymers of dinitrosocyclohexanes, and it is important to extend knowledge of such compounds by using further structural techniques.

Polymeric dinitrosoalkanes are reported in a patent92 and attention is directed to (28) (where n is claimed to be ≥2) and poly-2,7-dinitroso-2,7-dimethyloctane (29). It appears possible on the basis of the properties reported that these compounds may involve the formation of intramolecular cis-azodioxy groups.

The process of polymerization of m- and p-dinitrosobenzene may be contrasted with other addition polymerizations, where the initiation process is due to radical or cation or anion addition to the monomer. The polymerization of dinitrosobenzenes appears to be governed by the kinetic and thermodynamic characteristics of the system (eqn. (11)) and an initiator is not required.
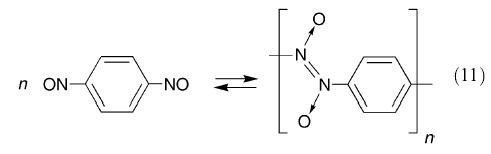 |
When the preparation is carried out at room temperature, the reaction goes rapidly to the right due to a low energy barrier coupled with a relatively high concentration of the monomer. All the evidence concerning polymeric p-dinitrosobenzene shows that it is stable only in the solid state. Donaruma93 was unable to detect any species other than the monomer when he attempted molecular weight measurements for the polymer in a range of solvent systems. This is paralleled by the fact that the mass spectrum of the heated polymer shows no peaks above that of the monomer unit.68,70 It thus appears that isolated polymer molecules of p-dinitrosobenzene in either the gaseous state or solution are thermodynamically unstable with respect to the monomer, and that the dissociation reaction is very rapid.
Although the degree of polymerization in polymeric dinitrosobenzenes is such that end group analysis cannot be done effectively by 13C NMR spectroscopy, we suggest94 that the C–N=O end groups, present to an extremely low degree, must presumably be available for further polymerization given the addition of more monomer. Thus the system should have the potential for behaving as a ‘living’ polymer in the presence of both air and hydroxylic solvents thereby enabling block copolymerization to take place. A further possibility, yet to be explored, is the direct copolymerization of two different dinitroso compounds. Addition of a monomeric C-nitroso compound should result in loss of the living polymer property due to a termination reaction forming azodioxy groups between the NO groups of the living polymer and the added RNO compound.
The effect of substituent groups on the properties of the polymers has received some attention but further detailed exploration is required. The effect of introducing methyl and isopropyl groups into the 2- and 5- positions, respectively, of p-dinitrosobenzene is the best documented case. 13C NMR spectroscopy shows that the solid polymer is totally dissociated to monomer in solution.70,71 The solubility of this polymer appears to be enhanced by the substituent groups to a greater extent than for the other mono- and di-methyl substituted p-dinitrosobenzenes that have been studied, and rapid depolymerization of the 2-methyl-5-isopropyl-1,4-dinitrosobenzene obviously ensues. In this manner, the polymeric solid behaves similarly to many substituted nitrosobenzenes which are trans-dimers in the solid state and which dissociate completely in solution at room temperature to give the monomer. Ruggli and Bartusch66 reported that the amorphous polymer of 2-methyl-5-isopropyl-1,4-dinitrosobenzene could be dissolved in benzene and that subsequent recrystallization produced white needles that could be subjected to further recrystallization from acetone, acetic acid or benzene. In such a colourless solid there are clearly no monomeric nitroso groups, but the solid is clearly different from the amorphous polymeric solid from which it was prepared. It is suggested therefore that the crystalline solid contains azodioxy groups. Kehrmann and Messinger78 had suggested, over a century ago, the formation of a dimer containing peroxidic links (30). It is more likely, however, that a dimer could be formed in which two cis-azodioxy groups bridge the two benzene rings in a structure analogous to that of a cyclophane (31). Such a compound implies the possibility of four isomeric structures.
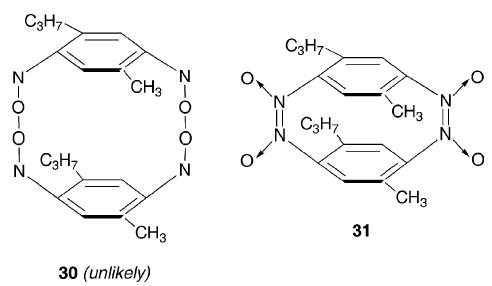
From consideration of the properties of other substituted nitrosobenzenes, it is possible to predict the structural properties of other substituted p-dinitrosobenzenes. Thus, it is known that 2,6-di-tert-butylnitrosobenzene is unable to self-dimerize due to steric effects, but that cross-dimerization is possible with unhindered nitroso compounds.95 This suggests, therefore, that 2,6-di-tert-butyl-1,4-dinitrosobenzene should be able to undergo head-to-tail self-polymerization (32) and tail-to-tail dimerization (33). In the second of these cases the molecule would possess two nitroso groups and one azodioxy group; further head-to-tail polymerization might also ensue with the hindered nitroso groups interacting with the unhindered tail nitroso groups of additional molecules of monomer. A further example of the steric effects of tert-butyl groups is provided by 2,5-di-tert-butyl-1,4-dinitrosobenzene (25 h). The compound 2-tert-butylnitrosobenzene is unable to self-dimerize96 and by analogy therefore (25 h) should exist as a non-polymerizing, non-dimerizing dinitroso compound. The strength of such an analogical argument is shown in the discovery71 that (25 h) is a green monomeric solid whereas the corresponding 2,5-diisopropyl-1,4-dinitrosobenzene exists as a polymer in the solid state which dissociates completely to the monomer in methylene chloride.

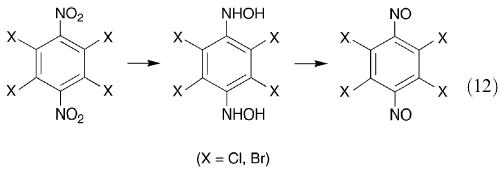
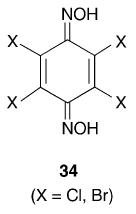
It is known97 that in the crystals of trans-dimeric 2,6-di-isopropylnitrosobenzene, the aromatic rings are virtually at right angles to the plane of the central C2N2O2 moiety, and that the 1H NMR spectrum shows that the molecule has a rigid structure. These considerations suggest that 2,3,5,6-tetraisopropyl-1,4-dinitrosobenzene should be able to polymerize, the resultant chain having the C6 rings alternately in plane and at right angles to the plane of the trans-azodioxy group. It has been claimed98 that oxidation of 2,3,5,6-tetrachloro- and 2,3,5,6-tetrabromo-1,4-diaminobenzene by 3-chloro- or 4-nitroperbenzoic acid gives the corresponding crystalline, green tetrahalogeno-1,4-dinitrosobenzene. This is unexpected in view of the formation of relatively stable trans-dimeric 2,6-dihalogenonitrosobenzenes. Attempted repetition of this work led to the production of the crystalline green tetrahalogeno-4-nitrosoanilines.74 This suggests that the best synthetic routes to the desired tetrasubstituted dinitrosobenzenes are either the oxidation of the p-benzoquinonedioxime (34) or the reduction of the 1,4-dinitro compound to the dihydroxylamine followed by mild oxidation to the dinitroso compound (eqn. (12)).
 |
It has already been noted that reactivity studies of substituted m-dinitrosobenzenes have yet to begin. An interesting combination of steric effects can be predicted for the dinitroso compounds (35–37) resulting from the corresponding tert-butyl-substituted 1,3-dinitrobenzenes using the dihydroxylamine preparative route. It is thus anticipated that 2-tert-butyl substitution (35) would lead to both nitroso groups being unable to undergo self dimerization reactions with both nitroso groups being forced into the anti-position, whereas 4-tert-butyl substitution (36) would cause this anti-effect to be confined to the 3-nitroso group only leaving the 1-nitroso group free to engage in dimerization reactions. For 5-substitution (37), it is predicted that neither of the nitroso groups would be affected by the bulky tert-butyl group leading to self polymerization.

2.4 Charged dinitroso compounds (nitrosolate anions)
In our discussion of dinitroso compounds thus far, we have confined our attention to electrically neutral species. There are, however, a number of reported examples of anionic dinitroso compounds derived from the various nitrosolic acids RC(NOH)NO first prepared by Wieland.99 The potassium salt99,100 of the aminomethylnitrosolic acid K+[H2NC(NO)2]− contains the anion (38; X = NH2) which possesses a planar structure (excluding the hydrogen atoms), the NO and CN bond lengths both being intermediate between those for single and double bonds. Further examples are provided by R = H,101 R = C6H5,102 and R = CH3.103,104 Both the acids and the anions are blue, and it has been shown105 that the aqueous solution of the anion where R = CH3 has an absorption maximum at 652 nm (ɛmax = 48 dm3mol−1cm−1).
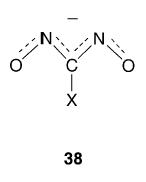
Schulz and coworkers have recently reported the preparation of new highly explosive alkali metal M+[HC(NO)2]− compounds (M = Li, Na, K, K(18-crown-6), Cs).106 The planarity of the anion (38) (where X = H) was determined by X-ray crystallographic characterization of the potassium and caesium salts, and suggests electronic delocalization over the whole anion. The calculated net atomic charges for O (−0.524), N (−0.043), C (0.008), and H (0.126) led the authors to suggest that the negative charge is located primarily on the O-atoms in the isolated anion.
3 Polynitroso compounds
Compounds such as 7-dimethylamino-4-nitrosobenzenefuroxan and the previously mentioned (20) and (21) may be considered relatives of polynitroso compounds.38 However, in this section, we will restrict the discussion to non-furoxan species.
Boyer6 has drawn attention to a possible preparation of a substituted 1,3,5-trinitrosobenzene from reduction of picrylazobenzene with potassium iodide in acetic acid; however, the noted citation107 only describes the reduction of one (and not three) of the nitro groups. Nevertheless, it is conceivable that studies of other 1,3,5-trinitrophenyl compounds could perhaps provide a starting point for the synthesis of trinitrosobenzene derivatives. In addition, the preparation108 of 1,3,5-trilithiobenzene and other trimetallated benzenes suggests a further synthetic route by reaction with nitrosyl chloride.

There are reports109,110 of the successful synthesis of p-nitrosated polystyrene (39). Dandge and Donaruma,111 and Yoneda et al.,110 have noted the tendency of p-nitrosated polystyrene (prepared from high molecular weight polystyrene) to gel, whereas that obtained from low molecular weight polystyrene is soluble in tetrahydrofuran. Drefahl et al.109 estimated that ~24% of the phenyl groups in the nitrosated polymer contained p-nitroso groups. The extent of interaction between the nitroso groups in different polymer chains to form azodioxy groups does not appear to have been studied. Other reactions of the nitroso groups are relatively unexplored. Condensation reactions with aniline using benzene as solvent, and with acetyl acetone and benzyl cyanide using THF solutions have been reported.109 In addition, the reductive acylation with zinc and acetic acid and acetic anhydride in benzene solution was carried out.109 Yoneda et al.110 have extended these observations to the condensation reactions with the monoamines aniline, p-chloro-, p-methyl- and o-methylaniline, and the diamines p-phenylenediamine, benzidine and o-toluidine. The diamines may therefore act as cross-linking between the polymer chains. Extension of these studies to encompass other reactions of the nitroso groups and to the synthesis of other polynitroso compounds is desirable. It may be possible to produce polynitroso compounds with 100% nitrosation of the pendant phenyl groups by polymerisation of p-nitrostyrene followed by reduction of the nitro group to either the amino or hydroxylamino group, followed further by mild oxidation to the nitroso group. A similar result could be achieved by the polymerisation of p-vinylaniline followed by mild oxidation of the amino groups to the nitroso group. At present, the extent of formation of azodioxy groups between the nitroso groups in different polymer chains is unknown. Such interaction could readily be revealed by the use of 13C CP MAS NMR spectroscopy, there being a difference of 25 ppm between the C-NO and C-N2O2-C signals. The available IR data110 imply that the monomeric nitroso group is present, but suggest the absence of the azodioxy group.
It has also been claimed112 that polymeric materials can be synthesized consisting of recurring p-dinitrosobenzene units linked by methylene groups between the 2 and 6 C-atoms of the individual rings. Presumably, any intermolecular polymerization through formation of azodioxy groups would lead to a cross-linked polymeric structure, but details of this do not appear to be given.
It has been established that polymers of three different nitrovinylbenzaldehydes can be converted to the respective acetals with alcohol and boron trifluoride-etherate and then subjected to UV irradiation in benzene solution to yield the corresponding o-nitrosobenzoate polymers (eqn. (13)).113 Information concerning the structural and spectroscopic properties of the nitroso and/or azodioxy groups in the low molecular weight polymeric materials obtained appears to be lacking.
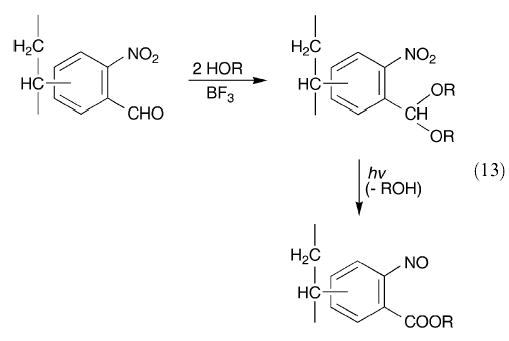 |
4 Theoretical studies of dinitroso compounds
In recent years, a small number of studies have been made on the structures of dinitroso compounds. Politzer and Bar-Adon114 calculated the optimal geometry (at the 6–31G level), the bond orders (with the STO-3G basis set), and electrostatic potentials (with the STO-5G basis set) of dinitrosoacetylene (40). The molecule is surprisingly non-linear, and the dihedral angle between the CCO planes is 87 ° which suggests that the NO groups are in perpendicular planes presumably due to each NO group conjugating individually with one of the CC π bonds. To the best of our knowledge, there is no experimental evidence for the existence of dinitrosoacetylene, and only a very small amount of evidence for mononitrosoacetylenes.115,116

Houk and coworkers117 investigated the dimerization of acetonitrile oxide and p-chlorobenzonitrile oxide to furoxans (eqn. (14)) by performing density functional theoretical calculations at the B3LYP/6-31G* level.
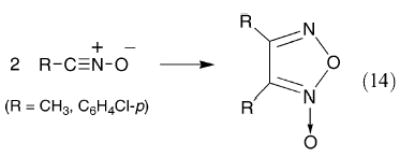 |
They conclude that the dimerization is stepwise via a dinitrosoalkene intermediate. The potential energy surface for furoxan formation from acetonitrile oxide is shown in Fig. 3. The formation of the first intermediate 2(ctc) is exothermic by 11.9 kcal mol−1 in the gas phase, and the intermediate is best represented as a dinitrosoalkene diradical (41) (actually a bis-iminoxy radical system).
Fig. 3.

Computed potential energy surface for the dimerization of acetonitrile oxide to furoxan in the gas phase. Reproduced with permission from Z.-H. Yu, P. Caramella, and K. N. Houk, J. Am. Chem. Soc. 2003, 125, 15420–15425.117 Copyright (2003) American Chemical Society.

This intermediate 2(ctc) forms via a singlet diradical TS1 in which the NO groups are trans presumably to avoid electrostatic repulsion between the two NO groups, and also between the N lone pair and the π electrons involved in forming the central carbon–carbon bond. Intramolecular rotation then forms the TS3 transition state and the non-planar 2(ccc) conformer (the non-planarity is ascribed to the O3–O6 repulsion). Intramolecular N…O bond formation then produces the stable furoxan. On the basis of these results, the authors propose that the experimentally observed tautomerizations of furoxans are likely to proceed via diradical transition states and dinitrosoalkene intermediates.
The related furoxan formation from p-chlorobenzonitrile oxide was computed similarly (Fig. 4),117 and also reveals a dinitrosoalkene diradical intermediate. The higher barrier for dimerization of this aromatic nitrile oxide was attributed to a smaller extent of conjugation between the C6-ring and the CNO moiety in the first transition state TS8 (analogous to TS1 in Fig. 3).
Fig. 4.
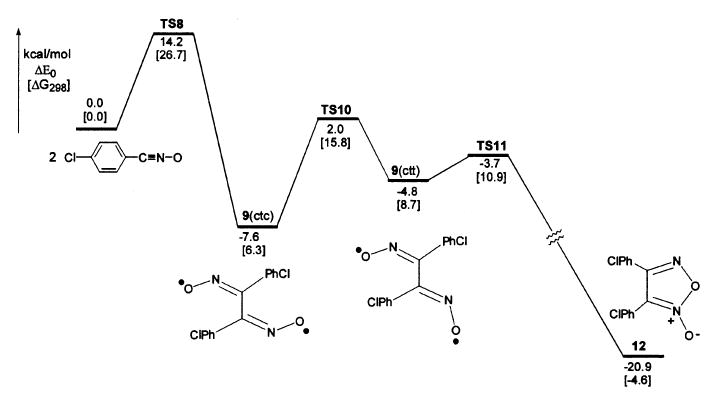
Computed potential energy surface for the dimerization of p-chlorobenzonitrile oxide to furoxan in the gas phase. Reproduced with permission from Z.-H. Yu, P. Caramella, and K. N. Houk, J. Am. Chem. Soc. 2003, 125, 15420–15425.117 Copyright (2003) American Chemical Society.
Other papers on ab initio calculations for 1,2-dinitrosoethylene at different levels of computation have been published.118–124 Interestingly, IR spectroscopic evidence for the formation of 2,3-dinitrosobut-2-ene as the photolabile intermediate during UV photolytic decomposition of dimethylfuroxan has been obtained,124 and the product was identified as the tct-2,3-DNB conformer with the help of theoretical calculations.
The calculations for three dinitrosobenzenes at the ab initio STO-3G and 3-21G levels give the bond distances (3–21G//3–21G) for the most stable conformers (42–44) presented in Table 1.122 It is apparent that the CC bond lengths are scarcely altered by disubstitution by the nitroso groups, and that the lengthening of the CN bond in the o- and p-isomers is in accord with a slight reduction in resonance interaction between the nitroso groups and the benzene ring as a result of the conjugation of the two π-electron withdrawing nitroso groups.
Table 1.
Geometries calculated for the most stable conformers of dinitrosobenzenes (3–21G//3–21G by ab initio orbital methods; bond lengths in Å)122
| Bond | Nitroso | 1,2-Dinitroso | 1,3-Dinitroso | 1,4-Dinitroso |
|---|---|---|---|---|
| NO | 1.223 | 1.220 | 1.222 | 1.221 |
| CN | 1.442 | 1.453 | 1.442 | 1.448 |
| C1C2 | 1.385 | 1.384 | 1.378 | 1.383 |
| C2C3 | 1.380 | 1.381 | 1.378 | 1.379 |
| C3C4 | 1.389 | 1.381 | 1.384 | 1.381 |
| C4C5 | 1.384 | 1.387 | 1.383 | 1.383 |
| C5C6 | 1.383 | 1.381 | 1.383 | 1.379 |
| C6C1 | 1.381 | 1.381 | 1.384 | 1.381 |

It has been demonstrated117 that theoretical calculations for dinitrosoalkenes and hence, by implication, for molecules containing two nitroso groups attached to π-bonded carbon frameworks, need to be performed at higher levels in order to to clarify the effects of linking the strong π-electron accepting nitroso groups to a conjugated system. It was suggested earlier125 that successful theoretical understanding of the benzofuroxan/o-dinitrosobenzene system is dependent upon the inclusion of electron correlation in the theoretical studies as illustrated by recent studies50–52 of the reaction mechanisms of benzofuroxans in which o-dinitrosobenzenes feature as important intermediates. It seems likely that the differences between the results obtained from earlier computational studies of dinitrosoalkenes are a result of working at lower computational levels.
5 Conclusions
The chemistry of dinitroso and polynitroso compounds remains relatively unexplored. It is reasonable to assume that the diversity of reactions exhibited by the mononitroso species should result in some unique chemistry associated with molecules containing more than one C-nitroso group. In particular, the use of dinitroso compounds as spin trapping agents and their chemical behavior in the presence of metals2,126 should prove to be topics of considerable interest. In addition, a need exists for synthetic routes to tri- and polynitroso compounds due the potential uses of these compounds in new materials.
Acknowledgments
We gratefully acknowledge the translation from the Japanese of reference 110 provided by Dr Benjamin Sklarz, and correspondence from Drs M. V. Lakshmikantham and L. G. Donaruma and Professor R. D. Topsom. GBRA is grateful to the National Institutes of Health for financial support.
Biographies
 Brian Gowenlock was born in Oldham, Lancashire, England in 1926. He received his B.Sc., M.Sc., and Ph.D. degrees from the University of Manchester. His Ph.D. studies in gas phase kinetics were carried out under the direction of Dr. Ernest Warhurst and Professor Michael Polanyi. He held posts at the University of Wales, Swansea, and the University of Birmingham before joining the Heriot-Watt University in Edinburgh as a Professor (1966–1990). He has been an honorary member of the University of Exeter since 1992 where he is a Visiting Professor. He is a Fellow of the Royal Society of Edinburgh (FRSE), and served on the University Grants Committee from 1976–1985 for services to which he was appointed Commander of the Order of the British Empire (CBE) in 1986.
Brian Gowenlock was born in Oldham, Lancashire, England in 1926. He received his B.Sc., M.Sc., and Ph.D. degrees from the University of Manchester. His Ph.D. studies in gas phase kinetics were carried out under the direction of Dr. Ernest Warhurst and Professor Michael Polanyi. He held posts at the University of Wales, Swansea, and the University of Birmingham before joining the Heriot-Watt University in Edinburgh as a Professor (1966–1990). He has been an honorary member of the University of Exeter since 1992 where he is a Visiting Professor. He is a Fellow of the Royal Society of Edinburgh (FRSE), and served on the University Grants Committee from 1976–1985 for services to which he was appointed Commander of the Order of the British Empire (CBE) in 1986.
 George Richter-Addo was born in Scotland in 1957 and grew up in Ghana (West Africa). He received his Honors B.Sc. and Dip. Ed. degrees in 1982 from the University of Cape Coast in Ghana, and his Ph.D. degree in Chemistry in 1988 under the direction of Professor Peter Legzdins at the University of British Columbia in Canada. Upon completion of his postdoctoral work with Professors Legzdins (Canada) and John Gladysz (University of Utah, USA), he joined the faculty at the University of Oklahoma in 1993, where he is now a University of Oklahoma Presidential Professor. His primary research interests lie in nitric oxide chemistry and its role in biology.
George Richter-Addo was born in Scotland in 1957 and grew up in Ghana (West Africa). He received his Honors B.Sc. and Dip. Ed. degrees in 1982 from the University of Cape Coast in Ghana, and his Ph.D. degree in Chemistry in 1988 under the direction of Professor Peter Legzdins at the University of British Columbia in Canada. Upon completion of his postdoctoral work with Professors Legzdins (Canada) and John Gladysz (University of Utah, USA), he joined the faculty at the University of Oklahoma in 1993, where he is now a University of Oklahoma Presidential Professor. His primary research interests lie in nitric oxide chemistry and its role in biology.
References
- 1.Gowenlock BG, Richter-Addo GB. Chem Rev. 2004;104:3315–3340. doi: 10.1021/cr030450k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee J, Chen L, West AH, Richter-Addo GB. Chem Rev. 2002;102:1019–1065. doi: 10.1021/cr0000731. [DOI] [PubMed] [Google Scholar]
- 3.S. R. Sandler and W. Karo, Organic Functional Group Preparations, 2nd ed.; Academic Press: Orlando, 1986, Vol. 2, Chapter 16.
- 4.D. L. H. Williams, Nitrosation, Cambridge University Press: Cambridge, U.K., 1988, Chapters 2 and 3.
- 5.Touster O. Org React. 1953;7:327–377. [Google Scholar]
- 6.J. H. Boyer, Methods of Formation of the Nitroso Group and Its Reactions, in The Chemistry of the Nitro and Nitroso Groups, Part 1; H. Feuer, Ed.; Interscience: New York, 1969, Chapter 5, p 215–299.
- 7.H. Metzger and H. Meier, in Methoden der Organischen Chemie; E. Müller, Ed.; Georg Thieme Verlag: Stuttgart, 1971, Vol. (Band) X/1, pp 891–1016.
- 8.W. Seidenfaden, in Methoden der Organischen Chemie; E. Müller, Ed.; Georg Thieme Verlag: Stuttgart, 1971, pp 1017–1090.
- 9.A. R. Katritzky, O. Meth-Cohn and C. W. Rees, Comprehensive Functional Group Transformations, Pergamon Press: London, 1995.
- 10.Adam W, Krebs O. Chem Rev. 2003;103:4131–4146. doi: 10.1021/cr030004x. [DOI] [PubMed] [Google Scholar]
- 11.P. McKenna, Ph.D. Thesis, Heriot-Watt University, 1986.
- 12.Müller E, Metzger H. Chem Ber. 1954;87:1282–1293. [Google Scholar]
- 13.Fabré R, Bertrand G. Revue Générale Caoutchouc Plastique. 1965;42:405–412. [Google Scholar]
- 14.Zakharkin LI, Zhigareva GG. J Gen Chem USSR. 1975;45:1268–1275. (Engl. Trans.). [Google Scholar]
- 15.Kotali A, Papageorgiou VP. Chimika Chronika (New Series) 1989;18:179–189. [Google Scholar]
- 16.Hütter K, Baader E, Frobel K, Zeeck A, Bauer K, Gau W, Kurz J, Karl W, Wendisch D. J Antibiotics. 1986;39:1193–1204. doi: 10.7164/antibiotics.39.1193. [DOI] [PubMed] [Google Scholar]
- 17.Kind R, Hütter K, Zeeck A, Schmidt-Base K, Egert E. J Antibiotics. 1989;42:7–13. doi: 10.7164/antibiotics.42.7. [DOI] [PubMed] [Google Scholar]
- 18.(a) A. Zeeck, K. Hütter and R. Kind, 4th European Carbohydrate Symposium, 1987, p. C-31; (b) A. Zeeck, personal communication to B.G.G.
- 19.Bannister B, Zapotocky BA. J Antibiotics. 1992;45:1313–1324. doi: 10.7164/antibiotics.45.1313. [DOI] [PubMed] [Google Scholar]
- 20.Moinet C, Simonneaux G, Autret M, Hindre F, Le Plouzennec M. Electrochim Acta. 1993;38:325–328. [Google Scholar]
- 21.Brunner H, Loskot L. J Organomet Chem. 1973;61:401–414. [Google Scholar]
- 22.Becker PN, Bergman RG. J Am Chem Soc. 1983;105:2985–2995. [Google Scholar]
- 23.Becker PN, Bergman RG. Organometallics. 1983;2:787–796. [Google Scholar]
- 24.Chiu WH, Cheung KK, Che CM. J Chem Soc, Chem Commun. 1995:441–442. [Google Scholar]
- 25.Leplyanin GV, Antonova LF, Rafikov SR, Faizullov RR, Zaev EE, Ponomareva VA, Grechishnikov YG. Izv Akad Nauk SSSR, Ser Khim. 1975:754–757. Chem. Abstr., 1975, 83, 59472. [Google Scholar]
- 26.Crawford GH., Jr Nitric oxide-olefin reaction products and thermoplastic and elastomeric materials therefrom, USA Patent 3,436,384. Chem Abstr. 1969;70:107017. [Google Scholar]
- 27.John EO, Kirchmeier RL, Shreeve JM. Inorg Chem. 1992;31:329–331. [Google Scholar]
- 28.Jackson YA, Parakka JP, Lakshmikantham MV, Cava MP. J Org Chem. 1997;62:2616–2618. doi: 10.1021/jo962209u. [DOI] [PubMed] [Google Scholar]
- 29.M. V. Lakshmikantham, personal communication to BGG (1997).
- 30.Vollmann H, Becker H, Corell M, Streeck H. Liebigs Ann Chem. 1937;531:1–73. [Google Scholar]
- 31.Errede LA, Hoyt JM. J Am Chem Soc. 1960;82:436–439. [Google Scholar]
- 32.Singh P. J Org Chem. 1975;40:1405–1408. [Google Scholar]
- 33.Snyder JP, Heyman ML, Suciu EN. J Org Chem. 1975;40:1395–1404. [Google Scholar]
- 34.Greene FD, Gilbert KE. J Org Chem. 1975;40:1409–1415. [Google Scholar]
- 35.Smith MA, Weinstein B, Greene FD. J Org Chem. 1980;45:4597–4602. [Google Scholar]
- 36.Boyer JH, Toggweiler U, Stoner GA. J Am Chem Soc. 1957;79:1748–1751. [Google Scholar]
- 37.Harries C, Jablonsky L. Ber. 1898;31:549–550. [Google Scholar]
- 38.Katritzky AR, Gordeev MF. Heterocycles. 1993;35:483–518. [Google Scholar]
- 39.Boulton AJ, Gray ACG, Katritzky AR. J Chem Soc. 1965;168:5958–5964. [Google Scholar]
- 40.Bailey AS, Case JR. Tetrahedron. 1958;3:113–131. [Google Scholar]
- 41.Grundmann C, Frommeld HD, Flory K, Datta SK. J Org Chem. 1968;33:1464–1466. [Google Scholar]
- 42.Mallory FB, Cammarata A. J Am Chem Soc. 1966;88:61–64. [Google Scholar]
- 43.Hwang KJ, Jo I, Shin YA, Yoo SE, Lee JH. Tetrahedron Lett. 1995;36:3337–3340. [Google Scholar]
- 44.Feelisch M, Schonafinger K, Noack E. Biochem Pharmacol. 1992;44:1149–1157. doi: 10.1016/0006-2952(92)90379-w. [DOI] [PubMed] [Google Scholar]
- 45.Wang PG, Xian M, Tang X, Wu X, Wen Z, Cai T, Janczuk AJ. Chem Rev. 2002;102:1091–1134. doi: 10.1021/cr000040l. [DOI] [PubMed] [Google Scholar]
- 46.Kaufman JVR, Picard JP. Chem Rev. 1959;59:429–461. [Google Scholar]
- 47.Boulton AJ, Ghosh PB. Adv Heterocycl Chem. 1969;10:1–41. [Google Scholar]
- 48.Gasco A, Boulton AJ. Adv Heterocycl Chem. 1981;29:251–340. [Google Scholar]
- 49.Hoffman R, Gleiter R, Mallory FB. J Am Chem Soc. 1970;92:1460–1466. [Google Scholar]
- 50.Eckert F, Rauhut G. J Am Chem Soc. 1998;120:13478–13484. [Google Scholar]
- 51.Eckert F, Rauhut G, Katritzky AR, Steel PJ. J Am Chem Soc. 1999;121:6700–6711. [Google Scholar]
- 52.Rauhut G, Eckert F. Sci Prog (Northwood, UK) 1999;82:209–231. [Google Scholar]
- 53.Takayama T, Kawano M, Uekusa H, Ohashi Y, Sugawara T. Helv Chim Acta. 2003;86:1352–1358. [Google Scholar]
- 54.Rauhut G, Eckert F. J Phys Chem A. 1999;103:9086–9092. and references therein. [Google Scholar]
- 55.Kirby GW. Chem Soc Rev. 1977;6:1–24. and references therein. [Google Scholar]
- 56.Gilchrist TL. Chem Soc Rev. 1983;12:53–73. [Google Scholar]
- 57.Bulacinski AB, Scriven EFV, Suschitzky H. Tetrahedron Lett. 1975:3577–3579. [Google Scholar]
- 58.Gallos JK, Malamidou-Xenikaki E. Heterocycles. 1994;37:193–198. [Google Scholar]
- 59.Sebban M, Goumont R, Halle JC, Marrot J, Terrier F. Chem Commun. 1999:1009–1010. [Google Scholar]
- 60.Dunkin IR, Lynch MA, Boulton AJ, Henderson N. J Chem Soc, Chem Commun. 1991:1178–1179. [Google Scholar]
- 61.Hacker NP. J Org Chem. 1991;56:5216–5217. [Google Scholar]
- 62.Murata S, Tomioka H. Chem Lett. 1992:57–60. [Google Scholar]
- 63.Nietzki R, Kehrmann F. Ber. 1887;20:613–636. [Google Scholar]
- 64.J. A. Brydson, Rubbery Materials and their Compounds, Elsevier; London, 1988, pp 370–371.
- 65.Ruggli P, Petitjean C. Helv Chim Acta. 1938;21:711–732. [Google Scholar]
- 66.Ruggli P, Bartusch G. Helv Chim Acta. 1944;27:1371–1384. [Google Scholar]
- 67.Khishchenko YS, Makarov MA, Gareev GA, Cherkashina NA, Koptina GS. Zh Prikl Khim. 1969;42:2384–2386. [Google Scholar]
- 68.Korsunskii BL, Nazin GM, Stepanov VR, Fedotov AA. Kinetics and Catalysis. 1993;34:539–542. [Google Scholar]
- 69.Ermakov OA, Komkova YF. J Org Chem, USSR. 1985;20:2053–2054. (Engl. Trans.). [Google Scholar]
- 70.Anderson L, Cameron M, Gowenlock BG, McEwen IJ. J Chem Soc, Perkin Trans 2. 1992:243–245. [Google Scholar]
- 71.Rathore R, Kim JS, Kochi JK. J Chem Soc, Perkin Trans 1. 1994:2675–2684. [Google Scholar]
- 72.Mehne P. Ber. 1888;21:729–735. [Google Scholar]
- 73.Nietzki R, Guiterman AL. Ber. 1888;21:428–434. [Google Scholar]
- 74.Anderson L, Boyd ASF, Cameron M, Gowenlock BG, Higginson CM, McEwen IJ, Smith GP. J Chem Res (S) 1994:245. J. Chem. Res. (M), 1994, 1477. [Google Scholar]
- 75.Best TT. Liebigs Ann Chem. 1889;255:176–188. [Google Scholar]
- 76.Pflug L. Liebigs Ann Chem. 1889;255:168–176. [Google Scholar]
- 77.Kehrmann F. Ber. 1915;48:2021–2035. [Google Scholar]
- 78.Kehrmann F, Messinger J. Ber. 1890;23:3557–3564. [Google Scholar]
- 79.Alway FJ, Gortner RA. Ber. 1905;38:1899–1901. [Google Scholar]
- 80.Lüttke W. Z Elektrochem. 1957;61:976–986. [Google Scholar]
- 81.Kokkinidis G, Papanastasiou G. J Electroanal Chem. 1988;257:239–255. [Google Scholar]
- 82.Roth WR, Hopf H, Horn C. Chem Ber. 1994;127:1765–1779. [Google Scholar]
- 83.Claus A. J Prakt Chem (Leipzig) 1890;41:483–514. [Google Scholar]
- 84.Schultz G, Ganguly KL. Ber. 1925;58:702–708. [Google Scholar]
- 85.Fischer O. Liebigs Ann Chem. 1895;286:145–186. [Google Scholar]
- 86.Borsche W, Feske E. Ber. 1926;59:815–821. [Google Scholar]
- 87.Khizhnyi VA, Goloverda GZ. Teor Eksperim Khim. 1988;24:161–167. [Google Scholar]
- 88.G. Bartusch, Ph. D. Thesis, University of Basel, 1944.
- 89.Orrell KG, Sik V, Stephenson D. Magn Reson Chem. 1987;25:1007–1011. [Google Scholar]
- 90.Childress WL, Donaruma LG. Macromolecules. 1974;7:427–430. [Google Scholar]
- 91.Dandge DK, Donaruma LG. Polym Sci Technol. 1984;25:173–183. Chem. Abstr., 1984, 101, 38947. [Google Scholar]
- 92.J. F. Pazos, Polymers coupled by nitroso groups, USA Patent 3,872,057, 1975, (Chem. Abstr., 1975, 83, 11401).
- 93.L. G. Donaruma, personal communication to BGG (1975).
- 94.Based on discussions between BGG and Professor K. G. Orrell.
- 95.Barclay LRC, Carson DL, Gray JA, Grossman M, Khazanie PG, Milton JR, Scott CE. Can J Chem. 1978;56:2665–2672. [Google Scholar]
- 96.Gowenlock BG, Cameron M, Boyd ASF, Al-Tahou BM, McKenna P. Can J Chem. 1994;72:514–518. [Google Scholar]
- 97.Gowenlock BG, McCullough KJ. J Chem Soc, Perkin Trans 2. 1989:551–553. [Google Scholar]
- 98.Hedayatullah M, Raoult JC, Denivelle L. Bull Soc Chim Fr. 1973:2702–2705. [Google Scholar]
- 99.Wieland H. Ber. 1906;38:1445–1461. [Google Scholar]
- 100.Berges P, Hinrichs W, von Schlabrendorff C, Klar G. J Chem Res (S) 1987:250. J. Chem. Res. (M), 1987, 2012. [Google Scholar]
- 101.Wieland H, Hess H. Ber. 1909;42:4175–4191. [Google Scholar]
- 102.Wieland H, Bauer H. Ber. 1906;39:1480–1488. [Google Scholar]
- 103.Wieland H. Liebigs Ann Chem. 1907;353:65–105. [Google Scholar]
- 104.Wiemer DF, Leonard NJ. J Org Chem. 1976;41:2985–2988. [Google Scholar]
- 105.Armand J, Minvielle RM. Compt Rend. 1965;260:2512–2515. [Google Scholar]
- 106.Brand H, Mayer P, Polborn K, Schultz A, Weigand JJ. J Am Chem Soc. 2005;127:1360–1361. doi: 10.1021/ja042877u. [DOI] [PubMed] [Google Scholar]
- 107.Willgerodt C. Ber. 1891;24:592–595. [Google Scholar]
- 108.Rot N, Bickelhaupt F. Organometallics. 1997;16:5027–5031. [Google Scholar]
- 109.Drefahl G, Horhold HH, Hofmann KD. J Prakt Chem (Leipzig) 1968;37:91–96. [Google Scholar]
- 110.Yoneda A, Sugihara K, Hayashi K, Tanaka M. Kobunshi Kagaku. 1973;30:180–185. Chem. Abstr., 1973, 32377a. [Google Scholar]
- 111.D. K. Dandge and L. G. Donaruma, Nitroso Polymers, in Encyl. Polym. Sci. Eng.; J. I. Kroschwitz, Ed.; Wiley: New York, 1987, Vol. 10, pp 185–191.
- 112.Lord Manufacturing Company. “Nitrogen-Containing Polymers”. British Patent 961, 097 (1964). USA Patent 19,610,906 (Chem. Abstr. 61:55454).
- 113.Kamogawa H, Takei M. J Polymer Sci: Part A. 1987;25A:967–977. [Google Scholar]
- 114.Politzer P, Bar-Adon R. J Am Chem Soc. 1987;109:3529–3534. [Google Scholar]
- 115.Motte JC, Viehe HG. Chimia. 1975;29:515–516. Chem. Abstr.584:58518. [Google Scholar]
- 116.Robson E, Tedder JM, Woodcock DJ. J Chem Soc (C) 1968:1324–1328. [Google Scholar]
- 117.Yu ZX, Caramella P, Houk KN. J Am Chem Soc. 2003;125:15420–15425. doi: 10.1021/ja037325a. [DOI] [PubMed] [Google Scholar]
- 118.Sedano E, Sarasola C, Ugalde JM, Irazabalbeitia IX, Guerrero AG. J Phys Chem. 1988;92:5094–5096. [Google Scholar]
- 119.Sedano E, Sarasola C, Ugalde JM, Fakultatea K, Unibertsitatea EH. Tetrahedron. 1989;45:6537–6544. [Google Scholar]
- 120.Sarasola C, Choi SC, Ugalde JM, Boyd RJ. J Phys Org Chem. 1990;3:143–146. [Google Scholar]
- 121.Stevens J, Schweizer M, Rauhut G. J Am Chem Soc. 2001;123:7326–7333. doi: 10.1021/ja010792c. [DOI] [PubMed] [Google Scholar]
- 122.Marriott S, Topsom RD, Gowenlock BG. J Phys Chem. 1990;94:5220–5221. [Google Scholar]
- 123.R. D. Topsom, private communication to BGG (1991).
- 124.Himmel HJ, Konrad S, Friedrichsen W, Rauhut G. J Phys Chem A. 2003;107:6731–6737. [Google Scholar]
- 125.Klenke B, Friedrichsen W. Tetrahedron. 1996;52:743–752. [Google Scholar]
- 126.Cameron M, Gowenlock BG, Vasapollo G. Chem Soc Rev. 1990;19:355–379. [Google Scholar]