

Abstract
Humans eating diets deficient in the essential nutrient choline can develop organ dysfunction. We hypothesized that common single nucleotide polymorphisms (SNPs) in genes involved in choline metabolism influence the dietary requirement of this nutrient. Fifty-seven humans were fed a low choline diet until they developed organ dysfunction or for up to 42 days. We tested DNA SNPs for allelic association with susceptibility to developing organ dysfunction associated with choline deficiency. We identified an SNP in the promoter region of the phosphatidylethanolamine N-methyltransferase gene (PEMT; −744 G→C; rs12325817) for which 18 of 23 carriers of the C allele (78%) developed organ dysfunction when fed a low choline diet (odds ratio 25, P=0.002). The first of two SNPs in the coding region of the choline dehydrogenase gene (CHDH; +318 A→C; rs9001) had a protective effect on susceptibility to choline deficiency, while a second CHDH variant (+432 G→T; rs12676) was associated with increased susceptibility to choline deficiency. A SNP in the PEMT coding region (+5465 G→A; rs7946) and a betaine:homocysteine methyl-transferase (BHMT) SNP (+742 G→A; rs3733890) were not associated with susceptibility to choline deficiency. Identification of common polymorphisms that affect dietary requirements for choline could enable us to identify individuals for whom we need to assure adequate dietary choline intake.—da Costa, K.-A., Kozyreva, O. G., Song, J., Galanko, J. A., Fischer, L. M., Zeisel, S. H. Common genetic polymorphisms affect the human requirement for the nutrient choline.
Keywords: choline deficiency, phosphatidylethanolamine N-methyltransferase, PEMT, choline dehydrogenase, CHDH, betaine:homocysteine methyltransferase, BHMT, genetic polymorphism
Choline is an essential nutrient needed for structural integrity and signaling functions of cell membranes, methyl group metabolism, and neurotransmitter synthesis (1). Humans eating diets deficient in choline develop fatty liver, liver damage, and muscle damage (2–4). These effects occur, in part, because a specific lack of phosphatidylcholine limits the export of excess triglyceride from liver (5, 6) and induces apoptosis and subsequent leakage of enzymes (e.g., AST, ALT, and CPK) from tissues of liver and muscle (3, 7, 8). Women’s dietary requirements for choline are of special interest because deficient maternal dietary intake of choline during pregnancy in humans was associated with a 4-fold increased risk of having a baby with a neural tube defect (9). In addition, offering pregnant rodents diets deficient in choline resulted in perturbed brain development in their fetuses (10–13).
We do not understand all of the factors that influence the dietary requirement for choline in humans, but we know that the requirement is modified by dietary availability of other methyl donors (1) and by endogenous de novo biosynthesis of choline moiety (14). (Fig. 1) The methylation of homocysteine can be accomplished by using a methyl group derived from one-carbon metabolism or by using a methyl group derived from choline. When choline is used as a methyl group source, it is first irreversibly oxidized to form betaine by choline dehydrogenase (CHDH) and is no longer available for synthesis of membrane phosphatidylcholine. Betaine, once formed, donates its methyl group to homocysteine via betaine:homocysteine methyltransferase (BHMT) to form methionine (1). Endogenous biosynthesis of choline occurs mainly in the liver, where phosphatidylethanolamine is methylated by phosphatidylethanolamine N-methyltransferase (PEMT) to form phosphatidylcholine. Pemt −/− mice develop fatty liver and liver damage (15) because they cannot form required amounts of phosphatidylcholine in membranes and they become choline deficient despite eating diets containing recommended amounts of choline (6, 8).
Figure 1.
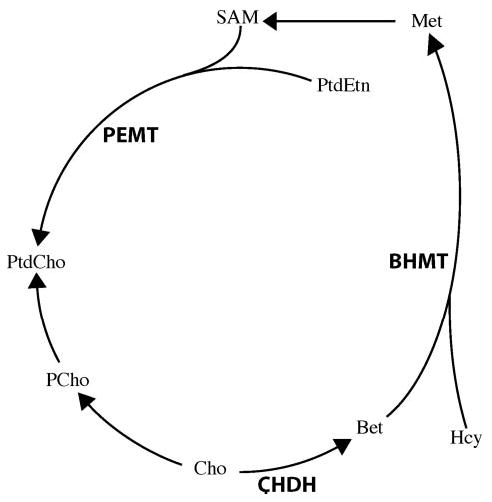
Three important genes in choline metabolism for which we identified single nucleotide polymorphisms. PEMT = phosphatidylethanolamine N-methyltransferase, which catalyzes the reaction to make phosphatidylcholine (PtdCho) from phosphatidylethanolamine (PtdEtn) using S-adenosylmethionine (SAM) to donate methyl groups; CHDH = choline dehydrogenase, which along with betaine aldehyde dehydrogenase irreversibly oxidizes choline (Cho) to form betaine (Bet); BHMT = betaine:homocysteine methyltransferase, which donates its methyl group to homocysteine (Hcy) to form methionine (Met); PCho = phosphocholine.
Genetic variations exist in these genes in humans, but those SNPs that have functional effects on the above enzymes of choline metabolism have not yet been completely identified. More than 98 polymorphisms exist in the PEMT gene (16), but only 1 SNP is known to be functionally significant (rs7946, +5465 G→A) (17). Two SNPs that produced an amino acid substitution in CHDH, rs9001 (318A→C; E40A) and rs12676 (432G→T; L78R) and one in BHMT, rs3733890 (742G→A; R239Q) were recently identified (18, 19) (Table 1). The latter may play a protective role in the risk of coronary heart disease (20). Nevertheless, there is no information at present about the possible functional effects of these polymorphisms. If decreased availability of methyl groups from choline is responsible for organ dysfunction in choline deficiency, then SNPs in CHDH or BHMT could alter susceptibility to developing organ dysfunction when fed a low choline diet. Alternatively, if organ damage is due to defective membrane formation, SNPs in PEMT could modify de novo phosphatidylcholine synthesis and SNPs resulting in decreased CHDH activity could decrease the use of choline as a methyl donor and make more substrate available for phosphatidylcholine synthesis from preexisting choline moiety, thereby altering susceptibility to developing organ dysfunction when fed a low choline diet.
TABLE 1.
SNPs studieda
| Gene | rs number | Base pair and sequence change |
|---|---|---|
| MTHFD1 | rs2236225 | +1958 G → A |
| PEMT | rs12325817 | −744 G → Cb |
| PEMT | rs2278952 | +164 C → Tb |
| PEMT | rs7946 | +5465 G → Ab |
| PEMT | not yet designated | −314 C→Tb |
| PEMT | not yet designated | +29 C→Gb |
| CHDH | rs9001 | +318 A → C |
| CHDH | rs12676 | +432 G → T |
| BHMT | rs3733890 | +742 G → A |
Each SNP is mapped to the genome and assigned a RefSNP accession ID (rs number). Base pair and sequence changes, also listed, are subject to revision when genes are resequenced.
PEMT SNP base pair numbers are numbered from transcription start site (25).
We conducted a study in which humans were fed a diet containing the Adequate Intake concentration for choline for 10 days, then fed a diet containing little choline for up to 6 wk, then fed a diet containing choline. We characterized susceptibility to developing liver and muscle dysfunction when fed the diet low in choline content and determined whether such susceptibility was influenced by common genetic polymorphisms.
MATERIALS AND METHODS
Study design
Healthy males (n=31) and females (n=35) were recruited by advertising. They ranged in age from 18 to 70 years and had body mass indices between 19 and 33. Informed consent was obtained from all participants after the nature and possible consequences of the study were explained; the criteria for subject selection and all details of the clinical protocol were approved by the Institutional Review Board of the University of North Carolina at Chapel Hill (UNC-CH). The ethnicity of the participants was Caucasians (65%), African-Americans (25%), Asians (5%), Native Americans (3%), and other heritages (2%) reflecting the local population characteristics of the Raleigh-Durham-Chapel Hill area. Inclusion was contingent on age-typical good state of health as determined by physical examination and standard clinical laboratory tests. Of the originally recruited 66 subjects, 61 completed at least the initial phase and the depletion phase. Of these 61, 1 subject was excluded due to 9 kg wt loss during the study and 3 subjects were excluded because they did not comply with diet restrictions, leaving 57 subjects included in analyses.
The participants were admitted to the UNC-CH General Clinical Research Center for the duration of the study and could leave only for brief periods under the supervision of study staff. The diets, which were composed of 0.8 g/kg high biological value protein, with 30% kcal coming from fat and the remaining kcal from carbohydrate, were prepared in-house to protocol specifications and are described in detail in another publication (21). Total food intake was adjusted to be isocaloric and provided adequate intakes of macro- and micronutrients. Initially, all participants received a diet of normal foods containing 550 mg choline/70 kg body wt/day, the current Adequate Intake (22). This diet contained 50 mg betaine/70 kg body wt/day. After 10 days (Fig. 2) the choline content of the diet was reduced to <50 mg/day (with 6 mg betaine/70 kg body wt/day), as confirmed by analysis of duplicate food portions (23, 24). Periodic determinations of urinary choline and betaine concentrations (24) were used to confirm compliance with the dietary restrictions. Subjects remained on this depletion diet until they developed organ dysfunction associated with choline deficiency, or for 42 days if they did not. Subjects were deemed to have organ dysfunction associated with choline deficiency if they had a >5-fold increase of serum creatine phosphokinase (CPK) activity (3) or if they had an increase in liver fat content by >28% while on the choline depletion diet, and if this increased CPK or increased liver fat resolved when choline was returned to the diet. After the depletion study, subjects were repleted by gradually increasing their choline intake to a final concentration of >550 mg per day and maintaining that concentration for at least 3 days. All 57 (54 for PEMT rs7946 and BHMT rs3733890) individuals were genotyped as described below for the following SNPs (the positions of the PEMT SNPs were enumerated with promoter B (25) as a reference): PEMT rs7946 (+5465 G→A), rs2278952 (+164 C→T), rs12325817 (−744 G→C) and for two new SNPs of PEMT that have not been previously reported at position −314C→T of the PEMT promoter, and +29C→G of exon 2, which adjoins the promoter; BHMT rs3733890; CHDH rs9001, and rs12676 (Table 1). Plasma collected at the end of the adequate choline intake phase and depletion phase were analyzed for concentrations of betaine, choline, and phosphatidylcholine (24).
Figure 2.
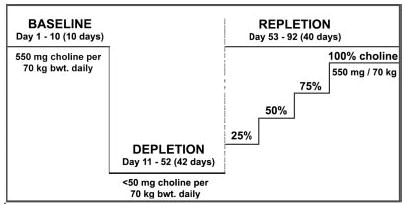
Study design. Healthy men and women were fed a baseline diet containing the choline Adequate Intake concentration for 10 days. They were then switched to a low choline diet (<50 mg choline) until they developed signs of organ dysfunction associated with choline deficiency or for up to 42 days. Subjects who developed signs of organ dysfunction were repleted with graded amounts of choline at 10 day intervals until their symptoms disappeared; those without signs of organ dysfunction were fed the 100% choline diet for at least 3 days before being discharged from the study. Some subjects were given a folic acid supplement (400 μg per day) during the depletion and repletion phases, but this did not affect their susceptibility to choline deficiency (from da Costa et al., Am J. Clinc. Nutr., in press).
Liver fat measurement
Liver fat was measured at the end of the 550 mg choline diet and at 21 and 42 days on the choline-deficient diet. Change in liver fat content was estimated by MRI with a Siemens Vision 41.5T clinical MR system using a modified “In and Out of Phase” procedure (26, 27). This approach utilizes the differences in transverse magnetization intensity after an ultra-brief time interval (FLASH; TE=2.2 ms and 4.5 ms, with a flip angle of 80°, and TR=140 ms). Processing of successive FLASH MRI images with software from Siemens Medical Solutions (Malvern, PA, USA) was used to estimate fat content. Organ fat content was derived from measurements across 3–5 liver slices per subject and standardized by relating the results to the fat content of similarly measured slices of spleen.
Laboratory analyses
Fasting blood samples were taken every 3–4 days for blood chemistries (including CPK analysis), after 10 days on the 550 mg/day choline diet (baseline), and at the end of the low choline diet (depletion phase) for choline and genotyping studies. Serum was analyzed using a dry slide colorimetric method for CPK activity by the McClendon Clinical Laboratories at University of North Carolina Hospitals, which is both Clinical Laboratory Improvement Act and College of American Pathologists accredited. Choline and its metabolites were analyzed and quantified directly by HPLC mass spectrometry (LC/ESI-IDMS) after the addition of internal standards labeled with stable isotopes that were used to correct for recovery (24).
Genotyping
Peripheral lymphocytes were isolated from blood by Ficoll-Hypaque gradient using Vacutainer® CPT™ tubes with sodium citrate (Becton Dickinson, Franklin Lakes, NJ, USA) (28,29) and genomic DNA extracted using PureGene (Gentra Systems, Minneapolis, MN, USA) according to the manufacturer’s instructions. SNP analyses were carried out as described below. Briefly, for the PEMT and CHDH genes, DNA sequencing was performed on double-stranded DNA templates obtained from genomic DNA by polymerase chain reaction (PCR) amplification. A negative control without DNA and a positive control with human DNA (PROMEGA Inc., Madison, WI, USA) for each PCR set were included. PCR products were purified with QIAquick® PCR Purification Kit 250 (QIAGEN Inc., Valencia, CA, USA) after electrophoresis in 0.8% or 3% agarose depending on the size of the fragment. Sequencing reactions were performed by the University of North Carolina at Chapel Hill Genome Analysis Facility, using a capillary sequencing machine (model 3100, Applied Biosystems, Foster City, CA, USA). Sequence results were interpreted using the programs Sequencher (Gene Code Corp., Ann Arbor, MI, USA). Basic local alignment search tool searches were performed using the National Center for Biotechnology (NCBI) program (http://www.ncbi.nlm.nih.gov/gorf/bl2.html).
Promoter of the PEMT gene
Successful amplification of the 1896 bp DNA fragment of the PEMT gene was performed using Takara Ex Taq polymerase (Fisher Scientific, Fair Lawn, NJ, USA) with an efficient 3′-5′ exonuclease activity for increased fidelity. Based on the GenBank sequence (accession number NC_000017), we designed a set of primers for amplification, as recommended by the manufacturers, and a set of primers for sequencing the overlapping segments in two directions (Table 2) using the Web primers design program (http://genome-www2.stanford.edu/cgi-bin/SGD/webprimer). The forward and reverse primers were 5′GAGCACGTGAGCTGTCAGTGCCTTTTG3′ and 5′CCAACCTCCTTCATACAACAGAGGTCC3′, respectively, and a three-step PCR was performed on an Applied Biosystems 2720 Thermal Cycler under the following conditions: 96°C for 2 min; 30 cycles (94°C for 30 s, 60°C for 1 min, 72°C for 2 min); extend 72°C for 7 min, and soak at 4°C. For the sequence determination of PEMT rs12325817, we used an additional primer (PEMT PRO seq-F2; Table 2) to verify the sequence in a region containing Alu repeats and a poly-A tail.
TABLE 2.
Primers for sequencing the PEMT promoter regiona
| Position of primer (bp) | Primer name | Sequence of primer 5′→3′ |
|---|---|---|
| 7728 - 7757 | PEMT PRO - F3 | GGAGTTATGGATCTAGGGAACTGGAGCAGC |
| 7711 - 7730 | PEMT PROseq - F1 | ATTTCACCCTCCTGAAAGGA |
| 8102 - 8082 | PEMT PROseq - R1 | TGACCAATCTAAGCCCAGGTT |
| 8092 - 8112 | PEMT PROseq - F2 | TAGATTGGTCATGGGAGGCTT |
| 8493 - 8474 | PEMT PROseq - R2 | ACAACATGGTGACACTCCGT |
| 8503 - 8522 | PEMT PROseq - F3 | TCTCGAACTCCTGACCATCA |
| 8878 - 8860 | PEMT PROseq - R3 | CCCGTAATCCCAGCACTTT |
| 8915 - 8935 | PEMT PROseq - F4 | GAGGAAAAAGACTCTGGCACA |
| 9300 - 9280 | PEMT PROseq - R4 | TTTACTCCATTGAGGGGTGCA |
| 9084 - 9101 | PEMT PROseq - F5 | TGATGGATCCCAGGAGGA |
| 9510 - 9490 | PEMT PROseq - R5 | GGCTTTCTGCTACCCAGTAAT |
| 9525 - 9498 | PEMT PRO - R1 | ACAACAGAGGTCCCGGCTTTCTGCTAC |
| 8438 - 8458 | PEMT PROseqMid - F3 | ACAACAGAGGTCCCGGCTTTCTGCTAC |
| 8824 - 8802 | PEMT PROseqMid - R3 | TCAGAGATCAGCCTGGCCAATAT |
| 8461 - 8480 | PEMT PROseqMid - F4 | ATTTTTAGTAGAGACGGAGT |
| 8840 - 8821 | PEMT PROseqMid - R4 | ATCACAAGGCCAGGAGTCAG |
| 8374 - 8394 | PEMT PRO SNP1- F | ACTTCCTGGGTTGAAGCGATT |
| 8597 - 8579 | PEMT PRO SNP1- R | TTTATTCTCTGGCCGTGCC |
Primers were used to sequence the PEMT gene as described in Materials and Methods.
PEMT
The coding region of PEMT containing the SNP rs7946 was amplified with the oligonucleotides 5′GGAGCACTTTGCCCCAGAATC3′ and 5′GACTTGGAGCCTTCAGAGCG3′ as forward and reverse primers, respectively (30). The sequences obtained were compared with ones stored in the NCBI database (http://www.ncbi.nlm.nih.gov/entrez/, accession number AF294467) using ClustalW multiple sequence alignment software http://www.ebi.ac.uk/Tools/sequence.html).
CHDH
Amplification of the 370 bp DNA fragment containing SNPs rs9001 and rs12676 in the CHDH gene was performed using BD Advantage GC Genomic PCR Kit (BD Biosciences, Mountain View, CA, USA). Based on the GenBank sequence (accession number NC_000003), we designed the following primers for amplification and sequencing: 5′AGTCATCTCATTCCCCTCCGTGGATCAGA3′ (forward) and 5′TAGCACCAGTTGTACCTGTCGTCGCACA3′ (reverse). A two-step PCR was done with the following conditions: 94°C for 1 min; 30 cycles (94°C for 30 s, 68°C for 3 min); extend 70°C for 5 min and soak at 15°C. The SNPs were numbered with the mRNA location (318 and 432, respectively; NT_018397).
BHMT
For the variant rs3733890 in exon 6 of BHMT (GenBank accession number NT_006713), the targeted DNA sequence: 5′GCCACTTTGACCCCACCATTAGT3′ and 5′TGGGAATTCTGGGAGATCGATG3′ as forward and reverse primers, respectively, were amplified by multiplex PCR, purified, then analyzed with matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (31). Samples were analyzed in duplicate.
Statistical analysis
Data for continuous variables are expressed as mean ± SE, and the statistical significance of differences between subjects on the two different diets were assessed using paired t test. A two-sample t test based on the differences between choline and metabolite concentrations in subjects on the baseline and depletion diets was used to compare the depleted with organ dysfunction and depleted without organ dysfunction groups. Genotype differences associated with organ dysfunction associated with choline deficiency were calculated using Fisher’s Exact Test to determine statistical significance (32). For P < 0.05, odds ratios and 95% confidence intervals were calculated as the odds of showing signs of deficiency for subjects with the risk allele divided by the odds of showing signs of deficiency for subjects without the risk allele. The Kruskal-Wallis test was used to compare differences in continuous variables by genotype (33).
RESULTS
Of the 57 participants, 68% developed organ dysfunction when fed the low choline diet and this resolved when choline was added back to their diets. Plasma betaine concentrations decreased almost 50% in response to the low choline diet (from 60±3 to 32±2 nmol/ml; P<0.001) and choline concentrations decreased almost 30% (from 9.8±0.3 to 7.1±0.2 nmol/ml; P<0.001). These decreases were irrespective of whether or not subjects developed organ dysfunction on the low choline diet. Plasma phosphatidylcholine concentrations were 9% lower when subjects were fed the low choline diet (1691±41 vs. 1868±45 nmol/ml than when on baseline diet; P<0.001); those subjects who developed organ dysfunction on the low choline diet had a 3-fold greater decrease in phosphatidylcholine (−228±47 nmol/ml) than those who did not (−64±31 nmol/ml; P<0.029 by t test). Plasma choline and phosphatidylcholine concentrations did not differ from baseline after subjects were repleted with choline-containing diets. We did not detect significant changes in choline, betaine, or phosphatidylcholine concentrations in plasma between genotypes (note that there was no significant change in plasma betaine concentrations associated with the BHMT genotype).
Gender was an important modifier of susceptibility to developing organ dysfunction when fed a low choline diet. When deprived of dietary choline, 77% of men and 80% of postmenopausal women developed fatty liver or muscle damage, whereas only 44% of premenopausal women developed such signs of organ dysfunction associated with choline deficiency (Fischer et al., Am. J. Clin. Nutr., under review). Note that within each gender grouping, a significant number of subjects were resistant to developing organ dysfunction, suggesting that other factors, such as genetic polymorphisms, may contribute to susceptibility to developing organ dysfunction when fed a low choline diet.
For each SNP, we tested for allelic association with susceptibility to developing organ dysfunction associated with eating a low choline diet. Although five tests were performed, no Bonferroni correction was done since small sample size made only very large effect sizes detectable. We identified two SNPs in the PEMT promoter region: rs12325817 (−774 G→C) and a new SNP that had not been previously reported at position −314 C→T. We found a SNP in exon 4 (+5465 G→A; rs7946) and two SNPs in exon 2, which adjoined the promoter: rs2278952 (+164, C→T) and + 29 C→G. The latter is a new genetic variant that has not been reported before. For rs12325817 (−774 G→C), the variant C allele was relatively common in our study population, where 18% were CC, 56% GC, and 26% GG genotype. The new SNPs at position −314 in the PEMT promoter and + 29 of exon 2 were rare, each occurring as a heterozygous allele in one subject (0.032 frequency); neither developed organ dysfunction when on the low choline diet. The rs2278952 SNP (+164 C→T) occurred at a frequency of 0.25 in women; it, too, was a heterozygous allele and was not associated with changes in susceptibility to developing organ dysfunction when on the low choline diet.
In all women, 18 of the 23 (78%) carriers of the PEMT–744C allele (rs12325817) developed organ dysfunction when fed a low choline diet (odds ratio 25, P=0.002; Table 3). In postmenopausal women, 11 of 12 (92%) of the allele carriers developed organ dysfunction when fed a low choline diet, and the 2 women without this allele did not. In the eight premenopausal women who were heterozygous for the allele (GC genotype), half developed organ dysfunction and half did not when fed a low choline diet, while the two premenopausal women who were homozygous for the allele developed organ dysfunction. Overall, the three women homozygous for this allele (CC genotype) developed organ dysfunction when fed a low choline diet, and seven of the eight females without this allele (GG genotype) did not (Table 3). There was no effect in men.
TABLE 3.
Effect of PEMT promoter SNP rs12325817 (−744 G→C) on susceptibility to organ dysfunction in humans eating a low choline dieta
| Signs of choline deficiency | GG | GC | CC | P value OR (95% CI) | |
|---|---|---|---|---|---|
| All subjects (57) | Yes | 7 | 25 | 7 | 0.10 |
| No | 8 | 7 | 3 | ||
| Men (26) | Yes | 6 | 10 | 4 | 0.49 |
| No | 1 | 2 | 3 | ||
| All women (31) | Yes | 1 | 15 | 3 | 0.002 |
| No | 7 | 5 | 0 | 25 (2, 256) | |
| Pre menopausal (16) | Yes | 1 | 4 | 2 | 0.10 |
| No | 5 | 4 | 0 | ||
| Postmenopausal (15) | Yes | 0 | 11 | 1 | 0.03 |
| No | 2 | 1 | 0 | 42 (1, 1348)b |
Subjects were fed a diet low in choline, and some developed signs of organ dysfunction (liver or muscle) that were reversed when choline was added back to their diets. Numbers of subjects are indicated for each genotype. Two-sided P values were calculated with a 2 × 3 Fisher exact test. For P < 0.05, odds ratios (OR) and 95% confidence intervals (CI) were calculated as the odds of showing signs of deficiency for subjects with the C allele divided by the odds of showing signs of deficiency for subjects without the C allele.
For postmenopausal and premenopausal women (where some cells were 0; see above), the odds ratio and 95% confidence intervals were computed after adding 0.5 to each cell, so these values underestimate the true values.
The first of two SNPs in the coding region of the CHDH gene (rs9001; +318 A→C) had a protective effect on susceptibility to developing organ dysfunction when fed a low choline diet in all subjects who carried the C allele (Table 4). We found no significant differences when the participants were grouped by gender or menopausal status. Although the second CHDH variant (rs12676; +432 G→T) was within 115 base pairs of SNP rs9001 (+318 A→C), it was formed independently (no difference in Hardy-Weinberg expected distribution for the population). Among all subjects, this SNP was not associated with susceptibility to developing organ dysfunction associated with choline deficiency. However, among premenopausal women, 5 of the 6 (83%) who were heterozygous for this variant developed organ dysfunction on a low choline diet compared with 2 of 10 (20%) who did so without this risk allele (odds ratio 20, P=0.04; Table 4).
TABLE 4.
Effect of choline dehydrogenase (CHDH) genotypes on susceptibility to organ dysfunction in humans eating a low choline dieta
|
CHDH rs9001, +318 A → C
|
CHDH rs12676, +432 G → T
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Signs of choline deficiency | AA | AC | CC | P value OR (95% CI) | GG | GT | TT | P value OR (95% CI) | |
| All subjects (57) | Yes | 34 | 4 | 1 | 0.03 | 19 | 19 | 1 | 0.23 |
| No | 10 | 6 | 2 | 0.2 (0.05, 0.7) | 13 | 5 | 0 | ||
| Men (26) | Yes | 18 | 2 | 0 | 0.22 | 10 | 9 | 1 | 1.00 |
| No | 4 | 2 | 0 | 3 | 3 | 0 | |||
| All women (31) | Yes | 16 | 2 | 1 | 0.13 | 9 | 10 | 0 | 0.07 |
| No | 6 | 4 | 2 | 10 | 2 | 0 | |||
| Pre menopausal (16) | Yes | 6 | 1 | 0 | 0.39 | 2 | 5 | 0 | 0.04 |
| No | 4 | 3 | 2 | 8 | 1 | 0 | 20 (1, 282) | ||
| Postmenopausal (15) | Yes | 10 | 1 | 1 | 0.52 | 7 | 5 | 0 | 1.00 |
| No | 2 | 1 | 0 | 2 | 1 | 0 | |||
Subjects were fed a diet low in choline and some developed signs of organ dysfunction (liver or muscle), which reversed when choline was added back to their diets. Numbers of subjects are indicated for each genotype. Two-sided P values were calculated with a 2 × 3 Fisher exact test. For P < 0.05, odds ratios (OR) and 95% confidence intervals (CI) were calculated as the odds of showing signs of deficiency for subjects with the C allele (T allele for CHDH 432) divided by the odds of showing signs of deficiency for subjects without the C allele (T allele for CHDH 432).
There was no association between the SNP in exon 4 of the PEMT gene (rs7946, +5465 G→A) and susceptibility to choline deficiency (Table 5), nor was the BHMT variant (rs3733890; +742G→A) associated with changes in susceptibility to choline deficiency (Table 5).
TABLE 5.
PEMT rs7946 (+5465 G→A ) and BHMT rs3733890 (+742 G→A ) genotypes were not associated with changes in susceptibility to organ dysfunction in humans eating a low choline dieta
|
PEMT rs7946, +5465 G → A
|
BHMT rs3733890, +742 G → A
|
||||||||
|---|---|---|---|---|---|---|---|---|---|
| Signs of choline deficiency | GG | GA | AA | P value | GG | GA | AA | P value | |
| All subjects (54) | Yes | 5 | 16 | 15 | 0.86 | 21 | 11 | 4 | 0.84 |
| No | 3 | 9 | 6 | 9 | 7 | 2 | |||
| Men (26) | Yes | 4 | 8 | 8 | 1.00 | 12 | 6 | 2 | 1.00 |
| No | 1 | 3 | 2 | 4 | 1 | 1 | |||
| All women (28) | Yes | 1 | 8 | 7 | 0.75 | 9 | 5 | 2 | 0.65 |
| No | 2 | 6 | 4 | 5 | 6 | 1 | |||
| Pre menopausal (16) | Yes | 0 | 5 | 2 | 0.63 | 4 | 2 | 1 | 0.79 |
| No | 2 | 4 | 3 | 3 | 5 | 1 | |||
| Postmenopausal (12) | Yes | 1 | 3 | 5 | 0.66 | 5 | 3 | 1 | 1.00 |
| No | 0 | 2 | 1 | 2 | 1 | 0 | |||
Subjects were fed a diet low in choline and some developed signs of organ dysfunction (liver or muscle) that reversed when choline was added back to their diets. Numbers of subjects are indicated for each genotype. Two-sided P values were calculated with a 2 × 3 Fisher exact test. PEMT = phosphatidylethanolamine N-methyltransferase; BHMT = betaine:homocysteine methyltransferase.
DISCUSSION
Common genetic polymorphisms have been reported to influence human requirements for nutrients. For example, a common SNP in the methyltetrahydrofolate reductase gene increases dietary requirements for the vitamin folic acid (34). However, these SNPs usually have very modest effects on nutrient needs. We recently reported that individuals who were carriers of the very common 5,10-methylenetetrahydrofolate dehydrogenase-1958A gene allele were more likely than noncarriers to develop signs of choline deficiency (35). We now report that we have identified common genetic variations in the PEMT and CHDH genes that may be associated with developing organ dysfunction when choline is removed from the diet of humans. While a larger study is needed to confirm these findings, it does appear that these polymorphisms influence the susceptibility to developing organ dysfunction when fed a low choline diet, and thus they increase the dietary requirement for choline needed to sustain optimal health.
In particular, women with a common variant in the promoter region of the PEMT gene rs12325817 (−774 G→C) were at significantly increased risk of developing organ dysfunction when dietary intake of choline is insufficient. PEMT activity is responsible for endogenous biosynthesis of choline moiety (1), and this activity is increased by estrogen treatment (36). We suggest that the promoter region of this gene is likely to have an estrogen response element (ERE). Indeed, the rs12325817 (−774 G→C) SNP is located within 50 bp of a putative ERE, which contains a perfect half-site consensus sequence (TGACC), but four of five bases differ in the other half-site (CGAAC vs. GGTCA). Given the sexually dimorphic effect of PEMT rs12325817 (−774 G→C), it is possible that this SNP alters the estrogen responsiveness of the promoter. Studies are under way in our laboratory to confirm this. We suggest that premenopausal women who are heterozygous for the PEMT rs12325817 (−774 G→C) C allele have sufficient estrogen to overcome the effects on estrogen-mediated transcription factor of the single allele, whereas post-menopausal women with lower estrogen levels are sensitive to the SNP. Men, with little estrogen, would be unaffected by an SNP that altered estrogen receptor complex binding.
The protective effect of the SNP in the CHDH gene (rs9001; 318 A→C) was more modest, and the frequency of this allele was relatively low (0.23). We observed a significant decrease in susceptibility to developing organ dysfunction on a low choline diet in all subjects, but a larger study would be needed to examine whether there are gender differences. More studies are also needed for the CHDH rs12676 (+432 G→T) SNP; first to confirm whether this SNP is associated with increased susceptibility to choline deficiency (we only achieved statistical significance in the subgroup of premenopausal women), then to explain this divergent response. It is not known whether these two SNPs have opposite effects on the activity of the CHDH enzyme.
The lack of effect of the SNP in exon 4 of the PEMT gene (rs7946, +5465 G→A) was unexpected, because we previously reported that this is a loss of function SNP and that persons with the variant A allele have increased risk of nonalcoholic fatty liver disease (30). Perhaps the modest decrease (30%) in activity of PEMT associated with this SNP was overshadowed by compensatory induction of the enzyme that is associated with choline deficiency in males (37, 38) and by estrogen-mediated activation of PEMT in females. The BHMT SNP effect was not surprising because, although this SNP has been reported to be protective against the risk of cardiovascular disease (20), the protein product of the gene variant did not differ in either catalytic activity or betaine binding when compared to the enzyme which did not contain the polymorphism (20, 39).
The SNPs we identified that increased susceptibility to developing organ dysfunction in humans fed low choline diets are likely to be of clinical importance. Humans fed intravenously (total parenteral nutrition) with solutions that deliver less choline than the adequate intake concentration often develop liver dysfunction that sometimes resolves when a choline source is added to their feeding solution (40). We suggest that humans with the identified SNPs are the ones most likely to be susceptible to this complication of parenteral nutrition. These SNPs, combined with poor dietary intake of choline, could contribute to adverse outcomes during pregnancy, a time when choline demand is high (9, 41). As noted earlier, deficient maternal dietary intake of choline during pregnancy in humans was associated with a 4-fold increased risk of having a baby with a neural tube defect (9). In rodent models, maternal dietary choline intake influenced brain development. More choline (~4× dietary levels) during days 11–17 of gestation in the rodent increased hippocampal progenitor cell proliferation (10, 11), decreased apoptosis in these cells (10, 11), enhanced long-term potentiation (LTP) in the offspring when they were adult animals (12, 42, 43), and enhanced visuospatial and auditory memory by as much as 30% in the adult animals through out their lifetime (13, 44–49). Mothers fed choline-deficient diets during late pregnancy have offspring with diminished progenitor cell proliferation and increased apoptosis in fetal hippocampus (10, 11), insensitivity to LTP when they were adult animals (12), and decremented visuospatial and auditory memory (13). For these reasons, identification of common polymorphisms that increase dietary requirements for choline during pregnancy could enable us to identify women for whom we need to assure adequate dietary choline intake. Further work with a larger sample size is warranted to replicate these important findings and to explore the mechanisms involved.
In summary, we report for the first time that SNPs in the phosphatidylethanolamine N-methyltransferase (PEMT) and choline dehydrogenase (CHDH) genes are associated with altered susceptibility to developing organ dysfunction on a low choline diet, and they likely affect dietary requirements for the nutrient choline. These SNPs are extremely common, and their effects should be considered when setting dietary reference intake levels. Studies determining the prevalence of these genetic polymorphisms in human populations of diverse composition should be conducted to facilitate such recommendations. In addition, since the genes of interest have many more polymorphisms than we tested, we cannot rule out the possibility that unmeasured but causal genetic variation is in linkage disequilibrium with the SNPs we genotyped.
Acknowledgments
This work was supported by NIH grants to S.Z. (DK55865, AG09525, and ES012997). Support for this work was also provided by grants from the NIH to the UNC Clinical Nutrition Research Unit (DK56350), the UNC General Clinical Research Center (RR00046), and the Center for Environmental Health and Susceptibility (ES10126). We thank Klaus Meyer and Per Magne Ueland, University of Bergen Norway, for the BHMT SNP analyses. The authors also thank Lester Kwock for the mass resonance imaging studies. K.D. participated in the study design, supervised sample collection and processing, participated in statistical analyses and data interpretation, and was responsible for writing the manuscript. O.G.K. designed the primers, and genotyped the samples for SNPs in the PEMT promoter and the CHDH gene. J.S. genotyped samples for the PEMT exon 4 SNP. J.A.G. carried out the genotype statistical analyses. L.M.F. participated in the design and supervision of the human study, and the recruitment of subjects. S.H.Z. was responsible for the funding, design, and conduct of the human study, participated in data interpretation, and provided significant input in the writing of the manuscript. There is no financial conflict of interest in relation to this study.
References
- 1.Zeisel SH, Blusztajn JK. Choline and human nutrition. Ann Rev Nutr. 1994;14:269–296. doi: 10.1146/annurev.nu.14.070194.001413. [DOI] [PubMed] [Google Scholar]
- 2.Zeisel SH, daCosta KA, Franklin PD, Alexander EA, Lamont JT, Sheard NF, Beiser A. Choline, an essential nutrient for humans. FASEB J. 1991;5:2093–2098. [PubMed] [Google Scholar]
- 3.da Costa KA, Badea M, Fischer LM, Zeisel SH. Elevated serum creatine phosphokinase in choline-deficient humans: mechanistic studies in C2C12 mouse myo-blasts. Am J Clin Nutr. 2004;80:163–170. doi: 10.1093/ajcn/80.1.163. [DOI] [PubMed] [Google Scholar]
- 4.Buchman A, Dubin M, Moukarzel A, Jenden D, Roch M, Rice K, Gornbein J, Ament M. Choline deficiency: a cause of hepatic steatosis during parenteral nutrition that can be reversed with intravenous choline supplementation. Hepatology. 1995;22:1399–1403. [PubMed] [Google Scholar]
- 5.Yao ZM, Vance DE. Head group specificity in the requirement of phosphatidylcholine biosynthesis for very low density lipoprotein secretion from cultured hepatocytes. J Biol Chem. 1989;264:11373–11380. [PubMed] [Google Scholar]
- 6.Yao ZM, Vance DE. The active synthesis of phosphatidylcholine is required for very low density lipoprotein secretion from rat hepatocytes. J Biol Chem. 1988;263:2998–3004. [PubMed] [Google Scholar]
- 7.Albright CD, da Costa KA, Craciunescu CN, Klem E, Mar MH, Zeisel SH. Regulation of choline deficiency apoptosis by epidermal growth factor in CWSV-1 rat hepatocytes. Cell Physiol Biochem. 2005;15:59–68. doi: 10.1159/000083653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Albright CD, Lui R, Bethea TC, da Costa KA, Salganik RI, Zeisel SH. Choline deficiency induces apoptosis in SV40-immortalized CWSV-1 rat hepatocytes in culture. FASEB J. 1996;10:510–516. doi: 10.1096/fasebj.10.4.8647350. [DOI] [PubMed] [Google Scholar]
- 9.Shaw GM, Carmichael SL, Yang W, Selvin S, Schaffer DM. Periconceptional dietary intake of choline and betaine and neural tube defects in offspring. Am J Epidemiol. 2004;160:102–109. doi: 10.1093/aje/kwh187. [DOI] [PubMed] [Google Scholar]
- 10.Albright CD, Friedrich CB, Brown EC, Mar MH, Zeisel SH. Maternal dietary choline availability alters mitosis, apoptosis and the localization of TOAD-64 protein in the developing fetal rat septum. Brain Res Dev. 1999;115:123–129. doi: 10.1016/s0165-3806(99)00057-7. [DOI] [PubMed] [Google Scholar]
- 11.Albright CD, Tsai AY, Friedrich CB, Mar MH, Zeisel SH. Choline availability alters embryonic development of the hippocampus and septum in the rat. Brain Res Dev. 1999;113:13–20. doi: 10.1016/s0165-3806(98)00183-7. [DOI] [PubMed] [Google Scholar]
- 12.Jones JP, Meck W, Williams CL, Wilson WA, Swartzwelder HS. Choline availability to the developing rat fetus alters adult hippocampal long-term potentiation. Brain Res Dev. 1999;118:159–167. doi: 10.1016/s0165-3806(99)00103-0. [DOI] [PubMed] [Google Scholar]
- 13.Meck WH, Williams CL. Choline supplementation during prenatal development reduces proactive interference in spatial memory. Brain Res Dev. 1999;118:51–59. doi: 10.1016/s0165-3806(99)00105-4. [DOI] [PubMed] [Google Scholar]
- 14.Bremer J, Greenberg D. Methyl transferring enzyme system of microsomes in the biosynthesis of lecithin (phosphatidylcholine) Biochim Biophys Acta. 1961;46:205–216. [Google Scholar]
- 15.Waite KA, Cabilio NR, Vance DE. Choline deficiency-induced liver damage is reversible in Pemt(−/−) mice. J Nutr. 2002;132:68–71. doi: 10.1093/jn/132.1.68. [DOI] [PubMed] [Google Scholar]
- 16.Saito S, Iida A, Sekine A, Miura Y, Sakamoto T, Ogawa C, Kawauchi S, Higuchi S, Nakamura Y. Identification of 197 genetic variations in six human methyltransferase genes in the Japanese population. J Hum Genet. 2001;46:529–537. doi: 10.1007/s100380170035. [DOI] [PubMed] [Google Scholar]
- 17.Song J, da Costa KA, Fischer LM, Kohlmeier M, Kwock L, Wang S, Zeisel SH. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD) FASEB J. 2005;19:1266–1271. doi: 10.1096/fj.04-3580com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.International Human Genome Sequencing Consortium. Finishing the euchromatic sequence of the human genome. Nature. 2004;431:931–945. doi: 10.1038/nature03001. [DOI] [PubMed] [Google Scholar]
- 19.Istrail S, Sutton GG, Florea L, Halpern AL, Mobarry CM, Lippert R, Walenz B, Shatkay H, Dew I, Miller JR, Flanigan MJ, Edwards NJ, Bolanos R, Fasulo D, Halldorsson BV, Hannenhalli S, Turner R, Yooseph S, Lu F, Nusskern DR, Shue BC, Zheng XH, Zhong F, Delcher AL, Huson DH, Kravitz SA, Mouchard L, Reinert K, Remington KA, Clark AG, Waterman MS, Eichler EE, Adams MD, Hunkapiller MW, Myers EW, Venter JC. Whole-genome shotgun assembly and comparison of human genome assemblies. Proc Natl Acad Sci U S A. 2004;101:1916–1921. doi: 10.1073/pnas.0307971100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Weisberg IS, Park E, Ballman KV, Berger P, Nunn M, Suh DS, Breksa AP, 3rd, Garrow TA, Rozen R. Investigations of a common genetic variant in betaine-homocysteine methyltransferase (BHMT) in coronary artery disease. Atherosclerosis. 2003;167:205–214. doi: 10.1016/s0021-9150(03)00010-8. [DOI] [PubMed] [Google Scholar]
- 21.Busby MG, Fischer L, Da Costa KA, Thompson D, Mar MH, Zeisel SH. Choline- and betaine-defined diets for use in clinical research and for the management of trimethylaminuria. J Am Diet Assoc. 2004;104:1836–1845. doi: 10.1016/j.jada.2004.09.027. [DOI] [PubMed] [Google Scholar]
- 22.Institute of Medicine, and National Academy of Sciences U. S. A. Dietary Reference Intakes for Folate, Thiamin, Riboflavin, Niacin, Vitamin B12, Panthothenic Acid, Biotin, and Choline, Vol. 1. National Academy Press; Washington D.C.: 1998. Choline; pp. 390–422. [PubMed] [Google Scholar]
- 23.Zeisel SH, Mar MH, Howe JC, Holden JM. Concentrations of choline-containing compounds and betaine in common foods. J Nutr. 2003;133:1302–1307. doi: 10.1093/jn/133.5.1302. [DOI] [PubMed] [Google Scholar]
- 24.Koc H, Mar MH, Ranasinghe A, Swenberg JA, Zeisel SH. Quantitation of choline and its metabolites in tissues and foods by liquid chromatography/electrospray ionization-isotope dilution mass spectrometry. Anal Chem. 2002;74:4734–4740. doi: 10.1021/ac025624x. [DOI] [PubMed] [Google Scholar]
- 25.Shields DJ, Agellon LB, Vance DE. Structure, expression profile and alternative processing of the human phosphatidylethanolamine N-methyltransferase (PEMT) gene. Biochim Biophys Acta. 2001;1532:105–114. doi: 10.1016/s1388-1981(01)00122-6. [DOI] [PubMed] [Google Scholar]
- 26.da Costa KA, Gaffney CE, Fischer LM, Zeisel SH. Choline deficiency in mice and humans is associated with increased plasma homocysteine concentration after a methionine load. Am J Clin Nutr. 2005;81:440–444. doi: 10.1093/ajcn.81.2.440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fishbein M, Gardner K, Potter C, Schmalbrock P, Smith M. Introduction of fast MR imaging in the assessment of hepatic steatosis. Magn Reson Imaging. 1997;15:287–293. doi: 10.1016/s0730-725x(96)00224-x. [DOI] [PubMed] [Google Scholar]
- 28.Fotino M, Merson E, Allen F. Micromethod for rapid separation of lymphocytes from peripheral blood. Ann Clin Lab Sci. 1971;1:131–133. [PubMed] [Google Scholar]
- 29.Ting A, Morris P. A technique for lymphocyte preparation from stored heparinized blood. Vox Sang. 1971:561–563. doi: 10.1111/j.1423-0410.1971.tb00469.x. [DOI] [PubMed] [Google Scholar]
- 30.Song J, da Costa KA, Fischer L, Kohlmeier M, Kwock L, Wang S, Zeisel SH. Polymorphism of the PEMT gene and susceptibility to nonalcoholic fatty liver disease (NAFLD) FASEB J. 2005;19:1266–1271. doi: 10.1096/fj.04-3580com. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Meyer K, Fredriksen A, Ueland PM. High-level multiplex genotyping of polymorphisms involved in folate or homocysteine metabolism by matrix-assisted laser desorption/ionization mass spectrometry. Clin Chem. 2004;50:391–402. doi: 10.1373/clinchem.2003.026799. [DOI] [PubMed] [Google Scholar]
- 32.Lowry R. Concepts and applications of inferential statistics. [accessed on 11/22/2004];2004 http://faculty.vassar.edu/lowry/webtext.html.
- 33.Kruskal W, Wallis W. Use of ranks in one-criterion variance analysis. J Am Statist Assoc. 1952;47:583–621. [Google Scholar]
- 34.Shelnutt KP, Kauwell GP, Chapman CM, Gregory JF, 3rd, Maneval DR, Browdy AA, Theriaque DW, Bailey LB. Folate status response to controlled folate intake is affected by the methylenetetrahydrofolate reductase 677C–>T polymorphism in young women. J Nutr. 2003;133:4107–4111. doi: 10.1093/jn/133.12.4107. [DOI] [PubMed] [Google Scholar]
- 35.Kohlmeier M, da Costa KA, Fischer LM, Zeisel SH. Genetic variation of folate-mediated one-carbon transfer pathway predicts susceptibility to choline deficiency in humans. Proc Natl Acad Sci U S A. 2005;102:16025–16030. doi: 10.1073/pnas.0504285102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Drouva SV, LaPlante E, Leblanc P, Bechet JJ, Clauser H, Kordon C. Estradiol activates methylating en-enzyme(s) involved in the conversion of phosphatidylethanolamine to phosphatidylcholine in rat pituitary membranes. Endocrinology. 1986;119:2611–2622. doi: 10.1210/endo-119-6-2611. [DOI] [PubMed] [Google Scholar]
- 37.Cui Z, Vance DE. Expression of phosphatidylethanolamine N-methyltransferase-2 is markedly enhanced in long term choline-deficient rats. J Biol Chem. 1996;271:2839–2843. doi: 10.1074/jbc.271.5.2839. [DOI] [PubMed] [Google Scholar]
- 38.Johnson PI, Blusztajn JK. Sexually dimorphic activation of liver and brain phosphatidylethanolamine N-methyltransferase by dietary choline deficiency. Neurochem Res. 1998;23:583–587. doi: 10.1023/a:1022470301550. [DOI] [PubMed] [Google Scholar]
- 39.Morin I, Platt R, Weisberg I, Sabbaghian N, Wu Q, Garrow TA, Rozen R. Common variant in betaine-homocysteine methyltransferase (BHMT) and risk for spina bifida. Am J Med Genet A. 2003;119:172–176. doi: 10.1002/ajmg.a.20115. [DOI] [PubMed] [Google Scholar]
- 40.Buchman AL, Ament ME, Sohel M, Dubin M, Jenden DJ, Roch M, Pownall H, Farley W, Awal M, Ahn C. Choline deficiency causes reversible hepatic abnormalities in patients receiving parenteral nutrition: proof of a human choline requirement: a placebo-controlled trial. J Parenter Enteral Nutr. 2001;25:260–268. doi: 10.1177/0148607101025005260. [DOI] [PubMed] [Google Scholar]
- 41.Zeisel SH, Mar MH, Zhou ZW, da Costa KA. Pregnancy and lactation are associated with diminished concentrations of choline and its metabolites in rat liver. J Nutr. 1995;125:3049–3054. doi: 10.1093/jn/125.12.3049. [DOI] [PubMed] [Google Scholar]
- 42.Pyapali G, Turner D, Williams C, Meck W, Swartzwelder HS. Prenatal choline supplementation decreases the threshold for induction of long-term potentiation in young adult rats. J Neurophysiol. 1998;79:1790–1796. doi: 10.1152/jn.1998.79.4.1790. [DOI] [PubMed] [Google Scholar]
- 43.Montoya DA, White AM, Williams CL, Blusztajn JK, Meck WH, Swartzwelder HS. Prenatal choline exposure alters hippocampal responsiveness to cholinergic stimulation in adulthood. Brain Res Dev. 2000;123:25–32. doi: 10.1016/s0165-3806(00)00075-4. [DOI] [PubMed] [Google Scholar]
- 44.Meck W, Williams C. Perinatal choline supplementation increases the threshold for chunking in spatial memory. NeuroReport. 1997;8:3053–3059. doi: 10.1097/00001756-199709290-00010. [DOI] [PubMed] [Google Scholar]
- 45.Meck W, Williams C. Characterization of the facilitative effects of perinatal choline supplementation on timing and temporal memory. NeuroReport. 1997;8:2831–2835. doi: 10.1097/00001756-199709080-00005. [DOI] [PubMed] [Google Scholar]
- 46.Meck W, Williams C. Simultaneous temporal processing is sensitive to prenatal choline availability in mature and aged rats. NeuroReport. 1997;8:3045–3051. doi: 10.1097/00001756-199709290-00009. [DOI] [PubMed] [Google Scholar]
- 47.Meck WH, Smith RA, Williams CL. Pre- and postnatal choline supplementation produces long-term facilitation of spatial memory. Dev Psychobiol. 1988;21:339–353. doi: 10.1002/dev.420210405. [DOI] [PubMed] [Google Scholar]
- 48.Meck WH, Williams CL. Metabolic imprinting of choline by its availability during gestation: Implications for memory and attentional processing across the lifespan. Neurosci Biobehav Rev. 2003;27:385–399. doi: 10.1016/s0149-7634(03)00069-1. [DOI] [PubMed] [Google Scholar]
- 49.Williams CL, Meck WH, Heyer DD, Loy R. Hypertrophy of basal forebrain neurons and enhanced visuo-spatial memory in perinatally choline-supplemented rats. Brain Res. 1998;794:225–238. doi: 10.1016/s0006-8993(98)00229-7. [DOI] [PubMed] [Google Scholar]