

Abstract
A high-throughput assay utilizing the voltage/ion probe reader (VIPR) technology identified salicylidene salicylhydrazide (SCS) as being a potent selective inhibitor of α2β1γ1θ GABAA receptors with a maximum inhibition of 56±5% and an IC50 of 32 (23, 45) nM.
Evaluation of this compound using patch-clamp electrophysiological techniques demonstrated that the compound behaved in a manner selective for receptors containing the β1 subunit (e.g. maximum inhibition of 68.1±2.7% and IC50 value of 5.3 (4.4, 6.5) nM on α2β1γ1 receptors). The presence of a β1 subunit was paramount for the inhibition with changes between α1 and α2, γ1 and γ2, and the presence of a θ subunit having little effect.
On all subtypes, SCS produced incomplete inhibition with the greatest level of inhibition at α1β1γ1θ receptors (74.3±1.4%). SCS displayed no use or voltage dependence, suggesting that it does not bind within the channel region. Concentration – response curves to GABA in the presence of SCS revealed a reduction in the maximum response with no change in the EC50 or Hill coefficient. In addition, SCS inhibited pentobarbitone-induced currents.
Threonine 255, located within transmembrane domain (TM) 1, and isoleucine 308, located extracellularly just prior to TM3, were required for inhibition by SCS.
SCS did not compete with the known allosteric modulators, picrotoxin, pregnenolone sulphate, dehydroepiandrosterone 3-sulphate, bicuculline, loreclezole or mefenamic acid. Neither was the inhibition by SCS influenced by the benzodiazepine site antagonist flumazenil.
In conclusion, SCS is unique in selectively inhibiting GABAA receptors containing the β1 subunit via an allosteric mechanism. The importance of threonine 255 and isoleucine 308 within the β1 subunit and the lack of interaction with a range of GABAA receptor modulators suggests that SCS is interacting at a previously unidentified site.
Keywords: GABAA receptor, salicylidene salicylhydrazide, β subunit, ion channel, transmembrane domain, mutagenesis, allosteric, modulator
Introduction
The majority of fast inhibitory neurotransmission within the mammalian brain is mediated by the neurotransmitter GABA acting on postsynaptic GABAA receptors. The GABAA receptor, which belongs to the superfamily of ligand-gated ion channels, is formed by an assembly of five homologous subunits. These subunits have been subclassified according to their degree of amino-acid homology as α1–6, β1–3, γ1–3, δ, ρ1–3, θ, ɛ and π, with the majority of receptors composed of two α, two β and a γ subunit (Chang et al., 1996; Farrar et al., 1999). Each subunit has a large extracellular N-terminal domain, four hydrophobic membrane-spanning domains that form the channel region and a small extracellular C-terminus (Korpi et al., 2002).
A large number of clinically used psychoactive drugs, for example, benzodiazepines, general anaesthetic agents, anticonvulsants and ethanol, exert their effects mainly or exclusively via an interaction with GABAA receptors. In addition, a number of other substances have been shown to interact with GABAA receptors, for example, loreclezole, avermectin, furosemide, zinc, picrotoxin and lanthanum (for a recent review, see Korpi et al., 2002). Electrophysiological and radioligand-binding studies using recombinant receptors have demonstrated that a number of these compounds display subunit selectivity. Meanwhile, the generation of chimeras and site-directed mutagenesis has enabled specific amino-acid residues that confer the selectivity to be identified, for example, selectivity of the anaesthetic etomidate for β2- and β3-containing receptors is due to an asparagine residue at positions 289 and 290, respectively (Belelli et al., 1997). Note an alternative numbering for this residue is B2/3 N265.
Within the CNS, the total number of GABAA receptor subtypes and the function of many of these are currently unknown (Sieghart & Sperk, 2002; Whiting, 2003). One way to dissect out the behavioural effects of the different GABAA receptor subtypes is to use a subtype-selective compound. In order to identify novel subunit selective compounds, a library of some 10,000 compounds was screened against three GABAA receptor subtypes (α2β1γ1θ, α3β3γ2s and α4β3γ2s). The subtypes were chosen from a large but not exhaustive repertoire of stable cell lines, with the initial aim of identifying compounds selective for as many subunits as possible. Herein we describe the identification and characterization of a salicylic acid derivative, salicylidene salicylhydrazide, with unique potency and selectivity for β1-containing receptors.
There are limited biological data with SCS; however, it is known to be a chelator of metal ions and in vitro has comparable cytotoxicity to cisplatin (IC50=1.8 μM) (Ainscough et al., 1999). To our knowledge, this is the first report to describe its interaction with GABAA receptors.
Methods
Voltage/ion probe reader assay (VIPR)
Primary screening data were generated using a Voltage/Ion Probe Reader assay (VIPRTM; Aurora Biosciences, CA, U. S.A.) as previously described (Adkins et al., 2001; Smith & Simpson, 2003). Briefly, Ltk− cells stably expressing GABAA receptor subunit combinations were seeded into black-sided Porvair 96-well plates and receptor expression was induced 24 h prior to experiment with 1 μM dexamethasone (Sigma, U.K.). Cells were washed in low-Cl− buffer (in mM sodium-D-gluconate 160, potassium-D-gluconate 4.5, CaCl2 2, MgCl2 1, D-glucose 10, HEPES 10, pH 7.4) and dye-loaded for 30 min to give final concentrations of 4 μM chlorocoumarin-2-dimyristoyl phosphatidylethanolamine (CC2-DMPE; FRET donor) and 1 μM bis(1,3-diethyl-2-thiobarbiturate)trimethineoxonol (DiSBAC2(3); FRET acceptor), with 0.5 mM tartrazine present extracellularly. Plates were then placed in a VIPR which performs automated additions using a Hamilton 2200 pipettor and records fluorescence emissions at 460 and 580 nM simultaneously from eight wells. A 400DF15 filter was used in the excitation pathway, and 460DF45 and 580DF60 filters in the respective emission pathways. Rapid ratiometric FRET measurements were made of GABA-evoked depolarizations in low-Cl− buffer as previously described (Adkins et al., 2001) and the ability of compounds to modulate an EC50 response to GABA was examined.
For each time point at each fluorescence emission wavelength, background fluorescence was subtracted (recorded from wells without cells in the same plate) and the ratio of fluorescence at 460–580 nm was calculated. GABA-evoked depolarizations were then expressed as a fractional change in this ratio. Algorithms written as Excel 97 (Microsoft Corp.) macros were used for automated calculation of fluorescence ratio and GABA responses.
Whole-cell patch clamp in mammalian cells
Whole-cell patch-clamp experiments were performed on the stable Ltk− cell lines expressing α1β1γ2s, α1β2γ2s, α2β1γ1, α2β3γ2s and α2β1γ1θ GABAA receptors as described elsewhere (Brown et al., 2002). In brief, cells were patch clamped using a pipette with a tip diameter of approximately 1.5–2.5 μm and a resistance of around 4 MΩ. The intracellular solution contained (in mM): CsCl 130, HEPES 10, BAPTA.Cs 10, ATP.Mg 5, leupeptin 0.1, MgCl2 1, NaVO3 0.1 (pH adjusted to 7.3) with CsOH and 320–340 mOsm by adding sucrose. Cells were voltage –clamped at −20 mV via an Axon 200B amplifier (Axon Instruments, Foster City, CA, U.S.A.) and perfused with artificial cerebrospinal fluid (aCSF) consisting of (in mM) NaCl 149, KCl 3.25, HEPES 10, MgCl2 2, CaCl2 2, D-glucose 11, sucrose 22, at pH 7.4. Drug solutions were applied to the cells via a multi-barrel drug-delivery system, which could pivot the barrels into place using a stepping motor. This ensured rapid application and washout of the drug. The measured agonist exchange time using this system was approximately 20–30 ms. GABA (1 mM and 1 μM) was applied to the cell (5 s on, 30 s washout) and the amplitude of the currents used to calculate an approximate EC20 concentration (individually determined for each cell and ranging from 8 to 35% of the response to 1 mM GABA). Noncumulative concentration–response curves examining the modulatory effects of SCS were constructed with SCS being applied for 30 s prior to co-application with the GABA EC20. Data were recorded and analysed using P-clamp (Version 8, Axon Instruments, Foster City, CA, U.S.A.).
Two-electrode voltage clamp in Xenopus oocytes
The cloning and sequencing of human α2, β1, β2, γ2s and the generation of the β1/β2 chimeras βΔ1.1, βΔ1.2 and βΔ1.4 (hereafter referred to as β21–237β1238–474, β11–236β2237–474, and β11–236β2237–334β1335–474, respectively) and the point mutants β1S290N, β2N289S, β1T255I, β1I308M have been described elsewhere (Wingrove et al., 1994, Hadingham et al., 1993a, 1993b). Site-directed mutagenesis, using standard techniques incorporating a diagnostic restriction site, was performed to generate the point mutants β1K334R, β2R333K, β2I254T and β2M307I (Wingrove et al., 1994). Oligonucleotide primers were synthesized by Sigma-Genosys (Cambridge, U.K.). Mutants were identified by the presence of the diagnostic site and this was confirmed by DNA sequencing using a CEQ 2000 Genetic Analysis System (Beckman-Coulter). The amino-acid numbering starts with the initiating methionine.
All aspects of animal care and use were conducted in accordance with the Animal (Scientific Procedures) Act 1986 and its associated guidelines. The isolation and injection of oocytes by this laboratory has been described in detail elsewhere (Whiting et al., 1995). In brief, stage V and VI oocytes were manually isolated, treated with collagenase (Type IA, 0.5 mg ml−1 for 6 min) and then injected with 10–20 nl of injection buffer (in mM NaCl 88, KCl 1, HEPES 15, at pH 7, filtered through nitro-cellulose 0.2 μm) containing different combinations of human GABAA subunit cDNAs engineered into the expression vector pCDM8 or pcDNA1.1Amp. The ratio of α2 : βy : γ2s (where y equals β1, β2 or chimeras or single-point mutations thereof) in these stock solutions was 1 : 1 : 1 or 1 : 0.1 : 1 with 1 corresponding to 6.6 ng μl−1.
Following incubation for 24–72 h, oocytes were placed in a 50 μl bath and perfused at 4–6 ml min−1 with modified Barth's medium (MBS) consisting of (in mM) NaCl 88, KCl 1, HEPES 10, MgSO4 0.82, Ca(NO3)2 0.33, CaCl2 0.91, NaHCO3 2.4, at pH 7.5. Cells were impaled with two 1–3 MΩ electrodes containing 2 M KCl, voltage-clamped at −70 mV using a Gene Clamp 500 amplifier (Axon Instruments, Foster City, CA, U.S.A.) and recordings visualized on a Gould Oscilloscope 1602, a Gould Windrograf chart recorder (Gould Medical Products Group, Valley View, OH, U.S.A.) and a Compaq Deskpro IBM compatible computer containing the software package ‘Oocyte' (Digitimer Ltd, Welwyn Garden City, Hertfordshire, U.K.).
After a period of stabilization (5–10 min), a maximal concentration of GABA (3 mM) was applied followed by a GABA EC20 concentration (individually determined for each oocyte and ranging from 15 to 30% of the response to 3 mM GABA). Noncumulative concentration–response curves examining the modulatory effects of SCS were constructed with SCS being applied for 45 s prior to co-application with the GABA concentration. Further experiments were designed to examine the interaction of SCS with other known GABAA receptor modulators. Experiments using GABAA receptor inhibitors were designed as follows: a GABA EC20 was applied and the response allowed to reach a plateau, a concentration of picrotoxin (PTX), pregnenolone sulphate (PS), dehydroepiandrosterone sulphate (DHEAS) or bicuculline, which was previously shown to inhibit a GABA EC20 response by approximately 70%, was then co-applied. Finally, when this inhibition reached a plateau, SCS 1 μM was co-applied. The effect of SCS on the GABA mimetic effect of pentobarbital and the modulatory effect of loreclezole and mefenamic acid were also examined. IV curves were constructed in a stepwise manner of 20 mV increments to determine the reversal potential for a GABA EC20 concentration in the absence and presence of SCS. Finally, to evaluate a possible use-dependent block, SCS or picrotoxin, for comparison, was continuously applied during repeated coapplications of a GABA EC20 concentration.
Analysis of data
Concentration–response curves were fitted using Prism (Version 3.03, Graph Pad Software Incorporated, San Diego, CA, U.S.A.) to a nonlinear least-square-fitting function represented by the equation y = min+[max - min)/[1+10(LogIC50−Logx)nH], where y is the response, min and max are the minimum and maximum responses, x is the drug concentration, IC50 is the concentration of drug eliciting a half-maximal inhibition of the response and nH is the Hill coefficient. The current–voltage plots were fitted to a second-order polynomial fit and the reversal potentials calculated (Prism). Data for the IC50 values are shown as the geometric mean (−s.e.m., +s.e.m.), whereas the Hill coefficients, maximum inhibition values and reversal potentials are shown as the arithmetic mean±s.e.m. Data were analysed for statistical significance using the Students t-test or a one-way ANOVA followed by the Tukey–Kramer post hoc test for multiple comparisons. A P-value less than 0.05 was considered significant.
Drugs and reagents
Electrophysiology
GABA, flumazenil, PTX, PS, DHEAS, bicuculline and mefenamic acid were purchased from Sigma Chemical Company (St Louis, MO, U.S.A.). SCS was initially synthesised by the Merck Sharp and Dohme Chemistry Department and later purchased from Lancaster Synthesis Ltd (Morecambe, Lancashire, U.K.). Pentobarbitone sodium (Sagatal) was purchased from Rhone Merieux Inc (Athens, GA, U.S.A.) and loreclezole from Tocris (Avonmouth, Bristol, U.K.). General salts and DMSO were from either VWR International Ltd (Lutterworth, Leicestershire, U.K.) or Sigma Chemical Company. Stock solutions of GABA (1 M in deionized H2O), flumazenil, PTX, bicuculline, loreclezole (10−2 M in DMSO) and PS, DHEAS and mefenamic acid (10−1 M in DMSO) were prepared and stored at −20°C. When using Xenopus oocytes, inhibition to SCS was only apparent if the stock solution (10−2 M in DMSO) was made fresh each day and dilutions in MBS made immediately prior to application.
VIPR
CC2-DMPE was obtained from Aurora Biosciences Corporation (CA, U.S.A.) and DiSBAC2(3) was from Molecular Probes Inc. (OR, U.S.A.) Tartrazine, gluconate salts and all other GABAA receptor modulators were obtained from Sigma. Compounds 1–5 were synthesized by the Merck Sharp and Dohme Chemistry Department, but are all commercially available from various sources.
Results
VIPR screening
Approximately 10,000 compounds from a structurally diverse screening library were screened for activity on α2β1γ1θ, α3β3γ2s and α4β3γ2s GABAA receptors at a single concentration (8 μM). The VIPR provides the opportunity to measure, with high throughput in 96-well format, changes in membrane potential elicited by activation of GABAA receptors. The ability of compounds to modulate the half-maximal response to GABA was examined. This screen identified structurally novel compounds with activity at each of these subtypes and, from this, active and apparently subtype-selective compounds were selected for confirmation and potency titrations. One of these, SCS, was confirmed in the follow-up experiments as being a partial inhibitor of GABA responses at α2β1γ1θ and α2β1γ1 receptors stably expressed in Ltk− cells. It was not, however, found to inhibit the responses at α1β3γ2s, α2β3γ2s, α3β3γ2s, or α4β3γ2s receptor subunit combinations.
In follow-up investigations, 150 compounds with structural similarity to SCS were tested in a single point screen for their ability to modulate GABA responses mediated by α2β1γ1θ receptors. Several active compounds were identified and tested at a range of concentrations using cells expressing α2β1γ1θ or α3β3γ2s receptors to confirm their activity and selectivity for α2β1γ1θ, notably compounds 1 (IC50 67 nM), 2 (54 nM) and 3 (207 nM). All of these had very modest maximal effects on GABA responses however. Two further compounds from this set, compounds 4 and 5, exhibited more pronounced maximal inhibition of GABA responses, although they were less potent and showed less selectivity (four-fold) than SCS or compounds 1–3. SCS was considered to possess a novel selectivity and activity profile deserving further evaluation (Table 1 ).
Table 1.
IC50 and maximum inhibition values for SCS and a range of structurally related compounds using FRET techniques and VIPR measurements at α2β1γ1θ and α3β3γ2s GABAA receptors expressed in Ltk− cells
|
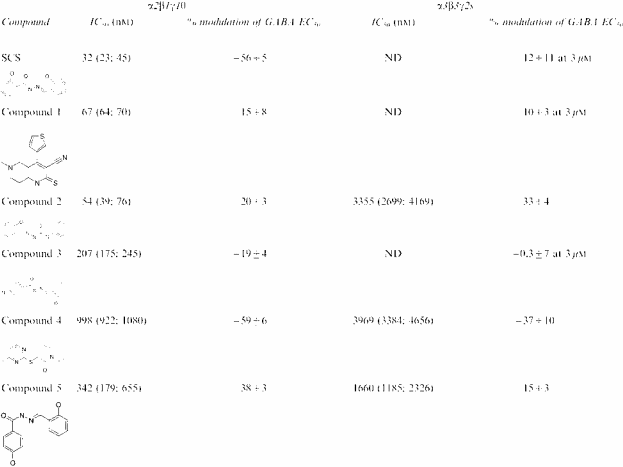
Data represent modulation of the response to an EC50 concentration of GABA, previously determined for each receptor subunit combination. Data for the IC50 values are presented as the geometric mean (−s.e.m.,+s.e.m.) and the maximum inhibition as the arithmetric mean±s.e.m. of three to eight determinations. ND=not determined.
SCS selectivity for β1-containing receptors using whole-cell patch clamp
Further characterization of SCS was performed using electrophysiological techniques. SCS (0.1 nM–3 μM) produced a concentration-dependent inhibition of GABA EC20 currents recorded from Ltk− cells expressing α2β1γ1θ, α2β1γ1 and α1β1γ2s receptors compared with α2β3γ2s and α1β2γ2s receptors upon which SCS had no effect (Figure 1 and Table 2 ). SCS was less potent, produced less inhibition and had a steeper Hill coefficient on α1β1γ2s receptors than α2β1γ1 and α1β1γ1θ (P<0.05). Whether this difference was due to the presence of the γ1 subunit within both receptor subtypes or the individual presence of a α2 and a θ subunit within each receptor subtype was not investigated.
Figure 1.

Concentration–response curves to SCS on Ltk− cells expressing α2β3γ2s, α2β1γ1θ, α2β1γ1, α1β1γ2s and α1β2γ2s receptors. Inhibition of the GABA EC20 response by SCS was only observed on α2β1γ1θ, α2β1γ1, α1β1γ2s receptors. Data are normalized to the control GABA EC20 response immediately prior to construction of the concentration–response curve. Data are mean±s.e.m. with the number of cells in each group indicated in brackets.
Table 2.
IC50, maximum inhibition and Hill coefficient values for SCS inhibition curves using whole-cell patch clamp on different GABAA receptors expressed in Ltk− cells
| Receptor | IC50 (nM) | Maximum inhibition (%) | Hill coefficient |
|---|---|---|---|
| α2β3γ2s | ND | 11.8±3.0 at 3 μM | ND |
| α2β1γ1θ | 4.5 (3.9, 5.1) | 74.3±1.4 | −0.75±0.02 |
| α2β1γ1 | 5.3 (4.4, 6.5) | 68.1±2.7 | −0.81±0.04 |
| α1β1γ2s | 7.9 (7.6, 12.6)* | 50.7±0.9* | −1.36±0.05* |
| α1β2γ2s | ND | 6.2±4.8 at 3 μM | ND |
Data represent modulation of the response to an EC20 concentration of GABA, individually determined for each cell. Data for the IC50 values are presented as the geometric mean (−s.e.m., +s.e.m.) and the maximum inhibition as the arithmetric mean±s.e.m. of six to seven cells. ND=not determined.
P<0.05 vs α2β1γ1 or α2β1γ1θ (one-way ANOVA followed by a Tukey–Kramer post hoc test).
Inhibition by SCS is not voltage or use dependent
The current–voltage relationship was examined for a GABA EC20 in the absence and presence of SCS (300 nM) in Xenopus oocytes expressing α2β1γ2s receptors. Comparable degrees of inhibition were observed at positive and negative holding potentials suggesting no voltage dependence (Figure 2a). The reversal potentials were not significantly different from one another (−24.4±3.5 (GABA) and −22.0±3.6 mV (GABA+SCS)), and are close to the predicted reversal potential for Cl− ions of −25.4 mV in Xenopus oocytes, with an external Cl− concentration of 89.91 mM (MBS used in this study) and an internal Cl− concentration of 33.4 mM (Barish, 1983). Picrotoxin, which is known to bind within the channel, displayed clear use dependence with successive GABA EC20 responses showing greater inhibition. Identical studies with SCS showed no use dependence, with successive GABA EC20 responses showing similar or slightly less inhibition (Figure 2b).
Figure 2.

(a) Current–voltage relationship for a GABA EC20 concentration in the absence and presence of SCS (300 nM) on Xenopus oocytes expressing α2β1γ2s receptors. Both IV curves (absence and presence of SCS) were performed in one oocyte and replicated four times. Data are normalized to the inward current obtained with the GABA EC20 concentration at −70 mV (=−100%). The inhibition by SCS showed no voltage dependence. Data are mean±s.e.m. (b) A representative trace illustrating the lack of use dependence of SCS on a Xenopus oocyte expressing α2β1γ2s receptors. Note the successive decrease in the degree of inhibition of the GABA EC20 from 52 to 41 to 35%. (c) A representative trace illustrating the use dependence of picrotoxin (PTX) on a Xenopus oocyte expressing α2β1γ2s receptors. Note the successive increase in degree of inhibition of the GABA EC20 from 53 to 69 to 75%.
Structural determinants necessary for the inhibition of GABAA receptors by SCS are located within the region arginine 238 and glycine 335 of the β1 subunit
The unique selectivity for β1-containing receptors prompted mutagenesis studies in Xenopus oocytes to identify the amino-acid residues responsible for this selectivity. Concentration–response curves to SCS on Xenopus oocytes expressing α2β1γ2s and α2β2γ2s receptors confirmed the data obtained with Ltk− cells. On α2β2γ2s receptors SCS, up to 3 μM, had no effect on the GABA EC20 response compared with a maximum inhibition of 32.8±4.9%, an IC50 of 4.36 (3.27, 5.81) nM and a Hill coefficient of −1.29±0.25 (n=4) on α2β1γ2s receptors (Figure 3b).
Figure 3.

Effect of SCS on the β1/β2 chimeras. (a) Diagrammatic illustration of the β1/β2 chimeras. β1 sequence is represented by clear boxes and β2 sequence by shaded boxes. SP=signal peptide, CL=cysteine link, TM=transmembrane. (b) Concentration–response curves to SCS on Xenopus oocytes expressing α2β1γ2s, α2β2γ2s, α2β21–237β1238–474γ2s, α2β11–236β2237–474γ2s and α2β11–236β2237–334β1335–474γ2s receptors. Data are normalized to the control GABA EC20 response immediately prior to construction of the concentration–response curve. Data are mean±s.e.m. with the number of cells in each group indicated in brackets. Inhibition by SCS is abolished when a small region encompassing TM1, TM2 and TM3 is replaced in β1 with the corresponding β2 sequence. (c) Schematic diagram illustrating the critical region responsible for SCS inhibition within the human β1 subunit as identified from the β1/2 chimeras. The TM regions are illustrated within the light grey area, and are in agreement with those proposed by Korpi et al. (2002). The four amino-acid residues that are different between β1 and β2/3 are circled. The darker two circles correspond to threonine 255 and isoleucine 308, the two residues critical for inhibition by SCS.
A series of chimeras between β1 and β2 were previously constructed (Wingrove et al., 1994) and used here to delineate the region of the β1 subunit that conferred inhibition by SCS (Figure 3a). Each chimera was co-expressed in Xenopus oocytes with α2 and γ2s cDNAs and inhibition curves to SCS constructed (Figure 3b). α2β21–237β1238–474γ2s gave similar results to wild type β1-containing receptors (maximum inhibition of 44.3±7.5%, an IC50 of 20.51 (15.70, 26.79) nM and a Hill coefficient of −1.26±0.15 (n=4)), whereas α2β11–236β2237–474γ2s and α2β11–236β2237–334β1335–474γ2s gave similar results to β2 wild type, that is, no inhibition by SCS up to 3 μM. These results indicated that this small region between K237 and G334 that included TM1–TM3 was responsible for the selectivity of SCS (Figure 3c).
T255 and I308 of the β1 subunit are required for inhibition by SCS
Alignment of the predicted amino-acid sequences of the β1, β2 and β3 subunits between amino acids 237 and 334 revealed four amino acids that were different in β1 compared to β2 and β3 (β1 T255, S290, I308, K334). These four residues were individually mutated in β1 to the β2/3 equivalent and in β2 to the β1 equivalent and co-expressed with α2γ2s. Similar to wild-type receptors, the single point mutations β1S290N and β1K334R were inhibited by SCS; IC50=39.5 (33.5, 46.5) nM, maximum inhibition=32.3±5.9%, Hill coefficient=−1.13±0.14 and IC50=15.8 (12.2, 20.6) nM, maximum inhibition=34.2±5.6%, Hill coefficient=−1.29±0.15, respectively. The maximum inhibition and Hill coefficient for α2β1S290Nγ2s and α2β1K334Rγ2s were not significantly different from wild type, whereas the IC50 values were significantly greater than wild type (P<0.05). Conversely β1T255I, β1I308M and all the four single point mutations within the β2 subunit (I254 T, N289S, M307I and R333 K) were unaffected by SCS similar to wild-type α2β2γ2s (Figure 4). These data demonstrate that both T255 and I308 within the β1 subunit confer the inhibition observed with SCS and mutation of either residue abolishes this inhibition. However, when these individual residues were mutated within β2 to the β1 counterpart inhibition was not conferred. Construction of a β2 cDNA containing both these point mutations was unsuccessful, so we were unable to determine if both residues could confer inhibition to β2.
Figure 4.

Effect of SCS on the β1 (a) and β2 (b) single point mutations co-expressed with α2 and γ2s in Xenopus oocytes. Data are normalized to the control GABA EC20 response immediately prior to construction of the concentration–response curve. Mutation of threonine 255 or isoleucine 308 in β1 to the β2 equivalent amino-acid was sufficient to abolish inhibition. Data are mean±s.e.m. with the number of cells in each group indicated in brackets.
White et al. (1995) have shown that the EC50 value generated from GABA concentration–response curves was not different in oocytes expressing α2β1γ2L and α2β2γ2L receptors. In agreement with this, our EC20 concentrations used for the two wild-type receptors were not significantly different. Wingrove et al. (1994) who expressed the β1/2 chimeras and some of the point mutations with α1 and γ2s reported that the affinities for GABA were unchanged. In this study, the EC20 values of most of the chimeras and point mutations were not significantly different from the wild-type receptors (the exceptions being α2β1γ2s vs α2β21–237β1238–474γ2s and α2β1γ2s vs α2β2N289Sγ2s one-way ANOVA with Tukey Kramer post hoc test). Caution has to be applied to any significance obtained, since firstly the number of cells expressing the various chimeras and point mutations were low (between 4 and 6) and secondly the EC20 concentration used was within the range of EC15–EC30 and not determined from a concentration–response curve.
Interaction with other GABAA receptor ligands
Pentobarbitone is known to directly activate GABAA receptors via a site distinct from the GABA-binding site (Thompson et al., 1996; Pistis et al., 1999). Similar to its effects on GABA-activated currents, SCS (1 μM) inhibited currents induced by a submaximal concentration of pentobarbitone (100 μM) on α2β1γ2s receptors. The degree of inhibition (39.3±3.4%, n=5) was comparable to that observed with GABA, suggesting that the inhibition is not competitive with GABA but allosteric in nature. To further confirm this hypothesis, a concentration–response curve to GABA in the absence and presence of SCS (300 nM) was constructed. As illustrated in Figure 5, SCS caused a significant reduction in the maximum response with no change in the pEC50 or Hill coefficient indicative of a noncompetitive mechanism. To verify that this reduction in maximum was a real effect and not simply run down, a maximum concentration of GABA (3 mM) was applied until reproducible responses were obtained and then repeated in the presence of SCS. As expected, the response to 3 mM GABA was reduced by 33.8±3.6% (n=3).
Figure 5.

Concentration–response curve to GABA in the absence and presence of SCS on Xenopus oocytes expressing α2β2γ2s receptors. Both concentration–response curves (absence and presence of SCS) were performed in one oocyte and replicated four times. Data were normalized to the current obtained with 3 mM GABA in the absence of SCS. The GABA pEC50 and Hill coefficient values were not significantly different (−4.2 vs −4.1 M and 1.1 vs 1.2, respectively), whereas the maximum response was significantly reduced in the presence of SCS (99.1 vs 85.9%, P=0.002). Data are mean±s.e.m.
Experiments were performed with the benzodiazepine site antagonist flumazenil to investigate a possible interaction of SCS via the benzodiazepine-binding site. Flumazenil (1 μM) alone elicited a small degree of inhibition of the GABA EC20 response (8.4±2.1%, n=3). This inhibition was further increased in the presence of SCS (1 μM) to 30±1.5% (n=3), demonstrating that SCS does not bind to the benzodiazepine-binding site.
Since SCS exhibits only partial block of the GABA response (36.3±3.9% at 1 μM), we hypothesized that if SCS interacted via the same binding site as a known antagonist, a reduction in the degree of inhibition would be observed. Conversely, if the two compounds interacted at separate sites, the degree of inhibition would be increased (additive effect). Competition experiments with SCS and a number of known GABAA receptor inhibitors (PTX, PS, DHEAS and bicuculline) were performed. Concentrations of each inhibitor that produced approximately 70% inhibition of a GABA EC20 response were determined. As shown in Figure 6, the degree of inhibition with all the four inhibitors was further increased upon co-application of SCS, suggesting additive effects.
Figure 6.

Competition of SCS with PTX, DHEAS, PS and bicuculline on Xenopus oocytes expressing α2β1γ2s receptors. The white bars represent the inhibition of a GABA EC20 by PTX (200 nM), DHEAS (10 μM), PS (10 μM) and bicuculline (6 μM), whereas the black bars represent the inhibition observed upon co-application of SCS (1 μM). Data are mean±s.e.m. of four oocytes for each inhibitor.
A number of allosteric modulators demonstrate β-subunit selectivity, for example, loreclezole, etomidate, furosemide (Wafford et al., 1994; Hill-Venning et al., 1997; Thompson et al., 1999). Regardless of whether the compound inhibits (e.g. furosemide) or potentiates (e.g. loreclezole) the GABA response, the selectivity reported has been for β2/3-containing receptors vs β1-containing receptors. More recently, Halliwell et al. (1999) have described the selectivity profile for the nonsteroidal anti-inflammatory agent mefenamic acid. Similar to loreclezole, this compound potentiated GABA-activated currents on α1β2γ2s receptors but not on α1β1γ2s. More relevant to this study was the observation that on α1β1 receptors, GABA-activated currents were inhibited by mefenamic acid compared with potentiation on α1β2. Experiments were performed to determine whether SCS influenced the functional response to loreclezole on α2β2γ2s receptors and mefenamic acid on α1β1 receptors. As illustrated in Figure 7, SCS (1 μM) had no significant effect on the potentiation of a GABA EC20 by loreclezole (10 μM) (256±40 vs 222±26%, n=4) or the inhibition of a GABA EC20 by mefenamic acid. SCS (1 μM) produced 57.5±1.9% inhibition at α1β1 receptors, and as previously reported; 300 μM mefenamic acid produced 82.7±2.9% inhibition. When combined, 300 μM mefenamic acid plus 1 μM SCS elicited 90.7±1.2% inhibition (n=4) slightly greater than mefenamic acid alone, again suggesting a purely additive effect.
Figure 7.

(a) A representative trace illustrating the effect of SCS on the potentiation of a GABA EC20 by the β2/3 selective modulator loreclezole on an oocyte expressing α2β2γ2s receptors. (b) A representative trace illustrating the effect of SCS on the inhibition of a GABA EC20 by mefenamic acid on an oocyte expressing α1β1 receptors. Loreclezole, mefenamic acid and SCS were applied 45 s before coapplication of GABA EC20. Current amplitude and time are indicated by the scale bars. A reduction in the amount of potentiation to loreclezole or inhibition to mefenamic acid by SCS would indicate competition and hence commonality of the two modulators at the binding or transduction level.
Discussion
SCS was initially identified from a high-throughput VIPR screen as being a potent partial subtype-selective inhibitor of GABAA receptors. This selectivity was not specific to SCS since a number of structurally similar compounds also displayed β1 selectivity. Detailed electrophysiological experiments identified SCS as having a unique pharmacology producing selective partial inhibition of β1-containing receptors.
Comparison with other β-selective compounds
In recent years, a number of modulators of the GABAA receptor have been reported that demonstrate β2/3 selectivity over β1, for example, loreclezole, etomidate, tracazolate, mefenamic acid, furosemide (Wafford et al., 1994; Hill-Venning et al., 1997; Halliwell et al., 1999; Thompson et al., 1999; 2002). In all cases, the potency of the modulator was reduced or abolished when serine was introduced into β2 or β3 (position 289 in human β2 and 290 in human β3) and increased or conferred when asparagine was introduced into β1 (position 290). SCS differed from these other modulators since although it inhibited α2β1S290Nγ2s receptors with reduced potency maximum efficacy was unaffected, and SCS had no effect on α1β2N289Sγ2s.
Mefenamic acid, loreclezole and SCS are broadly structurally similar in that they are composed of two aromatic or heteroaromatic rings linked by spacers of varying length. At physiological pH, the majority of SCS molecules will be nonionized, suggesting that the binding site is likely to exist in a lipid environment (the calculated pKa for deprotonation of the phenolic groups in SCS is 8.4–8.5), compared with mefenamic acid which exists mainly as the deprotonated carboxylic acid (pKa=3.7), suggesting an aqueous environment for the binding site and loreclezole which is nonionized (for protonation of the triazole pKa=1.8).
Importance of threonine 255 and isoleucine 308
Mutation of either threonine 255 or isoleucine 308 within the β1 subunit to the β2 counterpart was sufficient to abolish the inhibition; however, the converse individual mutations within β2 did not introduce any inhibition. To our knowledge, these residues have not been implicated in the action of other GABAA receptor modulators. A minor alteration to a compound's binding site or transduction pathway is often sufficient to abolish activity. It is much more difficult, however, to confer activity since this effect is often the net result of a number of processes involving many different regions of the protein. Although it is not certain if isoleucine 308 forms part of the TM2–TM3 loop or the TM3 domain itself (Bera et al., 2002; Korpi et al., 2002), the recent work of Bera et al. (2002) showed that in the α1 subunit the corresponding residue when mutated to cysteine was not accessible to MTS reagents, indicating that it is either tightly packed in the protein structure or that the covalent modification was functionally silent. Similarly, the residue corresponding to that of threonine 255 in the torpedo nicotinic acetylcholine receptor has been labelled with a hydrophobic probe (Blanton & Cohen, 1994).
From the data obtained, we suggest that SCS binds to a novel site on the GABAA receptor which may incorporate the residues threonine 255 and isoleucine 308. SCS may share a similar transduction mechanism to loreclezole and mefenamic acid, etc., involving serine/asparagine 290.
Possible role of metal chelation
An alternative possible mechanism of action could involve the ability of SCS to chelate metal ions, as recently demonstrated by Wilkins & Smart (2002) for the potentiating effect of dithiothreitol. The assumption here is that a contaminating metal ion within the buffers used could potentiate GABA responses to β1-containing receptors but not to β2- or 3- containing receptors. Chelation of this metal ion by SCS thereby removes this potentiation, which is revealed by an apparent inhibition. For this suggestion to be feasible, the metal ion would need to be more potent than SCS, be selective for β1-containing receptors and induce potentiation. Of the metal ions that interact with GABAA receptors, most have been shown to inhibit GABA responses with no β selectivity.
Experiments to probe the binding site and role of β1 subunits
We have demonstrated that SCS did not interact with any of the known allosteric modulators investigated in this study, suggesting an interaction at a novel binding site. The lack of voltage and use dependence would indicate that the binding site is not within the ion channel, but residues close to the channel are important for SCS inhibition. Although a tritiated form of SCS was synthesized, experiments designed to probe the binding site of SCS could not be performed due to very high nonspecific binding.
The β1 subunit is widely distributed in the brain (Pirker et al., 2000), making it difficult to speculate as to what therapeutic use a β1-selective inhibitor would have. Two recent reports using ‘knock-in' mouse lines have demonstrated that the clinical effects of the anaesthetic agents etomidate and propofol can be attributed to either the β2 (sedative-hypnotic effect) or the β3 (anaesthetic effect) subunit (Jurd et al., 2003; Reynolds et al., 2003). In the absence of β1 knockout mice, it was hoped that having identified a β1-selective compound we would be able to perform in vivo experiments in order to understand the role of β1-containing GABAA receptors. Unfortunately, these studies were precluded by poor in vivo pharmacokinetics, making it difficult to utilize this compound to determine the role of β1-containing GABAA receptors.
In conclusion, the present study describes the identification and characterization of SCS as a selective partial inhibitor of β1-containing GABAA receptors. SCS showed no interaction with any of the GABAA receptor modulators tested or any voltage or use dependence. Mutagenesis studies revealed the importance of threonine 255, located within TM1, and isoleucine 308, located just prior to TM3, as critical residues within β1 for conferring the inhibition. We hypothesize that SCS may interact at a previously unidentified site and exerts its effects via an allosteric mechanism.
Abbreviations
- DHEAS
dehydroepiandrosterone 3-sulphate
- FRET
fluorescence resonance energy transfer
- GABA
γ-aminobutyric acid
- GABAA
γ-aminobutyric acid type A
- I
isoleucine
- K
lysine
- M
methionine
- MBS
modified Barth's saline
- N
asparagine
- PS
pregnenolone sulphate
- PTX
picrotoxin
- R
arginine
- S
serine
- SCS
salicylidene salicylhydrazide
- T
threonine
- TM
transmembrane domain
- VIPR
voltage/ion probe reader.
References
- ADKINS C.E., PILLAI G.V., KERBY J., BONNERT T.P., HALDON C., MCKERNAN R.M., GONZALEZ J.E., OADES K., WHITING P.J., SIMPSON P.B. α4β3δ GABAA receptors characterized by fluorescence resonance energy transfer-derived measurements of membrane potential. J. Biol. Chem. 2001;276:38934–38939. doi: 10.1074/jbc.M104318200. [DOI] [PubMed] [Google Scholar]
- AINSCOUGH E.W., BRODIE A.M., DENNY W.A., FINLAY G.J., GOTHE S.A., RANFORD J.D. Cytotoxicity of salicylaldehyde benzoylhydrazone analogs and their transition metal complexes: quantitative structure–activity relationships. J. Inorg. Biochem. 1999;77:125–133. doi: 10.1016/s0162-0134(99)00131-2. [DOI] [PubMed] [Google Scholar]
- BARISH M.E. A transient calcium-dependent chloride current in the immature Xenopus oocyte. J. Physiol. (Lond). 1983;342:309–325. doi: 10.1113/jphysiol.1983.sp014852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BELELLI D., LAMBERT J.J., PETERS J.A., WAFFORD K., WHITING P.J. The interaction of the general anesthetic etomidate with the gamma-aminobutyric acid type A receptor is influenced by a single amino acid. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11031–11036. doi: 10.1073/pnas.94.20.11031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BERA A.K., CHATAV M., AKABAS M.H. GABAA receptor M2–M3 loop secondary structure and changes in accessibility during channel gating. J. Biol. Chem. 2002;277:43002–43010. doi: 10.1074/jbc.M206321200. [DOI] [PubMed] [Google Scholar]
- BLANTON M.P., COHEN J.B. Identifying the lipid–protein interface of the Torpedo nicotinic acetylcholine receptor: secondary structure implications. Biochemistry. 1994;33:2859–2872. doi: 10.1021/bi00176a016. [DOI] [PubMed] [Google Scholar]
- BROWN N., KERBY J., BONNERT T.P., WHITING P.J., WAFFORD K.A. Pharmacological characterization of a novel cell line expressing human α4β3δ GABAA receptors. Br. J. Pharmacol. 2002;136:965–974. doi: 10.1038/sj.bjp.0704795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG Y., WANG R., BAROT S., WEISS D.S. Stoichiometry of a recombinant GABAA receptor. J. Neurosci. 1996;16:5415–5424. doi: 10.1523/JNEUROSCI.16-17-05415.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FARRAR S.J., WHITING P.J., BONNERT T.P., MCKERNAN R.M. Stoichiometry of a ligand-gated ion channel determined by fluorescence energy transfer. J. Biol. Chem. 1999;274:10100–10104. doi: 10.1074/jbc.274.15.10100. [DOI] [PubMed] [Google Scholar]
- HADINGHAM K.L., WINGROVE P., LE BOURDELLES B., PALMER K.J., RAGAN C.I., WAFFORD K.A. Cloning of cDNA sequences encoding human α1 and α3 γ-aminobutyric acidA receptor subunits and characterization of the benzodiazepine pharmacology of recombinant α1-, α2-, α3-, and α5-containing human γ-aminobutyric acidA receptors. Mol. Pharmacol. 1993a;43:970–975. [PubMed] [Google Scholar]
- HADINGHAM K.L., WINGROVE P.B., WAFFORD K.A., BAIN C., KEMP J.A., PALMER K.J., WILSON A.W., WILCOX A.S., SIKELA J.M., RAGAN C.I., WHITING P.J. Role of the β subunit in determining the pharmacology of human γ-aminobutyric acid type A receptors. Mol. Pharmacol. 1993b;44:1211–1218. [PubMed] [Google Scholar]
- HALLIWELL R.F., THOMAS P., PATTEN D., JAMES C.H., MARTINEZ-TORRES A., MILEDI R., SMART T.G. Subunit-selective modulation of GABAA receptors by the non-steroid anti-inflammatory agent, mefenamic acid. Eur. J. Neurosci. 1999;11:2897–2905. doi: 10.1046/j.1460-9568.1999.00709.x. [DOI] [PubMed] [Google Scholar]
- HILL-VENNING C., BELELLI D., PETERS J.A., LAMBERT J.J. Subunit-dependent interaction of the general anaesthetic etomidate with the γ-aminobutyric acid type A receptor. Br. J. Pharmacol. 1997;120:749–756. doi: 10.1038/sj.bjp.0700927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORPI E.R., GRUNDER G., LÜDDENS H. Drug interactions at GABA(A) receptors. Prog. Neurobiol. 2002;67:113–159. doi: 10.1016/s0301-0082(02)00013-8. [DOI] [PubMed] [Google Scholar]
- JURD R., ARRAS M., LAMBERT S., DREXLER B., SIEGWART R., CRESTANI F., ZAUGG M., VOGT K.E., LEDERMANN B., ANTKOWIAK B., RUDOLPH U. General anesthetic actions in vivo strongly attenuated by a point mutation in the GABAA receptor β3 subunit. FASEB J. 2003;17:250–252. doi: 10.1096/fj.02-0611fje. [DOI] [PubMed] [Google Scholar]
- PIRKER S., SCHWARZER C., WIESELTHALER A., SIEGHART W., SPERK G. GABAA receptors: immunocytochemical distribution of 13 subunits in the adult rat brain. Neuroscience. 2000;101:815–850. doi: 10.1016/s0306-4522(00)00442-5. [DOI] [PubMed] [Google Scholar]
- PISTIS M., BELELLI D., MCGURK K., PETERS J.A., LAMBERT J.J. Complementary regulation of anaesthetic activation of human (α6β3γ2L) and Drosophila (RDL) GABA receptors by a single amino acid residue. J. Physiol. 1999;515:3–18. doi: 10.1111/j.1469-7793.1999.003ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REYNOLDS D.S., ROSAHL T.W., CIRONE J., O'MEARA G.F., HAYTHORNTHWAITE A., NEWMAN R.J., MYERS J., SUR C., HOWELL O., RUTTER A.R., ATACK J., MACAULAY A.J., HADINGHAM K.L., HUTSON P.H., BELELLI D., LAMBERT J.J., DAWSON G.R., MCKERNAN R., WHITING P.J., WAFFORD K.A. Sedation and anesthesia mediated by distinct GABAA receptor isoforms. J. Neurosci. 2003;23:8608–8617. doi: 10.1523/JNEUROSCI.23-24-08608.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SIEGHART W., SPERK G. Subunit composition, distribution and function of GABAA receptor subtypes. Curr. Top. Med. Chem. 2002;2:795–816. doi: 10.2174/1568026023393507. [DOI] [PubMed] [Google Scholar]
- SMITH A.J., SIMPSON P.B. Methodological approaches for the study of GABAA receptor pharmacology and functional responses. Anal. Bioanal. Chem. 2003;377:843–851. doi: 10.1007/s00216-003-2172-y. [DOI] [PubMed] [Google Scholar]
- THOMPSON S.A., ARDEN S.A., MARSHALL G., WINGROVE P.B., WHITING P.J., WAFFORD K.A. Residues in transmembrane domains I and II determine γ-aminobutyric acid type A receptor subtype-selective antagonism by furosemide. Mol. Pharmacol. 1999;55:993–999. doi: 10.1124/mol.55.6.993. [DOI] [PubMed] [Google Scholar]
- THOMPSON S.A., WHITING P.J., WAFFORD K.A. Barbiturate interactions at the human GABAA receptor: dependence on receptor subunit combination. Br. J. Pharmacol. 1996;117:521–527. doi: 10.1111/j.1476-5381.1996.tb15221.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMPSON S.A., WINGROVE P.B., CONNELLY L., WHITING P.J., WAFFORD K.A. Tracazolate reveals a novel type of allosteric interaction with recombinant gamma-aminobutyric acid(A) receptors. Mol. Pharmacol. 2002;61:861–869. doi: 10.1124/mol.61.4.861. [DOI] [PubMed] [Google Scholar]
- WAFFORD K.A., BAIN C.J., QUIRK K., MCKERNAN R.M., WINGROVE P.B., WHITING P.J., KEMP J.A. A novel allosteric modulatory site on the GABAA receptor β subunit. Neuron. 1994;12:775–782. doi: 10.1016/0896-6273(94)90330-1. [DOI] [PubMed] [Google Scholar]
- WHITE G., GURLEY D., HARTNETT C., STIRLING V., GREGORY J. Human α and β subunits contribute to the EC50 for GABA at the GABAA receptor expressed in Xenopus oocytes. Receptors Channels. 1995;3:1–5. [PubMed] [Google Scholar]
- WHITING P.J. GABA-A receptor subtypes in the brain: a paradigm for CNS drug discovery. Drug Discov. Today. 2003;8:445–450. doi: 10.1016/s1359-6446(03)02703-x. [DOI] [PubMed] [Google Scholar]
- WHITING P.J., WAFFORD K.A., PRIBILLA I., PETRI T.Channel cloning, mutagenesis, and expression Ion Channels. A Practical Approach 1995McLean, VA: IRL Press; 133–169.ed. Ashley, R.H. pp [Google Scholar]
- WILKINS M.E., SMART T.G. Redox modulation of GABAA receptors obscured by Zn2+ complexation. Neuropharmacology. 2002;43:938–944. doi: 10.1016/s0028-3908(02)00238-1. [DOI] [PubMed] [Google Scholar]
- WINGROVE P.B., WAFFORD K.A., BAIN C., WHITING P.J. The modulatory action of loreclezole at the γ-aminobutyric acid type A receptor is determined by a single amino acid in the β2 and β3 subunit. Proc. Natl. Acad. Sci. U.S.A. 1994;91:4569–4573. doi: 10.1073/pnas.91.10.4569. [DOI] [PMC free article] [PubMed] [Google Scholar]