

Abstract
The present study investigated the binding characteristics of various ligands to cannabinoid CB1 receptors in human neocortex and amygdala. In addition, the functionality of CB1 receptors in the human neocortex was assessed by examining the effects of CB1 receptor ligands on evoked [3H]-dopamine (DA) release in superfused brain slices and on synaptosomal cAMP accumulation.
Saturation-binding assays in human neocortical and amygdala synaptosomes using a radiolabelled cannabinoid receptor agonist ([3H]-CP55.940) revealed pKd values of 8.96 and 8.63, respectively. The numbers of binding sites (Bmax) were 3.99 and 2.67 pmol (mg protein)−1, respectively.
Various cannabinoid receptor ligands inhibited [3H]-CP55.940 binding with rank order potencies corresponding to those of previous studies in animal tissues.
Electrically evoked [3H]-DA release from human neocortical slices was inhibited by CP55.940 (IC50 6.76 nM, Imax 65%) and strongly enhanced by the cannabinoid receptor antagonist AM251. However, [3H]-DA release was not influenced in rat neocortex. In human tissue, the estimated endocannabinoid concentration in the biophase of the release-modulating CB1 receptors was 1.07 nM, expressed in CP55.940 units.
K+-evoked [3H]-DA release in the presence of tetrodotoxin (TTX) was strongly inhibited by CP55.940 in humans, but not in rats.
In human tissue, CP55.940 inhibited forskolin-stimulated cAMP accumulation (IC50 20.89 nM, Imax 35%). AM251 blocked this effect and per se increased forskolin-stimulated cAMP accumulation by ∼20%.
In conclusion, cannabinoids modulate [3H]-DA release and adenylyl cyclase activity in the human neocortex. CB1 receptors are located on dopaminergic nerve terminals and seem to be tonically activated by endocannabinoids.
Keywords: Anandamide, binding assay, cAMP accumulation, cannabinoid CB1 receptor, dopamine release, endocannabinoid, human neocortex
Introduction
The effects of Δ9-tetrahydrocannabinol (Δ9-THC) and related compounds in brain are mediated by cannabinoid CB1 receptors, which belong to the class of Gi protein-coupled receptors. Anandamide (N-arachidonylethanolamide, AEA) was first identified as an endogenous cannabinoid receptor ligand (Devane et al., 1992). This compound displays the prototype of a group of fatty acid amides, the so-called endocannabinoids, all exerting affinity for the cannabinoid receptor. Depolarization of neuronal cell membranes triggers the formation and release of AEA and other endocannabinoids (Di Marzo et al., 1994; Steffens et al., 2003a). Once released, the endocannabinoids diffuse to the presynaptic terminals to activate CB1 receptors, that is, they act as retrograde messengers (Wilson & Nicoll, 2001).
Activation of CB1 receptors leads to inhibition of adenylyl cyclase (Howlett et al., 1986) and to the inhibition of voltage-gated Ca2+ channels (Mackie & Hille, 1992; Twitchell et al., 1997), with subsequent depression of neurotransmitter release. For instance, cannabinoids have been shown to modulate acetylcholine (ACh) release in rat hippocampus or human neocortex (Gifford & Ashby, 1996; Steffens et al., 2003b), noradrenaline release in human or guinea-pig hippocampus (Schlicker et al., 1997), or dopamine release in guinea-pig retina or rat striatum (Schlicker et al., 1996; Cadogan et al., 1997).
Behaviourally, the impairment of cognition and memory by cannabinoids is commonly documented. A decreased cholinergic, GABAergic and glutamatergic neurotransmission in the hippocampus has been proposed to be mainly responsible for this phenomenon (for a review, see Sullivan, 2000). In addition, a decreased cortical dopaminergic neurotransmission may also contribute to the memory impairments elicited by cannabinoids, since (i) Δ9-THC has been shown to affect dopaminergic regulation of frontal cortical cognition (Jentsch et al., 1998) and (ii) an underlying decline in the function of the dopamine system projection to prefrontal cortex has been related to cognitive changes (for a review, see Braver & Barch, 2002). Besides, the selective D2 receptor agonist quinpirole impaired prefrontal cortical cognitive function in monkeys due to activation of D2 autoreceptors (Arnsten et al., 1995). In vitro experiments correspond to this finding since quinpirole strongly decreased the electrically evoked [3H]-DA release from human neocortical slices (IC50 23 nM, Imax approx. 80%; own data to be published; see also Fedele et al., 1993).
The first aim of the present study was to investigate whether cannabinoid CB1 receptor binding sites are present in human neocortex and amygdala and, in this context, to assess the binding affinity of various cannabinoid receptor ligands by their ability to compete for the binding of the tritiated synthetic compound (−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol (CP55.940). Secondly, we wanted to learn whether cannabinoid CB1 receptor activation diminishes evoked [3H]-DA release in superfused neocortical slices of humans and rats and to investigate whether these receptors are located presynaptically on dopaminergic nerve endings. Thirdly, a possible depolarization-induced endocannabinoid tone influencing human and/or rat neocortical [3H]-DA release was examined with the cannabinoid receptor antagonist N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide (AM251) and quantified by analysing the concentration–response curve of the exogenous agonist CP55.940.
Previous studies on the cannabinoid receptor-mediated signal transduction by using CB1 receptor-transfected cells commonly reported the inhibition of adenylyl cyclase with subsequent reduction of forskolin-stimulated cAMP accumulation (Felder et al., 1995; for a review, see Pertwee, 1997). However, in neocortical tissue preparations, the effect on cAMP accumulation is more controversial in the literature (Bidaut-Russell et al., 1990; Cadogan et al., 1997; Mato et al., 2002). Thus, the cannabinoid receptor signal transduction pathway was analysed in the human neocortex, investigating the effect of cannabinoids on basal and forskolin-stimulated cAMP accumulation in human neocortical synaptosomes.
Methods
Tissue sources
Fresh human non-epileptic neocortical tissue was obtained during surgical treatment of subcortical brain tumours or epilepsy. Amygdala tissue was only available from epilepsy surgery and, in contrast to neocortical tissue, had been assessed as epileptic according to electroencephalographic evaluations preceding surgery. The tissue derived from 51 patients of either sex (age 3–66 years). After premedication with midazolam or chlordiazepoxide, anaesthesia was performed with thiopental, fentanyl or flunitrazepam. Pancuronium was used for muscle relaxation. The tissue was removed in a gentle and atraumatic manner and was immediately placed in ice-cold saline to ensure viability. Before the operation, every patient was informed and signed a declaration of consent as requested by the local Ethics Committee of the University Hospital of Freiburg. Tissue macroscopically infiltrated with tumour was excluded, that is, tissue was used only when it appeared to be unaffected by the disease process to be operated on. The regions of human neocortical tissue included frontal, temporal and parietal areas. The white matter was separated (and discarded) from the grey matter, which contained all six neocortical layers after slicing. With respect to the amygdala, no attempt was made to enrich neuronal elements due to the little availability of this tissue.
Male Wistar rats (200–300 g, 8–12-weeks old) were maintained on a 12-h light/dark schedule with free access to food and water before they were decapitated under CO2 anaesthesia. All efforts were made to minimize both the suffering and the number of animals. Brains were quickly removed, the neocortex was dissected and immediately placed in ice-cold physiological buffer containing (in mM): NaCl 118, KCl 1.8, CaCl2 1.3, MgSO4 1.2, NaHCO3 25, KH2PO4 1.2, glucose 11, saturated with carbogene (5% CO2/95% O2), pH 7.4.
Further processing of the tissue depended on the experiment. For binding and cAMP experiments human tissue was frozen at −80°C until use; in case of superfusion experiments, human and rat neocortical tissues were processed directly (as described in the ‘Superfusion experiments' section).
Preparation of synaptosomes
Human brain samples were thawed and homogenized in 10 volumes (w v−1) of ice-cold sucrose (0.32 M)/HEPES (2.5 mM) buffer, pH 7.4. The following centrifugation steps were carried out in a Heraeus Biofuge 28RS (Osterode, Germany) at 4°C. The initial homogenate was centrifuged at 1000 × g for 10 min. The resulting supernatant was separated and centrifuged again at 10,000 × g for 10 min. The supernatant was discarded and the pellet was resuspended in ice-cold physiological buffer. Protein contents were determined based on the method of Lowry et al. (1951).
Saturation-binding experiments
Various concentrations of CP55.940 (0.032–10 nM) were used in order to determine saturation characteristics. For this purpose, the specific activity of [3H]-CP55.940 was decreased by adding different concentrations of unlabelled CP55.940. The binding reaction was initiated by adding 100 μl synaptosomal preparation to the assay which contained 880 μl assay buffer (50 mM Tris–HCl, 3 mM MgCl2, 1 mM EDTA, 0.1% bovine serum albumin (BSA), pH 7.4), 10 μl [3H]-CP55.940 and 10 μl unlabelled CP55.940, followed by a 60 min incubation at 30°C. Specific binding was defined as total binding minus binding in the presence of 10 μM CP55.940, and was determined for each concentration of [3H]-CP55.940/CP55.940.
Reactions were finished by rapid filtration through Whatman GF/C glass fibre filters soaked in buffer containing 0.1% polyethylenimine (PEI), using a 96-well harvester (Brandel M96, Gaithersburg, MD, U.S.A.). The filters were washed with 3 ml of ice-cold assay buffer and transferred into scintillation vials. After addition of 3 ml Ultima Gold liquid scintillation cocktail (Packard Bioscience, Groningen, Netherlands), the filters were shaken thoroughly for 1 h. Radioactivity in the filters was determined using a Tri-Carb 2100TR liquid scintillation analyzer (Packard Instruments, Meriden, CT, U.S.A.).
Competition-binding experiments
The cannabinoid receptor agonists CP55.940, WIN55212-2, Δ9-THC, AEA and the antagonist AM251 were evaluated for their ability to compete for the binding of [3H]-CP55.940. The assay was started by adding 100 μl synaptosomal preparation into each tube which contained 880 μl buffer, 10 μl [3H]-CP.55940 (20 pM final concentration) and 10 μl of the competing drug or vehicle. After a 60 min incubation at 30°C, further procedure was carried out as described above. In some experiments, 50 μM phenylmethylsulphonylfluoride (PMSF) was added to the preparation and binding buffer, in order to prevent AEA from hydrolytical degradation. Nonspecific binding was determined using 10 μM CP55.940.
Superfusion experiments
Fresh human or rat neocortical tissue specimens were sliced at a thickness of 350 μm perpendicularly, that is, the slices included all cortical layers (Mc Illwain tissue chopper, Bachofer, Reutlingen, Germany). The slices with a diameter between 2 and 4 mm were washed three times and then incubated with 2 ml buffer containing 0.1 μM [3H]-DA at 37°C for 45 min. After incubation, the slices were washed with buffer warmed up to 37°C, transferred into superfusion chambers (volume 100 μl, one slice per chamber) and then superfused with buffer (37°C) at a rate of 0.4 ml min−1. (+)-Oxaprotiline and fluvoxamine (each 1 μM) were present during incubation and throughout the superfusion experiment to prevent false labelling of serotonergic and noradrenergic nerve terminals with [3H]-DA (Feuerstein et al., 1986; Lupp et al., 1992). In some experiments, tetrodotoxin (TTX, 0.32 μM) was present throughout superfusion. BSA (0.1%) was routinely added to the superfusion medium in order to avoid adsorption of cannabinoids to the superfusion tubes.
After a pre-perfusion period of 60 min to equilibrate basal [3H]-outflow, the superfusate was collected in 5 min fractions (2 ml) in vials containing 3 ml scintillation fluid. At the end of superfusion, slices were removed from the chambers and solved in 0.5 ml Soluene (Packard Instruments). Ultima Gold (10 ml) was added after solvation. Radioactivity was determined by liquid scintillation spectrometry.
During the experiment, slices were depolarized three times after 70 min (S1), 165 min (S2) and 260 min (S3), with t=0 min being the start of superfusion. [3H]-DA release was stimulated either by applying a series of rectangular pulses (40 mA, 2 ms, 3 Hz, 90 pulses) or by elevating the K+ concentration of the superfusion buffer to 30 mM for 2 min. The concentration of Na+ in the buffer was reduced accordingly. Drugs to be tested were added at increasing concentrations 70 min before S2 and S3. The outflow of tritium was calculated as a fraction of the tritium content of a slice at the onset of the respective collection period (fractional rate). The stimulation-evoked [3H]-overflow was calculated as the difference between total [3H]-outflow from the onset of the respective stimulation to 20 min afterwards and the estimated basal outflow which was assumed to decline linearly from the collection period before to that 20 min after the onset of stimulation. The evoked [3H]-overflow was expressed as a percentage of the tritium content of the slice at the beginning of the respective stimulation.
If human tissue responded poorly to stimulation (S1 value of a slice <0.8% of tissue tritium), microscopic tumour penetration or otherwise affected tissue was assumed and the corresponding slices were excluded from further evaluation. To evaluate the drug effects, ratios S2/S1 and S3/S1 were calculated. Control experiments were always performed in parallel to drug experiments. In order to normalize the S2/S1 and S3/S1 ratios of the drug experiments, each ratio was divided by the mean ratio of the corresponding control chambers. The fractional rates before the three stimulations were represented by b1, b2 and b3. Drug effects on the basal [3H]-outflow were evaluated by using the b2/b1 and b3/b1 ratios.
cAMP assay
Freshly prepared synaptosomal aliquots (50 μl) were preincubated for 5 min at 37°C in 450 μl assay buffer (pH 7.4) containing 80 mM Tris–HCl, 2 mM MgCl2, 1 mM EDTA, 0.2 mM EGTA, 0.1 mM 3-isobutylmethylxanthine (IBMX), 0.1 mM GTP, 10 mM phosphocreatine, 30 U ml−1 creatinephosphokinase, 1 μM forskolin, 0.1% BSA and various concentrations of the drug to be tested. Some experiments were carried out in the absence of forskolin in order to determine basal cAMP accumulation in the absence or presence of drugs. After addition of 0.5 mM ATP, the tubes were incubated for 10 min at 37°C. Reaction was terminated by boiling the samples for 5 min at 95°C. After a 5 min centrifugation (10,000 × g, 4°C), cAMP accumulation was determined using a TRK 432 radioreceptor assay kit (Amersham Pharmacia Biotech).
Statistics
Unless stated otherwise, results are given as arithmetic means±s.e.m. of n separate individual data points (i.e. the number of assay tubes in binding and cAMP experiments or number of brain slices in superfusion experiments, respectively). To assess the significance of differences between two means, Student's t-test was applied. Note that statements on differences between means or other parameter estimates were always based on experiments involving at least four patients. The normal distribution of the data samples to be compared was tested using the Shapiro–Wilk test, which in the worst case displayed a P-value of 0.26; in other words, the error probability for the assumption of a non-normal distribution was always >25%.
Saturation-binding experiments were evaluated by nonlinear regression analysis using the logistic function
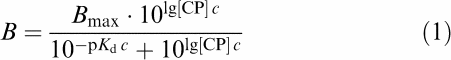 |
with B being the specific binding in pmol (mg protein)−1 (dependent variable) and lg[CP]=log10 of the concentration (M) of [3H]-CP55.940/CP55.940 (independent variable). The parameters to be estimated were Bmax, the asymptotic maximum of binding, that is, the number of binding sites per mg protein, and pKd, the negative log10 of the dissociation constant Kd between CP55.940 and the CB1 receptor. The estimate of the slope factor c served to decide whether a bimolecular reaction between the CP55.940 and its binding site occurred (Feuerstein & Limberger, 1999). Thus, an estimate of c near unity with a sufficiently narrow CI95 allowed assumption of a bimolecular reaction and a single Kd.
The following logistic function was used to evaluate data obtained from competition binding experiments (2a) and from superfusion experiments with human neocortex (2b):
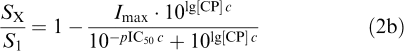 |
with Bnorm being normalized binding, SX/S1 being the normalized [3H]-DA release (dependent variable), and lg[cannabinoid, resp. CP]=log10 of the concentration (M, independent variable) of the corresponding drug to be evaluated with respect to an inhibitory property on [3H]-CP55.940 binding or on [3H]-DA release, respectively. The parameters to be estimated were Imax, the asymptotic maximum of relative inhibition [0,…, 1], and pIC50, the negative log10 of the inhibitor concentration leading to half-maximum inhibition. Again, the estimate of the slope factor c served to decide whether a bimolecular reaction between CB1 receptor and cannabinoids occurred. In this case, that is, c being near unity, another evaluation could take place, which yielded an estimate of the dissociation constant between receptor and inhibitor, Ki, considering the difference between the descriptive IC50 value and Ki, mechanistically interpretable as dissociation constant. Instead of applying the Cheng–Prusoff conversion of IC50 to Ki (IC50=Ki(1+[substrate]/Kd)=Ki+Ki/Kd[substrate]; Cheng & Prusoff, 1973), we preferred to introduce the concentration of the ligand directly into the function to be evaluated by nonlinear regression analysis (conversion of function (2a) with c=1 into function (3)):
with Bnorm being normalized binding (dependent variable) and with lg[cannabinoid]=log10 of the concentration (M, independent variable). Again, lg[CP] was log10 of the applied concentration of CP55.940. This concentration of CP55.940 was thus adjusted by the ratio Ki/Kd(= 10-pKi +pKd, since pKi=−lg Ki and pKd=−lg Kd) in order to account for different affinities of the receptor to CP55.940 and the various inhibitors. In this case, Kd or pKd was obtained from the estimation using the above-mentioned function (1). The advantage of this procedure was that an estimate of the CI95 of pKi was obtained directly.
Another more complicated logistic function (equation (14) of Feuerstein & Limberger, 1999) was used to fit by a second approach the concentration–response curve of CP55.940 in superfusion experiments with human tissue. This function reflects the condition that the extracellular level of an endogenous modulator, activating the same receptor as the exogenous agonist, is reduced by this exogenous agonist in a concentration-dependent manner:
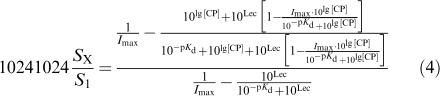 |
with SX/S1 being the normalized [3H]-DA release (dependent variable) and lg[CP]=log10 of the concentration of CP55.940 (M, independent variable). The estimated parameters of model function (4) were:
pKd=negative lg of the dissociation constant of the CP55.940/CB1 receptor complex, Lec=lg of the biophase concentration of endocannabinoids surrounding the CB1 receptors, Imax=maximum obtainable effect of CP55.940 under ‘endocannabinoid-free' conditions. This function allows an estimation of Lec in ‘CP-units' (see Results). A slope parameter ‘c' in function (4) was set to 1, assuming direct proportionality between receptor occupation and biological response. Instead of estimating the pKd, we included a fixed estimate of pKd value (8.96, see function (1)) in order to reduce the number of parameters to be estimated and, thereby, to increase the accuracy of the Imax and Lec estimates.
In order to facilitate comparisons of the present parameter estimates with those of other studies, all logarithmized estimates are given in molar concentrations.
Drugs
[Ring-2,5,6-3H]-DA (specific activity 60 Ci mmol−1) and [side chain-2.3.4(N)-3H]-CP55.940 (specific activity 158 Ci mmol−1) were purchased from Perkin-Elmer, U.S.A.; CP55.940, anandamide and AM251 were from Tocris/Cookson, Germany; WIN55212-2, Δ9-THC, PMSF, fluvoxamine maleate, forskolin, phosphocreatine, creatinephosphokinase, ATP-Na, GTP-Na, IBMX and TTX from Sigma, Germany; (+)-oxaprotiline HCl was kindly donated from Novartis Pharma, Switzerland. In all, 10 mM stock solutions of cannabinoids were either prepared in DMSO (CP55.940, AM251, WIN55212-2) or in EtOH (anandamide, Δ9-THC) and further diluted with the corresponding assay buffer containing 0.1% BSA; (+)-oxaprotiline was dissolved in HCl (10−3 M), PMSF was dissolved in EtOH, stock solutions of fluvoxamine, ATP, GTP, phosphocreatine, creatinephosphokinase, forskolin and TTX were prepared in buffer or DMSO (forskolin), respectively. The possible effects of all vehicles were controlled in each assay.
Results
Saturation-binding experiments
As shown in Figure 1, specific binding of CP55.940 in human neocortical and amygdala synaptosomes was saturable and of high affinity. Nonspecific binding, defined by using 10 μM unlabelled CP55.940, increased linearly over the range of radioligand concentrations studied. Nonlinear regression analysis revealed Bmax values of 3.99±0.22 pmol (mg protein)−1 (neocortex) and 2.67±0.14 pmol (mg protein)−1 (amygdala). The corresponding Kd values were 1.10±0.13 and 2.34±0.29 nM, respectively. As the slope factors c (i.e. Hill coefficients) were near unity (1.23±0.03, respectively, 1.20±0.04), a single binding site in both brain regions was assumed.
Figure 1.

Saturation characteristics of CP55.940 binding in human neocortical and amygdala synaptosomes. The specific activity of [3H]-CP55.940 (158 Ci mmol−1) was diluted with unlabelled CP55.940 to obtain final assay concentrations from 0.032 to 10 nM. Synaptosomes (∼100 μg protein) were incubated for 60 min at 30°C. Nonspecific binding was determined by using 10 μM CP55.940. (a, c) show saturation curves and parameter estimates of specific CP55.940 binding evaluated by nonlinear regression analysis. (b, d) show the corresponding Scatchard analyses of specific CP55.940 binding. Data points of each curve are means±s.e.m. of three experiments (i.e. tissue of 4–5 different patients), each performed in quadruplicate.
For comparison, Scatchard analyses were added to Figure 1, which revealed similar estimates of Kd and Bmax than the nonlinear regression analyses of the nonlinearized raw data.
Competition-binding experiments
The results of the competition assays are presented in Figure 2 and Table 1. All drugs tested effectively inhibited [3H]-CP55.940 binding in human neocortex and amygdala. The ranks of affinity for the human neocortical CB1 receptor were CP55.940=AM251>Δ9-THC≥anandamide (+PMSF)>WIN55212>anandamide (−PMSF). Similar results were found in the amygdala (CP55.940≥AM251>Δ9-THC>WIN55212-2≥anandamide (+PMSF)>anandamide (−PMSF)). All competition-binding curves yielded slope factors c around unity, suggesting a single binding site in both brain areas. In the presence of PMSF, the apparent affinity of anandamide was significantly enhanced, providing evidence for amidase activity in synaptosomal preparations of frozen human neocortical and amygdala tissue.
Figure 2.

Inhibition of specific [3H]-CP55.940 binding (20 pM) by CP55.940, AM251, Δ9-THC, WIN55212-2 (a, c) and anandamide±PMSF (50 μM; b, d) in human neocortical (upper panel) and amygdala (lower panel) synaptosomes. Synaptosomes (∼100 μg protein) were incubated for 60 min at 30°C with radiolabelled CP55.940 and various concentrations of drugs under study. Nonspecific binding was determined by using 10 μM CP55.940. The data points of each curve are means±s.e.m. of four experiments (i.e. tissue of 4–7 different patients), each performed in quadruplicate.
Table 1.
Affinities (Ki±s.e.m.) and slope parameters (c±s.e.m.) of various cannabinoids for the human neocortical and amygdala CB1 receptor
| Neocortex | Amygdala | |||
|---|---|---|---|---|
| Ki (nM) | c | Ki (nM) | c | |
| CP55.940 | 2.14±0.19 | 0.89±0.07 | 2.29±0.20 | 0.94±0.08 |
| AM251 | 2.14±0.15 | 0.85±0.07 | 3.09±0.41 | 0.91±0.11 |
| Δ9-THC | 19.5±1.7 | 0.84±0.07 | 17.8±0.3 | 0.98±0.15 |
| WIN55212-2 | 53.7±4.7 | 0.99±0.09 | 40.7±6.8 | 1.00±0.17 |
| Anandamide | ||||
| −PMSF | 209±27 | 0.89±0.07 | 182±27 | 1.17±0.18 |
| +PMSF (50 μM) | 25.7±3.3a | 0.76±0.08 | 54.0±7.2a | 0.85±0.11 |
P<0.05, compared to the corresponding Ki value in the absence of PMSF.
Superfusion experiments
In rat neocortical slices, evoked [3H]-overflow resulted in a mean S1 value of 2.99±0.18% of tissue tritium (n=33) when stimulated electrically and 4.13±0.74% (n=8) when stimulated with 30 mM K+. In concentrations up to 1 μM, the cannabinoid CB1 receptor agonist CP55.940 did not influence both electrically and K+ evoked overflow of tritium (data not shown). The cannabinoid receptor antagonist AM251 had no effect on electrically evoked [3H]-overflow in concentrations up to 1 μM (Figure 4).
Figure 4.

Effect of AM251 on electrically evoked [3H]-DA release from human or rat neocortical slices. Following incubation with [3H]-DA, slices were superfused and stimulated electrically (S1–S3; 90 pulses, 40 mA, 3 Hz). (+)-Oxaprotiline and fluvoxamine (1 μM each) were present in the incubation and superfusion medium. AM251 was either given alone at increasing concentrations 70 min before S2 and S3 or, when tested in combination with 0.1 μM CP55.940, present from 70 min before S2 until the end of superfusion (in this case, CP55.940 was given 70 min before S3). Inset: Effect of 0.1 μM CP55.940 in the absence or presence of AM251 in the human neocortex (taken from data points presented in Figures 3, 4). Effects are given as mean normalized SX/S1 ratios±s.e.m., obtained from the neocortical tissue of four different patients or two different rats each resulting in 4–15 single data points. *P<0.05, **P<0.01, compared to the corresponding controls. •P<0.05, compared to the effect of 0.1 μM CP55.940 in the absence of 0.1 μM AM251.
In human neocortical slices, the mean S1 value resulted in 2.28±0.10% of tissue tritium (n=62) when stimulated electrically and 3.28±0.32% (n=32) when stimulated with 30 mM K+. Drugs had no effect on basal [3H]-outflow since their b2/b1 and b3/b1 ratios were very similar to the corresponding control values (data not shown).
Both Ca2+ withdrawal and TTX (0.32 μM) strongly reduced the overflow of tritium: Ca2+ withdrawal, SX/S1=0.24±0.05, n=4; TTX, SX/S1=0.12±0.07, n=4, as compared to normalized controls of 1.00±0.12, n=4. Thus, we are assuming exocytotic and action potential-mediated [3H]-DA release in the following.
In contrast to the rat neocortex, CP55.940 depressed the electrically evoked [3H]-DA release in a concentration-dependent fashion with a maximum inhibition of ∼60%. Due to the release-enhancing effect of AM251 (see below), we assumed a marked inhibitory endocannabinoid tone on [3H]-DA release. Thus, in comparison, we applied both the logistic functions (2) and (4), which takes an endogenous tone into account (Figure 3).
Figure 3.

Effect of CP55.940 on electrically evoked [3H]-DA release from human neocortical slices. Following incubation with [3H]-DA, slices were superfused and stimulated electrically (S1–S3; 90 pulses, 40 mA, 3 Hz). (+)-Oxaprotiline and fluvoxamine (each 1 μM) were present in the incubation- and superfusion medium. CP55.940 was added in increasing concentrations 70 min before S2 and S3. The mean SX/S1 values (±s.e.m.) represent evoked tritium overflow, normalized to controls. The solid curve was obtained by using function (4), the dotted curve by using function (2). Data were obtained from a total of 12 patients.
The estimated parameters obtained from each function are presented in Table 2. As mentioned above, Lec can only be expressed in ‘CP units' of the dissociation constant of the CP55.940/CB1 receptor complex, since endocannabinoids were not the applied exogenous agonist.
Table 2.
Parameter estimates (±s.e.m.) obtained from the effect of CP55.940 on electrically evoked DA release in human neocortical slices (90 pulses, 40 mA, 3 Hz) using functions (2) or (4)
| Logistic function (2) | Function with endogenous tone (4) |
|---|---|
| Imax=0.65±0.06 | Imax derived=0.73±0.06 (Imax observed ∼0.60) |
| IC50=6.76±5.54 nM | pKd=8.96 (fixed value) |
| c=0.62±0.35 | ec==1.07±0.59 nM |
Since logarithmized estimates are given in molar concentrations, ‘ec'=10Lec M.
The next series of experiments was performed in order to examine the effects of the antagonist AM251 on electrically evoked [3H]-DA release. As shown in Figure 4, the inhibitory effect of CP55.940 was antagonized by AM251. Given alone, AM251 concentration-dependently enhanced [3H]-DA release with a maximum effect of ∼65% at 1 μM. Accordingly, in the presence of 0.1 μM CP55.940, the release-enhancing effect of AM251 was lower than in the absence of CP55.940.
Additional experiments with 0.32 μM TTX throughout the experiment were performed in order to block the impulse flow along the axons. In this case, [3H]-DA release was evoked by introduction of a K+-rich superfusion buffer (30 mM, 2 min). Compared to control values (SX/S1=1.00±0.06, n=12), 0.1 μM CP55.940 significantly reduced [3H]-DA release (SX/S1=0.30±0.10, n=8). This extent of inhibition is similar to that observed when slices were stimulated electrically (SX/S1=0.44±0.07, n=8).
cAMP assay
Basal cAMP accumulation in human neocortical synaptosomes resulted in 163.1±16.6 pmol (mg protein)−1 min−1 (n=10), and was increased by ∼180% in the presence of 1 μM forskolin. Neither CP55.940 (1 μM) nor AM251 (1 μM) had a significant effect on basal cAMP accumulation. The normalized values were 1.00±0.15 for controls, 0.86±0.03 for CP55.940 and 0.99±0.11 for AM251. The cAMP accumulation stimulated with 1 μM forskolin was concentration-dependently reduced by CP55.940, with a maximum inhibition of approx. 35% (Figure 5). The IC50 value was 20.9±12 nM and the slope factor c was around unity (0.91±0.33). The inhibitory effect of CP55.940 was fully reversed in the presence of AM251 (Figure 5, inset), which, given per se, slightly, but significantly increased forskolin stimulated cAMP accumulation by ∼20% in concentrations above 0.1 μM (Figure 6).
Figure 5.

Effect of CP55.940 on forskolin (1 μM)-induced cAMP accumulation in human neocortical synaptosomes. Drug effects are expressed as a fraction of forskolin-treated controls. Inset: Effect of 1 μM CP55.940 in the absence and presence of 1 μM AM251. Data represent means±s.e.m. of four experiments (i.e. tissue of four different patients) each performed in duplicate. **P<0.01, compared to the corresponding controls.
Figure 6.

Effect of AM251 (0.1–10 μM) on forskolin-stimulated cAMP accumulation in human neocortical synaptosomes. Drug effects are expressed as a fraction of forskolin-treated controls. Data represent means±s.e.m. of four experiments (i.e. tissue of four different patients) each performed in duplicate. *P<0.05, compared to the corresponding controls.
Discussion
The present study was carried out in order to characterize the actions of cannabinoids at human neocortical cannabinoid CB1 receptors, utilizing a receptor-binding assay and functional in vitro bioassays. First, it was necessary to demonstrate the existence of cannabinoid CB1 receptor-binding sites in the human neocortex, as commonly demonstrated in rat neocortical preparations by using, for example, radioligand-binding assays (Devane et al., 1988; Breivogel et al., 1997). The results of the saturation- and competition-binding experiments show that high-affinity cannabinoid receptors with a single binding site are present in synaptosomal preparations of frozen human neocortical and amygdala tissue. Saturation experiments using [3H]-CP55.940 revealed Kd values in the low nanomolar range and Bmax values in the low pmol (mg protein)−1 range. These values correspond in some degree to the results of earlier studies carried out either with human CB1 receptor-transfected cells (Felder et al., 1995), or with rat brain preparations (Devane et al., 1988; Breivogel et al., 1997).
There may be several reasons for varying Kd values between different studies. Irrespective of possible species differences, it has been shown that particularly the Kd estimates are influenced by various assay parameters (protein content, temperature, BSA concentration; Hillard et al., 1995). Furthermore, we preferred to use synaptosomes (i.e. predominantly presynaptic nerve terminals) instead of membrane preparations, which contain both pre- and postsynaptic binding sites. Thus, it was possible to compare more directly the Kd of CP55.940 with the IC50 estimate obtained from the superfusion experiments.
Concerning the ability of cannabinoid receptor ligands to compete for the binding of [3H]-CP55.940 to human neocortical and amygdala synaptosomes, our data reveal high affinities of CP55.940 and AM251 and moderate affinities of Δ9-THC, WIN55212-2 and AEA, confirming previous studies (Lan et al., 1999; for a review, see Howlett et al., 2002). With respect to WIN55212-2, one may speculate that this compound reveals species differences since it exerts considerably lower affinity to the human CB1 receptor (Ki∼54 nM in neocortex and ∼41 nM in amygdala tissue; see also Felder et al., 1995) than to the rat CB1 receptor (Ki∼2 nM; Kuster et al., 1993; Thomas et al., 1998).
The inhibition curves conducted with AEA in the absence or in the presence of PMSF showed an increased affinity when the enzyme inhibitor was present throughout the assay. Our results thus confirm the existence of AEA cleaving enzymes like fatty acid amidohydrolase (FAAH; for a review, see Di Marzo, 1998) in human brain tissue (Maccarrone et al., 1998).
After demonstrating the existence of cannabinoid CB1 receptor binding sites in human brain tissue, we were further interested to characterize the functionality of those receptors. Although it is widely accepted that activation of CB1 receptors in general leads to inhibition of neurotransmitter release (for a review, see Schlicker & Kathmann, 2001), no data exist concerning the modulating effects of cannabinoids on DA release in human and rat neocortex. However, knowledge of such a modulation seems to be important in terms of the impairing effects of cannabinoids on cognitition and memory (see below). As shown in Figure 3, CP55.940 markedly reduced electrically evoked [3H]-DA release with a potency and maximum inhibition similar to the effect of this compound on electrically evoked ACh release in rat and mouse hippocampal slices (Gifford et al., 1997; Kathmann et al., 2001). Although the slope factor c was only 0.62, a single site of action can nevertheless be assumed, since (i) the CI95 of c largely overlapped unity, (ii) may be influenced by an endocannabinoid tone (see below), and since (iii) c of the saturation-binding curve was near unity. The inhibitory effect of CP55.940 was probably mediated by the CB1 receptor, as the antagonist AM251 fully reversed the observed effect and the functional IC50 value (6.76 nM) obtained from the concentration–response curve was in the same order of magnitude of the Kd (1.10 nM) of the saturation-binding curve.
Surprisingly, CP55.940 did not influence electrically evoked [3H]-DA release from rat neocortical slices, suggesting the absence of a cannabinoid action on rat neocortical dopaminergic neurotransmission. This assumption is supported by a recent study in mouse brain, negating the coexpression of cannabinoid CB1 receptors and dopamine D1/2 receptors in the neocortex (Hermann et al., 2002).
In humans, the existence of CB1 receptors on dopaminergic terminals depressing neocortical [3H]-DA release was supported by the finding that the effect of CP55.940 on K+-evoked [3H]-DA release in the presence of TTX resembled the results when release was evoked electrically. Again, in the presence of TTX, we observed a considerable inhibitory effect of CP55.940 on K+-evoked [3H]-DA release in humans, but not in the rat. Since TTX blocks the pulse flow along the axons, one can assume that only human neocortical dopaminergic nerve terminals are endowed with CB1 receptors directly influencing DA release.
Next, we tested whether the antagonist AM251 given per se would have any effect on DA release. AM251 concentration-dependently increased DA release in the human neocortex, whereas it had no effect in the rat. The increasing effect was diminished by CP55.940, suggesting a CB1 receptor-mediated action. The enhanced release may be explained by the blockade of constitutively active CB1 receptors (Bouaboula et al., 1997) and/or by the disruption of an endocannabinoid tone leading to permanent depression of neurotransmitter release (Gifford & Ashby, 1996; Redmer et al., 2003). In our opinion, the latter alternative seems more likely, since we recently found evidence for a stimulation-dependent endocannabinoid tone at human neocortical CB1 receptors inhibiting ACh release (Steffens et al., 2003b). Indeed, application of function (4) yielded a 10Lec estimate of approx. 1 nM, representing the biophase concentration of endocannabinoids in ‘CP units'. To account for the different affinities of CP55.940 and the endogenous ligands, this 10Lec estimate has to be corrected by the affinity ratio Ki(endocannabinoids)/Ki(CP55.940) in order to converge to a more appropriate unit of the 10Lec value (Feuerstein & Limberger, 1999). Since the results of the present study revealed Ki estimates of approx. 2.1 nM (CP55.940) and approx 210 nM (AEA, representing endogenous ligands), a corrected 10Lec estimate of 1 nM*(210 nM/2.1 nM)=100 nM was obtained. Although this can only be a rough estimate of the actual endocannabinoid concentration surrounding the presynaptic CB1 receptors, a certain consistence with recently measured depolarization-induced AEA levels in the human neocortex (approx. 70 pmol g−1, equivalent to approx. 70 nM; Steffens et al., 2003a) is demonstrated.
Nevertheless, dealing with results from human tissue requires careful interpretation, for example, when comparing with data from animal experiments. For instance, human tissue derived from patients with different age. In addition, medications received for the underlying medical conditions, for example, epilepsy, might have altered the function of CB1 receptors in the human brain. We have also to admit that the number of different patients (≥4) yielding a parameter estimate (which, however, was obtained from much more data points) was rather small. Furthermore, microscopic tumour penetration cannot be fully eliminated by macroscopic inspection. Endocannabinoids have been shown to play an inhibitory role on tumour cell growth and on neurodegeneration (Hansen et al., 2001; Jacobsson et al., 2001). Therefore, endocannabinoids may apparently impact the results obtained with tissue from patients with brain tumours or epilepsy, which are known to be characterized, among others, by neurodegeneration. The viability of the human tissue, however, was clearly demonstrated by quantification of the evoked release (S1 values, see results), which were only slightly lower than the corresponding values obtained from rat experiments.
In a final series of experiments, the investigation of cAMP accumulation suggested some coupling of CB1 receptors to Gi proteins in human neocortical synaptosomes. CP55.940 concentration-dependently reduced forskolin-stimulated cAMP accumulation with a potency similar to that observed in guinea-pig hippocampal membranes (Schlicker et al., 1997). The action of CP55.940 was probably mediated via CB1 receptors, since the antagonist AM251 completely abolished the inhibitory effect. The IC50 values of CP55.940 for inhibiting electrically evoked [3H]-DA release and for inhibiting forskolin-stimulated cAMP accumulation (6.76 and 20.9 nM) were rather similar. One may speculate that, among other mechanisms (e.g. blockade of N-type Ca2+ channels), inhibition of adenylyl cyclase participates in the CB1 receptor-mediated inhibition of DA release in the human neocortex.
A similar degree of inhibition of the cAMP accumulation by cannabinoids was recently detected in neocortical membranes of post-mortem human brain. These results differ from the more pronounced inhibition in cells transfected with the human CB1 receptor (Felder et al., 1995; Ross et al., 1999). The difference is probably related to the use of tissue preparations where drug effects on cAMP accumulation in general are smaller.
According to the results of the superfusion study, AM251 given per se led to a stimulatory response although the percentage effect was markedly smaller than the effect on [3H]-DA release. Again, it cannot be ruled out that receptor-activating endogenous cannabinoids are present in the tissue, although the results of the AEA-binding curves give evidence for an AEA-degrading activity due to the right-shift of the concentration-inhibition curve in the absence of PMSF. Thus, endocannabinoids, which are not synthesized due to forskolin, should have a rather short half-life. On the other hand, one also has to consider a ligand-independent, constitutive CB1 receptor activity, as supposed recently due to the stimulating effects of the CB1 receptor antagonist/inverse agonist SR141716A on cAMP accumulation in human brain membranes (Mato et al., 2002). In this case, AM251 would behave as an inverse agonist (Vasquez et al., 2003). Furthermore, it may be possible that competitive CB1 receptor antagonists/inverse agonists act at different sites of the receptor (Sim-Selley et al., 2001), thereby producing combined inverse agonist and competitive antagonist effects (Meschler et al., 2000).
Irrespective of the mechanism leading to an enhanced DA release in the presence of AM251, this effect could be utilized for the treatment of cognitive deficits which are associated with a decreased dopaminergic neurotransmission in the cerebral cortex (for a review, see Braver & Barch, 2002).
In conclusion, the present study provides evidence for the presence of Gi protein-coupled cannabinoid CB1 receptors in the human neocortex. In contrast to the rat, these receptors seem to be located on dopaminergic nerve terminals and, furthermore, seem to be tonically activated by endogenous cannabinoids and/or exhibit constitutive activity. The knowledge of the modulatory effects of CB1 receptor agonists and competitive antagonists/inverse agonists on dopaminergic neurotransmission may help to detect targets for the treatment of cognitive or movement disorders.
Acknowledgments
We thank Dr M. Löffler for technical help, Dr R. Knörle and Dr P. Schnierle for protein measurements. This work was supported by the Deutsche Forschungsgemeinschaft (SFB 505, TP C8).
Abbreviations
- ACh
acetylcholine
- AEA
N-arachidonylethanolamide=anandamide
- AM251
N-(piperidin-1-yl)-5-(4-iodophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide
- BSA
bovine serum albumin
- CP55.940
(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl)cyclohexanol
- [3H]-DA
[3H]-dopamine
- PMSF
phenylmethylsulphonylfluoride
- Δ9-THC
Δ9-tetrahydrocannabinol
- TTX
tetrodotoxin
- WIN55212-2
R(+)-[2,3-dihydro-5-methyl-3-(4-morpholinylmethyl)pyrrolo [1,2,3,-de]-1,4-benzoxacin-6-yl]-1-naphtalenylmethanone mesylate
References
- ARNSTEN A.F., CAI J.X., STEERE J.C., GOLDMAN-RAKIC P.S. Dopamine D2 receptor mechanisms contribute to age-related cognitive decline: the effects of quinpirole on memory and motor performance in monkeys. J. Neurosci. 1995;15:3429–3439. doi: 10.1523/JNEUROSCI.15-05-03429.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BIDAUT-RUSSELL M., DEVANE W.A., HOWLETT A.C. Cannabinoid receptors and modulation of cyclic AMP accumulation in the rat brain. J. Neurochem. 1990;55:21–26. doi: 10.1111/j.1471-4159.1990.tb08815.x. [DOI] [PubMed] [Google Scholar]
- BOUABOULA M., PERRACHON S., MILLIGAN L., CANAT X., RINALDI-CARMONA M., PORTIER M., BARTH F., CALANDRA B., PECCEU F., LUPKER J., MAFFRAND J.P., LE FUR G., CASELLAS P. A selective inverse agonist for central cannabinoid receptor inhibits mitogen-activated protein kinase activation stimulated by insulin or insulin-like growth factor 1. Evidence for a new model of receptor/ligand interactions. J. Biol. Chem. 1997;272:22330–22339. doi: 10.1074/jbc.272.35.22330. [DOI] [PubMed] [Google Scholar]
- BRAVER T.S., BARCH D.M. A theory of cognitive control, aging cognition, and neuromodulation. Neurosci. Biobehav. Rev. 2002;26:809–817. doi: 10.1016/s0149-7634(02)00067-2. [DOI] [PubMed] [Google Scholar]
- BREIVOGEL C.S., SIM L.J., CHILDERS S.R. Regional differences in cannabinoid receptor/G-protein coupling in rat brain. J. Pharmacol. Exp. Ther. 1997;282:1632–1642. [PubMed] [Google Scholar]
- CADOGAN A.K., ALEXANDER S.P., BOYD E.A., KENDALL D.A. Influence of cannabinoids on electrically evoked dopamine release and cyclic AMP generation in rat striatum. J. Neurochem. 1997;69:1131–1137. doi: 10.1046/j.1471-4159.1997.69031131.x. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 per cent inhibition (I50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- DEVANE W.A., DYSARZ F.A., III, JOHNSON M.R., MELVIN L.S., HOWLETT A.C. Determination and characterization of a cannabinoid receptor in rat brain. Mol. Pharmacol. 1988;34:605–613. [PubMed] [Google Scholar]
- DEVANE W.A., HANUS L., BREUER A., PERTWEE R.G., STEVENSON L.A., GRIFFIN G., GIBSON D., MANDELBAUM A., ETINGER A., MECHOULAM R. Isolation and structure of a brain constituent that binds to the cannabinoid receptor. Science. 1992;258:1946–1949. doi: 10.1126/science.1470919. [DOI] [PubMed] [Google Scholar]
- DI MARZO V. Endocannabinoids and other fatty acid derivatives with cannabimimetic properties: biochemistry and possible physiopathological relevance. Biochim. Biophys. Acta. 1998;1392:153–175. doi: 10.1016/s0005-2760(98)00042-3. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., FONTANA A., CADAS H., SCHINELLI S., CIMINO G., SCHWARTZ J.C., PIOMELLI D. Formation and inactivation of endogenous cannabinoid anandamide in central neurons. Nature. 1994;372:686–691. doi: 10.1038/372686a0. [DOI] [PubMed] [Google Scholar]
- FEDELE E., ANDRIOLI G.C., RUELLE A., RAITERI M. Release-regulating dopamine autoreceptors in human cerebral cortex. Br. J. Pharmacol. 1993;110:20–22. doi: 10.1111/j.1476-5381.1993.tb13765.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FELDER C.C., JOYCE K.E., BRILEY E.M., MANSOURI J., MACKIE K., BLOND O., LAI Y., MA A.L., MITCHELL R.L. Comparison of the pharmacology and signal transduction of the human cannabinoid CB1 and CB2 receptors. Mol. Pharmacol. 1995;48:443–450. [PubMed] [Google Scholar]
- FEUERSTEIN T.J., HERTTING G., LUPP A., NEUFANG B. False labelling of dopaminergic terminals in the rabbit caudate nucleus: uptake and release of [3H]-5-hydroxytryptamine. Br. J. Pharmacol. 1986;88:677–684. doi: 10.1111/j.1476-5381.1986.tb10250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEUERSTEIN T.J., LIMBERGER N. Mathematical analysis of the control of neurotransmitter release by presynaptic receptors as a supplement to experimental data. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:345–359. doi: 10.1007/pl00005361. [DOI] [PubMed] [Google Scholar]
- GIFFORD A.N., ASHBY C.R., JR Electrically evoked acetylcholine release from hippocampal slices is inhibited by the cannabinoid receptor agonist, WIN 55212-2, and is potentiated by the cannabinoid antagonist, SR 141716A. J. Pharmacol. Exp. Ther. 1996;277:1431–1436. [PubMed] [Google Scholar]
- GIFFORD A.N., SAMIIAN L., GATLEY S.J., ASHBY C.R., JR Examination of the effect of the cannabinoid receptor agonist, CP 55,940, on electrically evoked transmitter release from rat brain slices. Eur. J. Pharmacol. 1997;324:187–192. doi: 10.1016/s0014-2999(97)00082-4. [DOI] [PubMed] [Google Scholar]
- HANSEN H.H., SCHMID P.C., BITTIGAU P., LASTRES-BECKER I., BERRENDERO F., MANZANARES J., IKONOMIDOU C., SCHMID H.H., FERNANDEZ-RUIZ J.J., HANSEN H.S. Anandamide, but not 2-arachidonoylglycerol, accumulates during in vivo neurodegeneration. J. Neurochem. 2001;78:1415–1427. doi: 10.1046/j.1471-4159.2001.00542.x. [DOI] [PubMed] [Google Scholar]
- HERMANN H., MARSICANO G., LUTZ B. Coexpression of the cannabinoid receptor type 1 with dopamine and serotonin receptors in distinct neuronal subpopulations of the adult mouse forebrain. Neuroscience. 2002;109:451–460. doi: 10.1016/s0306-4522(01)00509-7. [DOI] [PubMed] [Google Scholar]
- HILLARD C.J., EDGEMOND W.S., CAMPBELL W.B. Characterization of ligand binding to the cannabinoid receptor of rat brain membranes using a novel method: application to anandamide. J. Neurochem. 1995;64:677–683. doi: 10.1046/j.1471-4159.1995.64020677.x. [DOI] [PubMed] [Google Scholar]
- HOWLETT A.C., BARTH F., BONNER T.I., CABRAL G., CASELLAS P., DEVANE W.A., FELDER C.C., HERKENHAM M., MACKIE K., MARTIN B.R., MECHOULAM R., PERTWEE R.G. International Union of Pharmacology. XXVII. Classification of cannabinoid receptors. Pharmacol. Rev. 2002;54:161–202. doi: 10.1124/pr.54.2.161. [DOI] [PubMed] [Google Scholar]
- HOWLETT A.C., QUALY J.M., KHACHATRIAN L.L. Involvement of Gi in the inhibition of adenylate cyclase by cannabimimetic drugs. Mol. Pharmacol. 1986;29:307–313. [PubMed] [Google Scholar]
- JACOBSSON S.O., WALLIN T., FOWLER C.J. Inhibition of rat C6 glioma cell proliferation by endogenous and synthetic cannabinoids. Relative involvement of cannabinoid and vanilloid receptors. J. Pharmacol. Exp. Ther. 2001;299:951–959. [PubMed] [Google Scholar]
- JENTSCH J.D., VERRICO C.D., LE D., ROTH R.H. Repeated exposure to delta 9-tetrahydrocannabinol reduces prefrontal cortical dopamine metabolism in the rat. Neurosci. Lett. 1998;246:169–172. doi: 10.1016/s0304-3940(98)00254-7. [DOI] [PubMed] [Google Scholar]
- KATHMANN M., WEBER B., SCHLICKER E. Cannabinoid CB1 receptor-mediated inhibition of acetylcholine release in the brain of NMRI, CD-1 and C57BL/6J mice. Naunyn-Schmiedeberg's Arch. Pharmacol. 2001;363:50–56. doi: 10.1007/s002100000304. [DOI] [PubMed] [Google Scholar]
- KUSTER J.E., STEVENSON J.I., WARD S.J., D'AMBRA T.E., HAYCOCK D.A. Aminoalkylindole binding in rat cerebellum: selective displacement by natural and synthetic cannabinoids. J. Pharmacol. Exp. Ther. 1993;264:1352–1363. [PubMed] [Google Scholar]
- LAN R., LIU Q., FAN P., LIN S., FERNANDO S.R., MC CALLION D., PERTWEE R., MAKRIYANNIS A. Structure–activity relationships of pyrazole derivatives as cannabinoid receptor antagonists. J. Med. Chem. 1999;42:769–776. doi: 10.1021/jm980363y. [DOI] [PubMed] [Google Scholar]
- LOWRY O.H., ROSEBROUGH N.J., FARR A., RANDALL R.J. Protein measurements with the folin phenol reagent. J. Biol. Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- LUPP A., BÄR K.I., LÜCKING C.H., FEUERSTEIN T.J. Different effects of serotonin (5-HT) uptake blockers in caudate nucleus and hippocampus of the rabbit: role of monoamine oxidase in dopaminergic terminals. Psychopharmacology. 1992;106:118–126. doi: 10.1007/BF02253598. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., VAN DER STELT M., ROSSI A., VELDINK G.A., VLIEGENTHART J.F., AGRO A.F. Anandamide hydrolysis by human cells in culture and brain. J. Biol. Chem. 1998;273:32332–32339. doi: 10.1074/jbc.273.48.32332. [DOI] [PubMed] [Google Scholar]
- MACKIE K., HILLE B. Cannabinoids inhibit N-type calcium channels in neuroblastoma-glioma cells. Proc. Natl. Acad. Sci. U.S.A. 1992;89:3825–3829. doi: 10.1073/pnas.89.9.3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MATO S., PAZOS S., VALDIZAN E.M. Cannabinoid receptor antagonism and inverse agonism in response to SR141716A on cAMP production in human and rat brain. Eur. J. Pharmacol. 2002;443:43–46. doi: 10.1016/s0014-2999(02)01575-3. [DOI] [PubMed] [Google Scholar]
- MESCHLER J.P., KRAICHELY D.M., WILKEN G.H., HOWLETT A.C. Inverse agonist properties of N-(piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide HCl (SR141716A) and 1-(2-chlorophenyl)-4-cyano-5-(4-methoxyphenyl)-1H-pyrazole-3-carboxylic acid phenylamide (CP-272871) for the CB(1) cannabinoid receptor. Biochem. Pharmacol. 2000;60:1315–1323. doi: 10.1016/s0006-2952(00)00447-0. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Pharmacology of cannabinoid CB1 and CB2 receptors. Pharmacol. Ther. 1997;74:129–180. doi: 10.1016/s0163-7258(97)82001-3. [DOI] [PubMed] [Google Scholar]
- REDMER A., KATHMANN M., SCHLICKER E. Cannabinoid CB(1) receptor-mediated inhibition of hippocampal acetylcholine release is preserved in aged mice. Br. J. Pharmacol. 2003;138:1425–1430. doi: 10.1038/sj.bjp.0705194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ROSS R.A., GIBSON T.M., STEVENSON L.A., SAHA B., CROCKER P., RAZDAN R.K., PERTWEE R.G. Structural determinants of the partial agonist-inverse agonist properties of 6′-azidohex-2′-yne-delta8-tetrahydrocannabinol at cannabinoid receptors. Br. J. Pharmacol. 1999;128:735–743. doi: 10.1038/sj.bjp.0702836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHLICKER E., KATHMANN M. Modulation of transmitter release via presynaptic cannabinoid receptors. Trends Pharmacol. Sci. 2001;22:565–572. doi: 10.1016/s0165-6147(00)01805-8. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., GÖTHERT M. Cannabinoid receptor-mediated inhibition of dopamine release in the retina. Naunyn-Schmiedeberg's Arch. Pharmacol. 1996;354:791–795. doi: 10.1007/BF00166907. [DOI] [PubMed] [Google Scholar]
- SCHLICKER E., TIMM J., ZENTNER J., GÖTHERT M. Cannabinoid CB1 receptor-mediated inhibition of noradrenaline release in the human and guinea-pig hippocampus. Naunyn-Schmiedeberg's Arch. Pharmacol. 1997;356:583–589. doi: 10.1007/pl00005093. [DOI] [PubMed] [Google Scholar]
- SIM-SELLEY L.J., BRUNK L.K., SELLEY D.E. Inhibitory effects of SR141716A on G-protein activation in rat brain. Eur. J. Pharmacol. 2001;414:135–143. doi: 10.1016/s0014-2999(01)00784-1. [DOI] [PubMed] [Google Scholar]
- STEFFENS M., FEUERSTEIN T.J., VAN VELTHOVEN V., SCHNIERLE P., KNÖRLE R. Quantitative measurement of depolarization-induced anandamide release in human and rat neocortex. Naunyn-Schmiedeberg's Arch. Pharmacol. 2003a;368:432–436. doi: 10.1007/s00210-003-0817-1. [DOI] [PubMed] [Google Scholar]
- STEFFENS M., SZABO B., KLAR M., ROMINGER A., ZENTNER J., FEUERSTEIN T.J. Modulation of electrically evoked acetylcholine release through cannabinoid CB1 receptors: evidence for an endocannabinoid tone in the human neocortex. Neuroscience. 2003b;120:455–465. doi: 10.1016/s0306-4522(03)00318-x. [DOI] [PubMed] [Google Scholar]
- SULLIVAN J.M. Cellular and molecular mechanisms underlying learning and memory impairments produced by cannabinoids. Learn. Mem. 2000;7:132–139. doi: 10.1101/lm.7.3.132. [DOI] [PubMed] [Google Scholar]
- THOMAS B.F., GILLIAM A.F., BURCH D.F., ROCHE M.J., SELTZMAN H.H. Comparative receptor binding analyses of cannabinoid agonists and antagonists. J. Pharmacol. Exp. Ther. 1998;285:285–292. [PubMed] [Google Scholar]
- TWITCHELL W., BROWN S., MACKIE K. Cannabinoids inhibit N- and P/Q-type calcium channels in cultured rat hippocampal neurons. J. Neurophysiol. 1997;78:43–50. doi: 10.1152/jn.1997.78.1.43. [DOI] [PubMed] [Google Scholar]
- VASQUEZ C., NAVARRO-POLANCO R.A., HUERTA M., TRUJILLO X., ANDRADE F., TRUJILLO-HERNANDEZ B., HERNANDEZ L. Effects of cannabinoids on endogenous K+ and Ca2+ currents in HEK293 cells. Can. J. Physiol. Pharmacol. 2003;81:436–442. doi: 10.1139/y03-055. [DOI] [PubMed] [Google Scholar]
- WILSON R.I., NICOLL R.A. Endogenous cannabinoids mediate retrograde signalling at hippocampal synapses. Nature. 2001;410:588–592. doi: 10.1038/35069076. [DOI] [PubMed] [Google Scholar]