

Abstract
Orotidine 5′-monophosphate decarboxylase catalyzes the conversion of orotidine 5′-monophosphate to uridine 5′-monophosphate, the last step in biosynthesis of pyrimidine nucleotides. As part of a Structural Genomics Initiative, the crystal structures of the ligand-free and the6-azauridine 5′-monophosphate-complexed forms have been determined at 1.8 and 1.5 Å, respectively. The protein assumes a TIM-barrel fold with one side of the barrel closed off and the other side binding the inhibitor. A unique array of alternating charges (Lys-Asp-Lys-Asp) in the active site prompted us to apply quantum mechanical and molecular dynamics calculations to analyze the relative contributions of ground state destabilization and transition state stabilization to catalysis. The remarkable catalytic power of orotidine 5′-monophosphate decarboxylase is almost exclusively achieved via destabilization of the reactive part of the substrate, which is compensated for by strong binding of the phosphate and ribose groups. The computational results are consistent with a catalytic mechanism that is characterized by Jencks's Circe effect.
Orotidine 5′-monophosphate decarboxylase (ODCase) (EC 4.1.1.23) formally catalyzes the exchange of CO2 for a proton at the C6 position to form uridine 5′-monophosphate (UMP) (1). The intermediate implied by this description consists of a C6-carbanion, the conjugate base of the UMP carbon acid. The ODCase reaction is unique in biological decarboxylation reactions in that the carbanion intermediate is not stabilized by conjugation interactions and, thus, the reaction rate is exceptionally slow in aqueous solution (2). The remarkable catalytic power of ODCase, which accelerates the reaction by 17 orders of magnitude over the aqueous process, has fascinated chemists and biochemists alike, leading to a number of proposals of mechanisms with novel features (3–7). However, as more results accumulated for this class of enzymes, possibilities for the mechanism became increasingly limited as cofactors and catalytic groups continued to be excluded from consideration (8–10). The high-resolution x-ray structure of ODCase from Methanobacterium thermoautotrophicum reveals that the mechanism is almost fully characterized by the formal description, along with electrostatic features of the enzyme's active site that provide selective destabilization of the orotidine group. In what follows, we report the results from a joint experimental and theoretical investigation, providing a mechanism that involves significant ground state destabilization effects in enzyme catalysis (11).
The key to ODCase's catalytic power is its ability to utilize a phenomenon, which we classify as electrostatic stress [following Fersht's description of “stress” in catalysis (12)]. Although binding of the orotidine 5′-monophosphate (OMP) results in significant stabilizing interactions with the phosphate and ribose in the active site as revealed by the x-ray structural analysis, electrostatic interactions between the orotate group and ODCase is strongly destabilizing. In fact, the C6-carboxylate anion is held in a repulsive relationship with Asp70. This combination of attraction and repulsion in enzyme–substrate interactions has been described by Jencks as the “Circe effect,” which refers to the ability of an enzyme to use strong attractive forces to lure a substrate into the active site to provide destabilization of the reactive group that undergoes chemical transformation (11). Our computational analyses yield compelling evidence to support Jencks's hypothesis.
Materials and Methods.
ODCase was expressed in Escherichia coli and was purified with the help of a His-tag. Se-Met protein was produced in the same way by using an auxotroph strain. Crystals adopt space group P41212 (a = b = 56.9 Å, c = 124.5 Å) and P21 (a = 73.0 Å, b = 98.6 Å, c = 73.3 Å; β = 104.03°) for native and complexed crystals, the structures were determined by using the multiple anomalous dispersion method, and models were refined at 1.8 Å and 1.5 Å resolution, respectively. A detailed description is given elsewhere (ref. 13; N.W., A. Cristendat, A. Dharamsi, and E.F.P., unpublished work).
Structure determination.
Diffraction data were processed with denzo/scalepack (14) or mosflm (15). The structure was determined by multiple anomalous dispersion phasing at the Se absorption edge using the peak (0.9799 Å), edge (0.9575 Å), and remote wavelength (0.9802 Å) data sets. Initial positions of Se atoms were determined with the program solve (16) and were refined with the program sharp (17). All nine sites were found, and the overall figure of merit was 0.84 for data to 2Å resolution. The experimental map from sharp showed density of high quality and was traced by using the program o (18). Molecular replacement calculations were done with the program package amore (19). The refinements and addition of water molecules of both models were done with the program package cns (20).
Calculations.
The potential of mean force for the decarboxylation reaction in aqueous solution was obtained by using umbrella sampling in Monte Carlo simulations of the 1-methylorotate anion plus 735 water molecules along with periodic boundary conditions in the isothermal-isobaric ensemble at 25°C and 1 atm (1 atm = 101.3 kPa). The solute molecule was treated quantum mechanically by using the Austin Model 1 theory throughout. The three-point charge TIP3P model was adopted for water (21). A total of 15 simulation windows were used, spanning a range of the reaction coordinate from 1.4 to 7 Å. Each simulation consisted of at least 2 × 106 configurations for equilibration, followed by averaging for 4 × 106 configurations. The configurations were chosen with Metropolis and preferential sampling.
For the calculation of the orotidylate decarboxylation in ODCases, the orotate group was treated quantum mechanically along with the generalized hybrid orbital method to represent the covalent bond division between the QM and MM region (22). The 1′ carbon atom was chosen as the boundary atom. Thus, the calculation in the enzyme system utilized the same quantum-mechanical fragment as that in the aqueous simulation. The protein system and the classical region of the substrate were represented by the CHARMM22 force field (23). The carboxyl group of OMP was initially docked into the active site of the x-ray crystal structure for the enzyme–inhibitor complex, followed by 100 steps of energy minimization to remove close contacts. Then, the enzyme–substrate system was immersed into a 24 Å sphere of water centered at the average position of the C6 and carboxyl carbon atoms. Water molecules that were within 2.5 Å from any nonhydrogen protein–substrate atoms were removed. There were two water molecules left in the active site, forming hydrogen bonds with the OMP carboxylate ion. The system was initially equilibrated for at least 100 ps, followed by a series of eight umbrella sampling molecular dynamics simulations. Each simulation consisted of ≈50 ps for equilibration, followed by 50 ps for averaging. Because the reaction coordinate spans a much shorter range in the enzyme, only eight simulations were needed.
Computation of free energy of solvation.
The electrostatic stress, or free energy of transfer from water to the enzyme active site, was determined by free energy perturbation calculations using the electrostatic decoupling scheme for the GS and TS (Rc = 2.4 Å) in water and in ODCase. Each free energy calculation involved 10 windows with double-wide sampling, and each window consisted of 2 × 106 configurations/50 ps of equilibration, followed by 4 × 106 configurations/50 ps averaging. A cutoff distance of 14 Å was used throughout all calculations. As a rough approximation to the long-range electrostatics neglected by the cutoff scheme, a Born energy correction was made, assuming a dielectric constant of 4 for the protein from 14 to 16 Å from the active site, and 78 for bulk water beyond 16 Å. The entire calculation consisting of more than 200 million electronic structure calculations was accomplished in 1 week, making use of a 28-processor IBM-SP2 computer at Buffalo, while the crystal structure is being further refined.
Analysis and Description of the Structure.
As part of a Structural Genomics Initiative (24), we have solved the crystal structure of ODCase from M. thermoautotrophicum. The analysis was based on a multiple anomalous dispersion data set collected from a crystal of the Se-methionine-substituted apo-form of the enzyme (apoE). The resulting coordinate set was used in molecular replacement calculations to solve the ODCase/6-azauridine (6-aza) UMP complex structure. The final apoE structure, which represents one monomer of the dimeric ODCase molecule with a total of 212 residues and 235 water molecules, was refined at 1.8 Å resolution with good stereochemistry to an Rcryst of 21.0% and an Rfree of 23.8% (Table 1). The corresponding numbers for the inhibitor-complexed enzyme are Rcryst = 20.0%; Rfree = 20.6%; a total of 900 residues, representing the two dimers found in the asymmetric unit, and 770 water molecules. There are no residues in the disallowed region of the Ramachandran plots for either structure.
Table 1.
Data collection and refinement statistics
| Native
|
MAD (Se-Met) of apoE
|
||||
|---|---|---|---|---|---|
| ApoE | E-azaUMP | Remote | Peak | Edge | |
| Diffraction data | |||||
| Wavelength, Å | 1.0 | 1.0 | 0.9575 | 0.9799 | 0.9802 |
| Resolution, Å | 1.8 | 1.5 | 2.0 | 2.0 | 2.0 |
| Temperature, K | 100 | 100 | 100 | 100 | 100 |
| Measured reflections, n | 153,588 | 1,127,371 | 417,754 | 399,788 | 400,747 |
| Unique reflections, n | 20,729 | 156,498 | 14,614 | 14,514 | 14,526 |
| Completeness, % | 99.1† | 95.6‡ | 99.4 | 98.6 | 98.6 |
| R*sym | 0.040 | 0.058 | 0.060 | 0.070 | 0.080 |
| Space group | P41212 | P21 | P41212 | P41212 | P41212 |
| Se sites, n | — | — | 9 | 9 | 9 |
| Figure of merit, before solvent flattening | — | — | 0.84 | ||
| Refinement statistics | |||||
| Resolution, Å | 30–1.8 | 30–1.5 | |||
| Reflections, total, n | 19,510 | 153,676 | |||
| Reflections used for Rfree, n | 1,980 | 7,760 | |||
| Rcryst | 0.210 | 0.200 | |||
| Rfree | 0.238 | 0.206 | |||
| rmsd bond length, Å | 0.005 | 0.005 | |||
| rmsd bond angle, ° | 1.2 | 1.2 | |||
| Average B-factor, Å2 | 20.8 | 21.1 | |||
rmsd, root mean square deviation.
Rsym = ⋜|I − 〈I〉|/ΣI, where I is the observed intensity and 〈I〉 is the average intensity from multiple observations of symmetry-related reflections.
† 99.7% complete in 1.94–1.76Å shell.
‡ 87.9% complete in 1.58–1.50Å shell.
The overall fold of the ODCase monomer is a 9 α-helix/8 β-strand TIM barrel (Fig. 1). The smaller opening of the barrel is covered by the N-terminal loop that has clear density in the apoE whereas it remains disordered in the 6-azaUMP complex form.
Figure 1.

Ribbon diagram of the ODCase dimer. (A) The 2-fold axis runs vertically. Monomer A is colored in red and green (for helices and strands of β-sheets, respectively) while monomer B is colored in blue and yellow. The extra helix from the pET15b vector is colored in purple. The barrel opening is facing up in monomer B. (B) Side view of the dimer. The 2-fold axis is now perpendicular to the page. (C) Stereoview of the active site with 2Fo-Fc map (contoured at 1.5 σ). The map was calculated before 6-azaUMP was added to the model, providing a totally unbiased representation of the bound inhibitor.
For the complex structure, an extra helix at the C terminus was observed for two of the four molecules in the asymmetric unit. This helix, with sequence EDPAANKARKEAELAAATA, was encoded by the pET15b vector DNA between the BamHI site and the T7 terminator. Apparently, the stop codon of the ODCase gene itself was skipped by the E. coli protein expression machinery. Crystal contacts are the most probable reason for the formation of these helices. The dimer interface involves 18% of the surface area of each monomer and consists of both ionic and hydrophobic interactions. The two monomers superimpose well with an rms deviation of 0.3 Å for 211 Cα-atoms (residues 14–224). There are no major differences between the apo-dimer and the inhibitor-bound dimer (rms deviation between 401 Cα-atoms is 0.85 Å).
The Active Site.
The active site is located at the top of the larger opening and is completed by the second monomer. It features two distinctive characteristics: (i) an extensive electrostatic interaction and hydrogen bonding network with the ribose and phosphate groups, and (ii) a hydrophobic pocket surrounding the pyrimidine ring. In addition, there is a novel Lys42-Asp70-Lys72-Asp75B charge network beneath the ribose ring and the base, providing key functional roles in binding, catalysis, and product release (Fig. 2A).
Figure 2.

(A) Structural view of the active site, with the important elements and interactions of catalysis. Monomer A is colored in light gold and monomer B in pale blue. Even with 6-azaUMP bound, there is a cavity in the active site [purple contour, calculated with a van der Waals probe of 1.2 Å radius. This cavity is big enough to accommodate the C6 carboxylate of OMP or the product CO2. Hydrophobic residues lining the active site are shown in pale yellow. (B) Schematic view of the interactions between the active site and 6-azauridine. (C) Schematic view of ionic interactions and H-bonds associated with the phosphate group. All of the distances given are averages of values found in the four molecules in the asymmetric unit. The various corresponding distances differ by less than 0.3 Å from each other.
Most of the enzyme–inhibitor complex's stabilization energy is contributed by the phosphate and ribose binding. The phosphate group is clamped by a stretch of residues around Arg203 and the loop formed by amino acids 180–190 (180GVGAQGGDPG190) (Fig. 2 B and C). This loop is totally disordered in the apoE structure. Both the guanidinium group and the backbone amide of Arg203 interact directly with the phosphate, as does the amide of Gly202. Several water molecules, which form the first hydration shell of the phosphate group, mediate the remaining contacts to backbone carbonyls (Ile200, Val182) and amides (Gly181, Ser204). This clearly is a favorable mode of binding because it avoids the cost of desolvation to be paid if the first hydration shell is completely removed in exchange for enzyme–substrate interactions. One water molecule bridges the 5′-phosphate group and the C2 oxygen of 6-azaUMP. Each of the ribose hydroxyls is held by two hydrogen bonds to the enzyme. Asp20 and Lys42 interact with the 3′ hydroxyl whereas Asp75B and Thr79B bind to the 2′ hydroxyl. All four residues as well as Arg203 are highly conserved across species. The ribose ring is in the 2′ endo form with the base in the syn conformation; i.e., the 2′ hydrogen occupies the axial position and the C2 oxygen sits above the ribose ring. This is a somewhat less favorable conformation of free 6-azaUMP, although not inaccessible (25).
The base interacts loosely with the enzyme active site (Fig. 2B), especially when compared with the tight binding of phosphate and ribose. On one side, the C2 oxygen of the base shares a water molecule with the phosphate group and the C4 oxygen hydrogen bonds to a water molecule that interacts with the backbone carbonyl of Glu125 and with the backbone amide of Ser127. N3 is recognized by the Ser127 hydroxyl side chain, which in turn is hydrogen-bonded to Gln185. Electron density for the side chain of this residue indicates alternate positions with the water molecule shared between the phosphate and the C2 oxygen of 6-azaUMP (Fig. 1C). On the opposite side, four groups carrying alternate charges constitute a delicate hydrogen bonding network. A multiple sequence alignment of 17 ODCases shows 100% conservation of Lys42, Asp70, Lys72, and Asp75 (Lys72 corresponds to Lys93 in the yeast ODCase). Lys72 bridges the two anionic residues Asp70 and Asp75B whereas the second oxygen atom of Asp70 also interacts with Lys42. The binding pocket around the base features an array of hydrophobic residues (Ile96, Leu123, Val155, Ile178, Phe180, and Ile200). The orientation of the aromatic ring of the inhibitor 6-azaUMP is strongly influenced by the electrostatic interaction between N6 and the proton donor Lys72. In the case of barbituric acid monophosphate, the best inhibitor of ODCase known (4), the distance from Lys72 to the negatively charged oxygen on C6 would only be ≈2.3 Å instead of 3 Å to N6 of 6-azaUMP, assuming barbituric acid monophosphate binds to the ODCase active site in an analogous manner. The product UMP, however, is a weak inhibitor [Ki = 4.6 × 10−4M (4)], most probably because its C6 carbon does not allow such a favorable interaction.
As shown in Fig. 2A, there is a partially hydrophobic cavity in the active site that can easily accommodate the C6 carboxylate or the reaction product CO2. However, if OMP is directly modeled in the same conformation as 6-azaUMP in the active site, the C6-carboxylate group is in overlap with Asp70. Therefore, some conformational changes of the enzyme should take place to accommodate the substrate binding. This is corroborated by molecular dynamics simulation.
Combined QM/MM Calculations.
These intriguing observations prompted us to perform computational studies to unravel the catalytic mechanism of ODCase. To this end, we have carried out statistical mechanical Monte Carlo and molecular dynamics simulations of the decarboxylation reaction of orotidylate in water and in the enzyme environment. The computational approach features a combined quantum mechanical and molecular mechanical (QM/MM) technique in which part of the substrate, the orotate group in the active site, is treated quantum mechanically and the surrounding solvent–enzyme system is approximated classically by the CHARMM22 force field (23, 26–29). Because the electronic structure of the substrate is determined through Hartree-Fock molecular orbital calculations in the presence of the solvent–enzyme electric field throughout the fluid simulations, the enzyme action on the chemical reactivity in the bond dissociation process can be adequately investigated (29). In the present study, we have used the semiempirical quantum mechanical Austin Model 1 theory (30) to describe the orotate ion along with the generalized hybrid orbital method to treat the boundary between the QM and MM region (22). The Austin Model 1 calculations yield an energy of reaction of 35.5 kcal/mol, in excellent agreement with the prediction of 36.4 and 36.7 kcal/mol at the ab initio MP2/6-31G* and the B3LYP/6-31+G* level (7). This indicates that Austin Model 1 is reasonable for describing the present decarboxylation reaction.
Key results are shown in Fig. 3, which depicts the computed potentials of mean force for the decarboxylation reactions in water and in ODCase. In Fig. 3, the reaction coordinate, RC, is defined as the distance between the orotate C6 and the carboxylate carbon atom. The computed activation free energy in aqueous solution is ΔGaq‡ = 37.2 kcal/mol (Fig. 3), which may be compared with the experimental value of 38.5 kcal/mol (2). The potential of mean force levels off beyond RC = 4.5 Å, resulting in a calculated free energy of reaction of 30.5 kcal/mol, 5 kcal/mol less endothermic than the gas phase process. Therefore, consistent with the findings by Lee and Houk (7), the orotate decarboxylation reaction is not extremely sensitive to solvation effects. This is in contrast to other decarboxylation reactions involving delocalization stabilization (31, 32). Remarkably, the activation free energy for the decarboxylation is predicted to be ΔGcat‡ = 14.8 kcal/mol in ODCase, again in quantitative accord with the experimental value of 15.2 kcal/mol (2). This corresponds to a reduction of 22.4 kcal/mol in ΔG‡, which translates to a rate acceleration of kcat/kaq = 2.6 × 1016 for the OMP decarboxylation by ODCase. The experimental value is 1.7 × 1017 (2). The good agreement between computation and experiment, both in the absolute and relative activation free energies, supports the validity of the combined QM/MM method.
Figure 3.

Computed free energy reaction profiles for the decarboxylation reactions of 1-methylorotate to 1-methyluracil anion and CO2 in water, and OMP to form the UMP anion and CO2 in ODCase. PMF, potential of mean force; R, the distance between the C6-atom of orotidine and the carbon of the leaving group, CO2.
Reaction Mechanism.
Both transition state (TS) stabilization (ΔΔGTS) and ground state (GS) destabilization (ΔΔGb) may contribute to the increase in kcat, which corresponds to a lowering in the activation barrier (ΔGact‡) (11, 12). Although the role of GS destabilization on enzyme catalysis has been frequently discussed (11, 12), computational studies have focused primarily on the effects of TS stabilization. Indeed, in a recent article, Warshel emphasized that an enzyme would not gain much catalytic benefit from GS destabilization because, under the evolutionary pressure of increasing kcat/KM, changing the energy of GS without changing the TS energy will only alter kcat, but not kcat/KM (33). Warshel's analysis provides a major insight by making two main assumptions: (i) ground state destabilization increases the energy of the entire enzyme–substrate complex ES (Fig. 1 of ref. 33), and (ii) the total energy of the TS (ES‡) is not changed. He concluded that enzymes attain a large kcat by stabilizing the charges of the TS more than the corresponding stabilization in water (33). However, from the limited interactions between the ODCase and 6-azauracil, the remarkable rate enhancement of the ODCase reaction does not seem to be entirely attributable to TS stabilization effects because both GS and TS are charged species, and the latter has an even more dispersed charge distribution. Thus, any strong TS stabilization would inevitably stabilize almost equally the GS. As an alternative, Jencks's Circe effect provides a rational to the mechanism of the ODCase reaction (11). In this proposal, Jencks emphasized the fact that it is only the reactive group undergoing bond breaking and formation that needs to be destabilized. Therefore, by luring the substrate into the active site with strong attractive forces for the nonreactive part of the substrate, an enzyme utilizes some of this binding energy to directly destabilize the reactive group for chemical transformation, thereby lowering the observed activation barrier (11). Whereas Warshel's electrostatic picture of TS stabilization works for a variety of enzymes (33, 34), we provide compelling evidence, through computational analyses, demonstrating that the Circe effect is a viable mechanism, at least for the ODCase reaction.
For many enzymes, the difficulty in analyzing concepts like the Circe effect is to separate the reactive group from the binding part in a substrate (11). Fortunately, the simplicity of the ODCase reaction allows us to readily identify the reactive group in OMP as the orotate unit, SR, whereas the remaining ribose and phosphate groups constitute the binding block, Sb (Scheme 1). 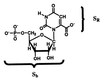 With this definition, the total free energy of binding, ΔGb, can be decomposed into an effective binding term, ΔGbeff, and an electrostatic stress component, ΔGES, such that ΔGb = ΔGbeff + ΔGES. Here, we use the term “electrostatic stress” in the same sense as that defined by Fersht (12). Specifically, the effective binding energy specifies the interaction between the “nonreactive” part of the substrate and the enzyme (11) whereas the electrostatic stress, ΔGES, refers to the electrostatic destabilization imposed on the reacting group by the enzyme relative to that in aqueous solution. The electrostatic stress thus corresponds to the free energy of transfer for the reactive group SR on going from water into the enzyme's active site in the presence of the rest of the substrate. Technically, the computational analysis involves determination of free energies of charge-annihilation of the reactive orotate group (SR) in water and in the enzyme, using the free energy perturbation method (35, 36). The same energy decomposition analysis can be applied to the binding of the TS (SR‡).
With this definition, the total free energy of binding, ΔGb, can be decomposed into an effective binding term, ΔGbeff, and an electrostatic stress component, ΔGES, such that ΔGb = ΔGbeff + ΔGES. Here, we use the term “electrostatic stress” in the same sense as that defined by Fersht (12). Specifically, the effective binding energy specifies the interaction between the “nonreactive” part of the substrate and the enzyme (11) whereas the electrostatic stress, ΔGES, refers to the electrostatic destabilization imposed on the reacting group by the enzyme relative to that in aqueous solution. The electrostatic stress thus corresponds to the free energy of transfer for the reactive group SR on going from water into the enzyme's active site in the presence of the rest of the substrate. Technically, the computational analysis involves determination of free energies of charge-annihilation of the reactive orotate group (SR) in water and in the enzyme, using the free energy perturbation method (35, 36). The same energy decomposition analysis can be applied to the binding of the TS (SR‡).
Table 2 lists the electrostatic components of the computed free energies of solvation for the orotate ion (SR) at the GS and the TS in water as well as in ODCase, from which the electrostatic stress is estimated to be ΔGES = 17.8 kcal/mol at the GS, and ΔGES‡ = 15.6 kcal/mol in the TS. At the GS, the large, positive electrostatic stress implies that, of the observed ΔGb = −8.3 kcal/mol total binding free energy for OMP (37), ΔGbeff is necessarily −26.1 (−8.3 − 17.8) kcal/mol. Making use of the experimental ΔGb (37) and the computed ΔGaq‡ (2) and ΔGcat‡, we estimate the binding affinity of OMP at TS by ODCase to be −30.7 kcal/mol, which is the energy difference between E′S‡ and E + S‡ (Fig. 4). This may be compared with the experimental value of −31.6 kcal/mol (2) from kaq/(kcat/KM), where kaq = exp(−ΔGaq‡/RT).
Table 2.
Calculated electrostatic free energies (in kcal/mol) for a 1-methylorotate anion in water and in orotidine monophosphate decarboxylase
| Ground state
|
Transition state
|
|||||
|---|---|---|---|---|---|---|
| Water | ODCase | ΔGES | Water | ODCase | ΔGES‡ | |
| ΔGFEP | −50.3 | −37.5 | 12.8 | −41.1 | −30.5 | 10.6 |
| ΔGBorn | −16.4 | −11.4 | 5.0 | −16.4 | −11.4 | 5.0 |
| ΔGtot | −66.7 | −48.9 | 17.8 | −57.5 | −41.9 | 15.6 |
The error bars for values from the free energy perturbation calculation, ΔGFEP, (±1 σ) were obtained by computing separate averages over blocks of 105 configurations (or 10 ps) during the simulation. They are ∼0.3 kcal/mol for the computed free energy changes. ΔGBorn is the Born free energy to account for the long range electrostatic effects attributable to the use of a spherical cutoff in the computation.
Figure 4.

A schematic representation of the Circe-effect energy-decomposition in the GS and TS. Computed binding and activation free energies (kcal/mol) are shown, along with the corresponding experimental values given in parentheses. The hidden thermodynamics of the overall observed binding energies for GS is revealed by considering the reactive orotate group (SR) and the ribose and phosphate binding part (Sb) of the substrate separately. A similar energy decomposition is also shown for the TS. KM is the Michelis-Menten dissociation constant, and kcat and kaq are rate constants for the catalyzed and noncatalyzed aqueous reaction, respectively. KTS = kaq/(kcat/KM) is an apparent equilibrium constant for the dissociation of the transition state from the enzyme (34). E and E′ denote different conformational states of the enzyme when the substrate is at the GS and TS, respectively.
The electrostatic stress analysis is illustrated in Fig. 4, which shows the decomposition (in boxes) of the total binding free energy for the ground state (ΔGb) and the transition state (ΔGTS) of OMP. The transition state of the substrate in ODCase, E′S‡, is placed at 14.8 kcal/mol above the ground state ES‡, corresponding to ΔGcat‡ in Fig. 3. Evidently, as the substrate binds in the enzyme, the reactive component of OMP, SR, is activated by 17.8 kcal/mol. Consequently, only an additional 14.8 kcal/mol is needed to rupture the C-C bond, which is reflected by the observed activation barrier, ΔGcat‡. By comparison with the aqueous background reaction (Fig. 3), the effect of TS stabilization can be determined as the difference between the “effective” activation barrier in the enzyme (ΔGES + ΔGcat‡) and that in water (ΔGaq‡). This gives a predicted TS stabilization of −4.6 kcal/mol. Alternatively, TS stabilization may be estimated by taking the difference of the computed electrostatic stress for the GS and TS, which is −2.2 kcal/mol. These two values are in reasonable agreement. Note that the small difference between these two estimates is attributable to computational uncertainties in the free energy perturbation calculation. Thus, the remarkable catalytic power of ODCase is achieved both by ground state destabilization of the reactive group of OMP and by transition state stabilization. However, the former (17.8 kcal/mol) provides a much greater contribution to the reduction of ΔGaq‡ than the latter (4.6 kcal/mol).
Molecular dynamics simulation shows the ground state with the orotate unit locked in a mostly hydrophobic pocket with its carboxylate group in close proximity to Asp70 at an O-O distance of only 3.5 Å. Such a hydrophobic environment coupled with strong electrostatic repulsion from Asp70 is markedly different from that in aqueous solution, where 1-methylorotate is found to form 12 strong hydrogen bonds (an average of 7.5 nearest neighbors for the carboxyl group) with solvent water molecules. Hydrophobic interactions and electrostatic repulsion between the carboxylate groups impose electrostatic stress on the orotate group, which also increases the energy of the enzyme itself. Thus, decarboxylation is accompanied by enzyme conformational change and the formation of a carbanion intermediate in which the negative charge is located further away from Asp70. The formation of a localized carbanion intermediate has been proposed previously for the elimination of benzaldehyde from thiamin (38). Lys72, which forms bridging hydrogen bonds to Asp70 and Asp75b, is also hydrogen-bonded to the ribose 2′-OH group in the GS. At the TS/product state, Lys72 migrates by 2 Å, breaking the Lys72-OH hydrogen bond, to form an ion-pair with the newly created C6 carbanion.
Although the hydrophobic environment of the orotate binding pocket is perfectly suited to accommodate the nonpolar product CO2, Lys72 protonates the strongly basic orotidyl carbanion to form UMP, a result that is supported by biochemical and site-specific mutagenesis studies (39). This, in turn, provides a mechanism, which may be termed as electrostatic expulsion, for product release from the enzyme active site: neutralization of Lys72 leads to increased electrostatic repulsion between Asp70 and Asp75B, which were originally held together by two salt bridges from Lys72. This causes conformational changes in the enzyme active site expelling the products from the binding pocket. Lys72 is then reprotonated by the solvent, restoring the catalytic cycle. Scheme 2 summarizes the overall mechanism of the ODCase reaction. ![]()
In summary, we have determined the crystal structure of orotidine monophosphate decarboxylase and, based on it, propose a reaction mechanism consistent with Jencks's Circe effect. Combined quantum mechanical and molecular mechanical (QM/MM) simulations indicate that both ground state destabilization and transition state stabilization contribute to the reduction of the activation barrier. However, in the case of ODCase, it is the ground state destabilization that plays the predominant role.
Acknowledgments
The authors thank Ronald Kluger for his chemical insight, valuable discussions, assistance in the interpretation of results, and bringing ref. 11 to our attention. We acknowledge Dinesh Christendad for help with the synthesis of 6-azaUMP and for preliminary kinetic data, and the staff at the BioCARS beamlines, APS, for their generous time commitments and support. N.W. is most grateful to Matthew Kimber for support. This work was supported by the PMH/OCI Foundation (E.F.P.) and the National Institutes of Health (J.G.).
Abbreviations
- ODCase
orotidine 5′-monophosphate decarboxylase
- UMP
uridine 5′-monophosphate
- 6-azaUMP
6-azauridine UMP
- OMP
orotidine 5′-monophosphate
- GS
ground state
- TS
transition state
- QM
quantum mechanical
- MM
molecular mechanical
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org [PDB ID codes 1DV7 (ligand-free form of ODCase) and 1DVJ (6-azaUMP complex)].
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.050417797.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.050417797
References
- 1.Lieberman I, Kornberg A, Simms E S. J Biol Chem. 1955;215:403–415. [PubMed] [Google Scholar]
- 2.Radzicka A, Wolfenden R. Science. 1995;267:90–93. doi: 10.1126/science.7809611. [DOI] [PubMed] [Google Scholar]
- 3.Beak P, Siegel B. J Am Chem Soc. 1976;98:3601–3606. doi: 10.1021/ja00428a035. [DOI] [PubMed] [Google Scholar]
- 4.Levine H L, Brody R S, Westheimer F H. Biochemistry. 1980;19:4993–4999. doi: 10.1021/bi00563a010. [DOI] [PubMed] [Google Scholar]
- 5.Acheson S A, Bell J B, Jones M E, Wolfenden R. Biochemistry. 1990;29:3198–3202. doi: 10.1021/bi00465a007. [DOI] [PubMed] [Google Scholar]
- 6.Cleland W W, Kreevoy M M. Science. 1994;264:1887–1890. doi: 10.1126/science.8009219. [DOI] [PubMed] [Google Scholar]
- 7.Lee J K, Houk K N. Science. 1997;276:942–945. doi: 10.1126/science.276.5314.942. [DOI] [PubMed] [Google Scholar]
- 8.Cui W, DeWitt J G, Miller S M, Wu W. Biochem Biophys Res Commun. 1999;259:133–135. doi: 10.1006/bbrc.1999.0737. [DOI] [PubMed] [Google Scholar]
- 9.Miller B G, Smiley J A, Short S A, Wolfenden R. J Biol Chem. 1999;274:23841–23843. doi: 10.1074/jbc.274.34.23841. [DOI] [PubMed] [Google Scholar]
- 10.Smiley J A, Saleh L. Bioorg Chem. 1999;27:297–306. [Google Scholar]
- 11.Jencks W P. Adv Enzymol Relat Areas Mol Biol. 1975;43:219–410. doi: 10.1002/9780470122884.ch4. [DOI] [PubMed] [Google Scholar]
- 12.Fersht A. Structure and Mechanism in Protein Science. New York: Freeman; 1999. pp. 372–375. [Google Scholar]
- 13.Brody R S, Westheimer F H. J Biol Chem. 1979;251:4238–4244. [PubMed] [Google Scholar]
- 14.Otwinowski Z. In: Proceedings of the CCP4 Study Weekend. Sawyer L, Isaacs N, Bailey S, editors. Warrington, U.K.: Science and Engineering Research Council Daresbury Laboratory; 1993. pp. 56–62. [Google Scholar]
- 15.Leslie A G W. In: Crystallographic Computing 5: From Chemistry to Biology. Moras D, Podjarny A D, Thierry J C, editors. Oxford: Oxford Univ. Press; 1990. pp. 50–61. [Google Scholar]
- 16.Terwilliger T C, Berendzen J. Acta Crystallogr D. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.de La Fortelle E, Bricogne G. Methods Enzymol. 1997;276:472–494. doi: 10.1016/S0076-6879(97)76073-7. [DOI] [PubMed] [Google Scholar]
- 18.Jones T A, Zou J Y, Cowan S W, Kjeldgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 19.Navaza J. Acta Crystallogr A. 1994;50:157. [Google Scholar]
- 20.Brünger A T, Adams P D, Clore G M, DeLano W L, Gros P, Grosse-Kunstleve R W, Jiang J S, Kuszewski J, Nilges M, Pannu N S, et al. Acta Crystallogr D. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- 21.Jorgensen W L, Chandrasekhar J, Madura J D, Impey R W, Klein M L. J Chem Phys. 1983;79:926–935. [Google Scholar]
- 22.Gao J, Amara P, Alhambra C, Field M J. J Phys Chem A. 1998;102:4714–4721. [Google Scholar]
- 23.MacKerell A D, Jr, Bushford D, Bellott M, Dunbrack R L, Evanseck J O, Field M J, Fischer S, Gao J, Guo H, Ha S, et al. J Phys Chem B. 1998;102:3586–3616. doi: 10.1021/jp973084f. [DOI] [PubMed] [Google Scholar]
- 24.Edwards A, Arrowsmith C. Abstracts, American Crystallographic Association Annual Meeting. Buffalo, NY: Am. Crystallogr. Assoc.; 1999. p. 30. [Google Scholar]
- 25.Saenger W, Suck D, Knappenberg M, Dirkx J. Biopolymers. 1979;18:2015–2036. doi: 10.1002/bip.1979.360180814. [DOI] [PubMed] [Google Scholar]
- 26.Warshel A, Levitt M. J Mol Biol. 1976;103:227–49. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 27.Field M J, Bash P A, Karplus M. J Comput Chem. 1990;11:700–733. [Google Scholar]
- 28.Gao J, Xia X. Science. 1992;258:631–635. doi: 10.1126/science.1411573. [DOI] [PubMed] [Google Scholar]
- 29.Gao J. Acc Chem Res. 1996;29:298–305. [Google Scholar]
- 30.Dewar M J S, Zoebisch E G, Healy E F, Stewart J J P. J Am Chem Soc. 1985;107:3902–3909. [Google Scholar]
- 31.Kemp D S, Paul K G. J Am Chem Soc. 1975;97:7305–7312. [Google Scholar]
- 32.Gao J. J Am Chem Soc. 1995;117:8600–8607. [Google Scholar]
- 33.Warshel A. J Biol Chem. 1998;273:27035–27038. doi: 10.1074/jbc.273.42.27035. [DOI] [PubMed] [Google Scholar]
- 34.Cannon W R, Benkovic S J. J Biol Chem. 1998;273:26257–26260. doi: 10.1074/jbc.273.41.26257. [DOI] [PubMed] [Google Scholar]
- 35.Zwanzig R. J Chem Phys. 1961;34:1931–1935. [Google Scholar]
- 36.Gao J. J Phys Chem. 1992;96:537–540. [Google Scholar]
- 37.Bell J B, Jones M E. J Biol Chem. 1991;266:12662–12667. [PubMed] [Google Scholar]
- 38.Kluger R, Lam J F, Kim C. Bioorg Chem. 1993;21:275–283. [Google Scholar]
- 39.Smiley J A, Jones M E. Biochemistry. 1992;31:12162–12168. doi: 10.1021/bi00163a027. [DOI] [PubMed] [Google Scholar]