

Abstract
Clinical observations with the selective serotonin reuptake inhibitor (SSRI), S-citalopram, indicate that S-citalopram is more efficacious and produces earlier symptom relief than RS-citalopram. Since R-citalopram is at least 20-fold weaker than S-citalopram as inhibitor of the 5-HT transporter (SERT) in preclinical studies, the clinical data suggest an unexpected antagonistic interaction between the two enantiomers. We therefore characterised the interaction of R- and S-citalopram with the SERT in in vivo and in vitro assays.
In both behavioural (potentiation of 5-hydroxytryptophan (5-HTP)-induced behaviour) and electrophysiological studies (inhibition of 5-HT-elicited ion currents in Xenopus oocytes expressing the human SERT (hSERT) R-citalopram inhibited the effects of S-citalopram in a dose-dependent manner. With S-citalopram : R-citalopram ratios of 1 : 2 and 1 : 4, 5-HTP potentiation was significantly smaller than with S-citalopram alone.
emsp;R-citalopram did not antagonise the effects of another SSRI (fluoxetine) in either behavioural or electrophysiological studies.
In oocytes, inhibition of hSERT-mediated currents by R-citalopram was almost completely reversible and characterised by fast on- and off-sets of action. In contrast, the off-set for S-citalopram was 35-fold slower than for R-citalopram.
Kinetic analysis of the oocyte experiments suggests that S-citalopram binding to SERT induces a long-lasting, inhibited state of the transporter and that coapplication of R-citalopram partially relieves SERT of this persistent inhibition.
We propose that the kinetic interaction of R- and S-citalopram with SERT is a critical factor contributing to the antagonistic effects of R-citalopram on S-citalopram in vitro and in vivo.
Keywords: Citalopram, escitalopram, functional antagonism, 5-HTP, behaviour, Xenopus oocytes, in vivo binding, kinetic interaction
Introduction
Selective serotonin reuptake inhibitors (SSRIs) have been the drugs of choice for the treatment of depression for the last two decades. Although the administration of an SSRI is followed by an immediate increase in the concentration of extracellular serotonin (5-HT) in the brain, the antidepressant action does not appear until after 4–6 weeks. The mechanism underlying this delay in clinical efficacy is still enigmatic, although it has been hypothesised that the rate of increase in 5-HT levels may be a determining factor (Artigas et al., 1994, 1996; Blier & de Montigny, 1994; Blier et al., 1997). The SSRI S(+)-citalopram (escitalopram) is the therapeutically active enantiomer of R(−),S(+)-citalopram (RS-citalopram). Since citalopram has a high selectivity for 5-HT reuptake inhibition and R-citalopram has a very low affinity for the serotonin transporter (SERT) (Sánchez et al., 2003), it was expected that S-citalopram would be similar in efficacy to, but twice as potent as RS-citalopram. However, recently conducted clinical studies indicated that S-citalopram has a significantly faster onset for its antidepressant action than the RS-citalopram (Lepola et al., 2003). This difference is also observed in animal models predictive of antidepressant effects, that is, reversal of chronic mild stress-induced hedonic deficits (Montgomery et al., 2001) and antidepressant-induced increase of agonistic behaviour (Mitchell and Hogg, personal communication). Furthermore, coapplication of R- and S-citalopram produced a lower increase in extracellular 5-HT levels in a microdialysis study in rat frontal cortex when compared to S-citalopram alone (Mørk et al., 2003). These data indicate that R-citalopram antagonises the effect of S-citalopram in functional assays.
The molecular target of the citalopram enantiomers is the 5-HT transporter (SERT), a member of the Na- and Cl-dependent neurotransmitter transporter family, which also includes the transporters for dopamine, noradrenaline, GABA, glycine and other small molecules (Amara & Kuhar, 1993). Typically for this family, SERT couples the high-affinity uptake of 5-HT to the cotransport of sodium and chloride ions. In more recent electrophysiological studies, it has been shown that SERT and related transporters have complex permeation properties, mediating not only the transport of substrates but also substrate-dependent as well as -independent ion fluxes (reviewed by Sonders & Amara, 1996). For example, SERT mediates a small inward current (referred to as leakage current), which can be blocked both by substrates and by nontransported inhibitors such as SSRIs. However, the most prominent SERT-mediated current is transport associated – a voltage-dependent, inwardly directed ion flux, which is elicited by transported substrates, but blocked by SSRIs or other nontransported ligands such as cocaine or tricyclic antidepressants (Mager et al., 1994). We have used these transport-associated currents as a sensitive read-out to study the interaction of R- and S-citalopram with human SERT (hSERT).
The present report further characterizes the functional interaction between the two citalopram enantiomers at the SERT, and thereby attempts to provide a molecular mechanism for the apparent inhibitory action of R-citalopram on the pharmacological effects of S-citalopram in preclinical and clinical studies. We have complemented this study with experiments exploring whether a similar functional interaction exists for R-citalopram and the SSRI, fluoxetine.
Methods
Ethical permission for the studies was granted by the animal welfare committee, appointed by the Danish Ministry of Justice and all animal procedures were carried out in compliance with the EC Directive 86/609/EEC and with Danish law regulating experiments on animals.
hSERT cloning and in vitro transcription
The hSERT cDNA was cloned from human placenta (Mortensen et al., 1999) and subcloned into pGEMHE (Liman et al., 1992) using XhoI/HindIII.
hSERT cRNA was transcribed and capped in vitro (mMessage mMachine T7 kit, Ambion, Inc., Austin, TX, U.S.A.) from the NotI-linearised hSERT-pGEMHE construct, and further purified using RNeasy Mini columns (Qiagen GmbH, Hilden, Germany).
Oocyte isolation and injection
Xenopus laevis females were anaesthetised with 0.4% tricaine (aminobenzoic acid ethyl ester, Sigma, St Louis, MO, U.S.A.), before stage V and VI oocytes were removed through a small abdominal incision. Following manual isolation with fine forceps, the oocytes were treated with a mild collagenase solution for 6 min (Sigma: type IA (0.5 mg ml−1)). The oocytes were injected with 23 nl of hSERT cRNA (0.05–0.1 μg μl−1) and incubated at 19°C in sterile-filtered MBS buffer (88.0 mM NaCl; 1.0 mM KCl; 15.0 mM HEPES, pH 7.5; 2.4 mM NaHCO3; 0.41 mM CaCl2; 0.82 mM MgSO4; 0.3 mM Ca(NO3)2) supplemented with 100 μg ml−1 penicillin, 100 μg ml−1 streptomycin and 2 mM sodium pyruvate. Oocytes were used in two-electrode voltage-clamp experiments 2–5 days after injection.
Two-electrode voltage clamping
Oocytes were placed in a 200 μl bath continuously superfused (4–6 ml min−1) with Ringer buffer (115 mM NaCl; 2.5 mM KCl; 10 mM HEPES, pH 7.5; 1.8 mM CaCl2; 0.1 mM MgCl2). In two-electrode voltage-clamp experiments, oocytes were impaled with two 1–3 MΩ electrodes containing 3 M KCl and voltage clamped at −70 mV using a GeneClamp 500B (Axon Instruments, Inc., Union City, CA, U.S.A.).
Drugs were applied in the perfusate until a steady current was observed (less than 1 min for 5-HT and 4–10 min for coapplication of 5-HT and the SSRIs), with 6 min between each 5-HT application to allow recovery from desensitisation. Currents were recorded using pCLAMP 8 software (Axon Instruments, Inc.). Experiments were initiated when consecutive applications of 5-HT produced responses with less than 5% variation.
Data were obtained from three to seven oocytes from at least two different oocyte batches.
Housing of mice
Male NMRI/BOM mice (18–25 g; Bomholtgaard, Denmark) were used for the 5-hydroxytryptophan (5-HTP) potentiation studies and in vivo binding studies. The mice were housed in groups of 10 in plastic cages (35 × 30 × 12 cm3). Animals were habituated to the animal facilities for at least 1 week before testing. The room temperature (21±2°C), relative humidity (55±5%) and air exchange (16 vol h−1) were automatically controlled. The animals had free access to commercial food pellets and tap water before test sessions.
Potentiation of 5-HTP-induced behaviour in mice
The test procedure is described in detail by Hyttel et al. (1992). Briefly, 30 min after subcutaneous (s.c.) administration of test compounds, mice were given 5-HTP (100 mg kg−1, intravenous (i.v.)). Thereafter, the animals were evaluated in their home cage during a 15 min observation period with respect to stereotypy (lateral head movements), tremor and hind limb abduction. The behavioural changes were scored as 0=not present, 1=present in mild to moderate degree (i.e. occurring intermittently during the observation period), 2=present in a marked degree (i.e. present during the entire observation period). A total of five to 10 mice were used per dose. Each dose–response experiment was conducted twice with overlapping doses. All behavioural scoring was carried out under blinded conditions by a rater unaware of the treatment.
In vivo binding affinity
In agreement with a previously published method (Larsen et al., 2004), mice were treated with drug s.c. 30 min before receiving 4 μCi [3H]MADAM (N,N-dimethyl-2-(2-amino-4-methylphenyl thio)benzylamine) (200 μl) i.v. via the tail vein. In vivo binding was carried out according to Andersen et al. (1987) with a few modifications. After 15 min, the animals were decapitated and the cerebral cortex was quickly dissected and homogenised in ice-cold buffer (50 mM Tris, 120 mM NaCl, 5 mM KCl, pH 7.4). Homogenate (0.5 ml) was filtered through 0.1% polyethyleneimine-soaked Whatman GF/C filters. Filters were washed twice with 5 ml ice-cold buffer and counted in a scintillation counter. The process of decapitation, dissection, homogenisation and filtration was completed within 60 s. A group of three saline-treated animals was used to determine total [3H]MADAM binding and a group of three animals that was pretreated with 30 mg kg−1 escitalopram before injection of [3H] MADAM was used to determine the level of nonspecific binding. Using this paradigm, a level of specific binding of 15 fmol mg−1 protein in saline-treated animals was obtained.
The protein content of the homogenate was measured using the assay described by Smith et al. (1985).
Drugs
R- and S-citalopram (enantiomeric purity >99.9%) and fluoxetine were synthesised at H. Lundbeck A/S and dissolved in saline (in vivo experiments) or Ringer (oocyte studies). In animal studies, the injection volume was 10 ml kg−1 body weight. All other compounds were obtained from normal commercial sources. Further details on dosages and concentrations used in the study are summarised in figure legends.
Data analysis and statistical assessment
EC50 values for each individual oocyte were determined using nonlinear iterative curve fitting by the program GraFit 5.04 (Erithacus software, Ltd, Surrey, U.K.) fitting the logistic equation
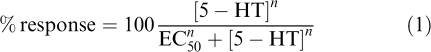 |
where EC50 is the concentration producing half-maximum response and n is a slope factor.
IC50 values for each individual oocyte were determined using nonlinear iterative curve fitting by the program GraFit 5.04 (Erithacus software) fitting the logistic equation:
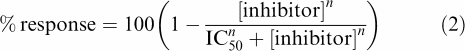 |
where IC50 is the concentration reducing the response to 50% of the initial response and n is a slope factor. IC50 values were converted to Ki values using the Cheng–Prusoff equation
 |
where the EC50 and IC50 values were determined as described above.
The response of 5-HT (assuming a constant concentration of this agonist during the experiment) in the presence of a competitive inhibitor can be calculated by Equation (4):
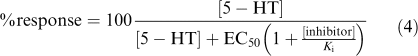 |
In the presence of two competitive inhibitors, the response can be described by Equation (5):
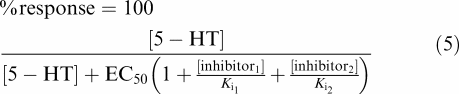 |
In oocyte traces from kinetic experiments, single exponential curves were analysed fitting the association (6) and dissociation (7) equations:
where It is the current at a given time, t, I0 is the current at t=0, τ is a time constant corresponding to 1/k and C is an arbitrary constant.
ED50 values from in vivo binding studies were determined using nonlinear regression by the program Prism (GraphPad, San Diego, CA, U.S.A.).
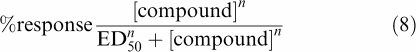 |
where ED50 is the average dose yielding 50% of the maximal displacement of [3H]-MADAM and n is the slope factor.
Percent occupancy was calculated as
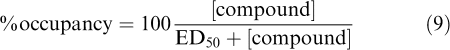 |
Oocyte and behavioural data were analysed by one-way analysis of variances followed by a post hoc comparison of means by t-test and considered significant if P<0.05.
Results
The potentiating effect of S-citalopram, R-citalopram, RS-citalopram and fluoxetine on 5-HT-induced behaviour by the 5-HT precursor, 5-HTP, was used as a measure of 5-HT reuptake inhibition. In this mouse model, clear differences in the potencies of the compounds have been observed. Thus, S-citalopram was approximately twice as potent as RS-citalopram, which was approximately 10 times as potent as R-citalopram, giving a potency ratio of 20 (Sánchez, 2003). With such a difference in potency, little or no interaction between R- and S-citalopram should be expected. However, when S-citalopram (0.5, 1.0 and 2.0 mg kg−1) was coapplied with R-citalopram in ratios of 1 : 2 and 1 : 4 in this study, there was – except for 2 mg S-citalopram + 3.9 mg R-citalopram – a statistically significant reduction in the effect of S-citalopram (Figure 1). In contrast, when R-citalopram was coapplied with fluoxetine a significant and dose-dependent increase of the fluoxetine-induced behaviour was observed, suggesting that the antagonistic behaviour of R-citalopram does not extend to fluoxetine, but appears to be specific for S-citalopram (Figure 2).
Figure 1.

Effect of S-citalopram, 0.50, 1.0 and 2.0 mg kg−1, alone and in combination with R-citalopram in the mouse 5-HTP potentiation test. Pretreatment time was 30 min. Results are expressed as the percent of maximum score and shown as mean±s.e.m. For further details refer to Methods. *P<0.05, **P<0.01 and ***P<0.001 compared to corresponding S-citalopram-treated group (one-way analysis of variance followed by post hoc comparisons of means by t-test). Data for R-citalopram administered alone are shown in Figure 2.
Figure 2.

Effect of R-citalopram alone, fluoxetine, 4.5, 9.0 or 18 mg kg−1, alone and in combination with R-citalopram in the mouse 5-HTP potentiation test. Pretreatment time was 30 min. Results are expressed as the percent of maximum score and shown as mean±s.e.m. For further details refer to Methods. *P<0.05, **P<0.01 and ***P<0.001 compared to corresponding fluoxetine-treated group (one-way analysis of variance followed by post hoc comparisons of means by t-test).
In order to test the hypothesis that the functional effect of R-citalopram was a consequence of binding to the SERT, in vivo binding potencies of R- and S-citalopram were determined from [3H]MADAM binding in mouse brain. [3H]MADAM, which is a highly selective ligand for SERT (Chalon et al., 2003), has previously been used to label the SERT in vivo, so the potencies obtained for the citalopram enantiomers are a reflection of their binding affinity for SERT.
The ED50 values of S- and R-citalopram for displacing [3H]MADAM from mouse SERT were 0.070 and 4.7 mg kg−1, respectively (data not shown), yielding a potency ratio of 67. Assuming that the binding and binding kinetics of R- and S-citalopram are independent of the presence of the other enantiomer, and that the interaction of each enantiomer with SERT is competitive, it is possible to calculate the occupancy of SERT. At the citalopram doses used in the behavioural part of the present study, the corresponding occupancies of SERT for S-citalopram were calculated to be 88% (0.5 mg kg−1), 93% (1.0 mg kg−1) and 97% (2.0 mg kg−1), with corresponding values for R-citalopram of 18% (1.0 mg kg−1), 30% (2.0 mg kg−1), 45% (3.9 mg kg−1) and 63% (7.8 mg kg−1). Similarly, an ED50 value of 2.0 mg kg−1 was found for fluoxetine and at the doses used in the 5-HTP studies (4.5, 9.0 and 18 mg kg−1); the corresponding occupancies were calculated to be 69, 82 and 90%, respectively (Larsen et al., 2004).
In order to characterise the interaction of R- and S-citalopram at the transporter level, the hSERT was expressed in Xenopus oocytes and the two-electrode voltage-clamp technique used to record SERT-mediated currents and to characterise the pharmacological profile of each enantiomer.
An initial characterisation of hSERT-expressing Xenopus oocytes showed that 5-HT induces an inwardly directed and concentration-dependent current with an EC50 value of 0.7 μM and a slope factor of 1.2 (data not shown), which is similar to that previously described by Mager et al. (1994). Maximal currents were typically 30–40 nA (holding potential −70 mV). In order to obtain a sufficiently high current, so that fractional currents could be quantified, 10 μM 5-HT was used in all experiments.
Concentration-inhibition studies for R- and S-citalopram were performed using a paradigm in which the oocyte was pretreated with the SSRI for 5 min before exposure to 5-HT. Inhibition of the 5-HT-induced current by increasing concentrations of R- or S-citalopram showed that S-citalopram (Ki: 5 nM) is approximately 60 times more potent than R-citalopram (Ki: 330 nM) (data not shown), which is also in good agreement with the potency ratio obtained from the in vivo [3H]MADAM binding experiment.
The interaction of the two enantiomers with the hSERT was subsequently characterised using a physiologically relevant paradigm, in which a fixed concentration of R- and/or S-citalopram was continuously applied, and 5-HT was coapplied for approximately 2–5 min at 6 min intervals. The rationale for using ratios of <1 : 1 (S-citalopram : R-citalopram) is based on a faster metabolic breakdown of S-citalopram in vivo, leading to steady-state brain and serum levels of up to 1 : 3 (S : R) in rats (Kugelberg et al., 2001). The results from these oocyte experiments (illustrated in Figure 3) revealed that 0.15 μM S-citalopram decreased the 5-HT-elicited current to approximately 30%, whereas 0.45 and 5 μM R-citalopram inhibited the currents to approximately 80 and 54%, respectively (numeric values presented in Table 1 ).
Figure 3.

Inhibition of 5-HT currents by R- and S-citalopram in Xenopus oocytes expressing hSERT. Following a stable response to 10 μM 5-HT, R- or S-citalopram and combinations thereof were constantly applied and the oocyte exposed three times to 5-HT at 6 min intervals. The stable response in the presence of R- and S-citalopram was measured and the percent response, relative to the response to 10 μM 5-HT alone, was calculated. (a) Effect of 0.45 μM R- and 0.15 μM S-citalopram plus the combination. (b) Effect of 5 μM R- and 0.15 μM S-citalopram plus the combination. Values are mean values±s.e.m. obtained from three to seven oocytes from at least two different batches of oocytes. ns=no statistically significant difference, **P<0.01, ***P<0.001 compared to oocytes treated with 0.15 μM S-citalopram alone (one-way analysis of variance followed by post hoc comparisons of means by t-test).
Table 1.
Percent response to 10 μM 5-HT in the continuous presence of S- and/or R-citalopram
| 0 μM S | 0.15 μM S | |
| 0 μM R | 100 | 30 |
| 0.45 μM R | 80 | 42 |
| 5 μM R | 54 | 29 |
For experimental details, see legend to Figure 3
Using Equation (4) describing competitive interaction between an agonist and a competitive antagonist (Gaddum, 1937), assuming constant concentrations of 5-HT throughout the experiment and purely competitive interactions, an additional estimate of the potencies of R- and S-citalopram was calculated. With continuous 5-HT application (Figure 3), Ki values for S- and R-citalopram were estimated to be 5 and 430 nM, respectively, which is consistent with results from the concentration-inhibition studies where oocytes were preincubated with inhibitor. In order to determine if the effect of R- and S-citalopram when present simultaneously could be modelled as the sum of two antagonists acting at the same site, the expected current was calculated using Equation (5).
As shown in Figure 3 and Table 1, coapplication of different concentrations of R-citalopram with 0.15 μM S-citalopram resulted in less inhibition of the 5-HT-elicited currents than predicted (R0.45 μM: measured 42±4.6%, calculated 30%, P<0.05; R5 μM: measured 29±2.2%, calculated 23%, P<0.05). A possible explanation for the nonadditive behaviour of R- and S-citalopram could be that they either bind to different populations or conformational states of hSERT, or induce a shift between different conformational states. In such a model, S-citalopram, might bind irreversibly to hSERT or induce a long-lasting inhibited state of the transporter. In contrast, R-citalopram, although binding to SERT, may not induce the same conformational changes as S-citalopram, and therefore may keep SERT in a conducting/transport-competent state. In addition, the association and dissociation kinetics of the drug-transporter interaction may be different for distinct populations or conformational states of the carrier.
However, any mathematical model describing molecular interactions is based on simplifying assumptions and assumes ideal behaviour of its components. As the observed discrepancies between measured and calculated values were significant, but not dramatic, a more robust experimental paradigm to demonstrate the nonadditive effects of R- and S-citalopram was designed. The protocol was modified so that differences in the off-set of action would be measured by changes in 5-HT-elicited currents. Following a stable current amplitude to 10 μM 5-HT, the oocytes were pretreated for 5 min with R-, S-citalopram or fluoxetine alone, or combinations thereof. Immediately thereafter, the SSRI(s) were coapplied with 10 μM 5-HT until a steady current was observed (usually after 4–10 min). Subsequently, 5-HT was reapplied three times allowing a 6 min washout period between each application. This should enable the detection of differences in the off-set of action of R- and S-citalopram. As shown in Figure 4a, 0.45 μM R-citalopram, when coapplied with 0.15 μM S-citalopram, was able to increase significantly the recovery of the 5-HT-elicited current (P<0.05), an effect that was even greater when the R-citalopram concentration was raised to 5 μM (Figure 4b, P<0.001). Under these conditions, the effect of R-citalopram alone was very small. Even though a ratio of 1 : 33 (S : R) is not physiologically relevant, the rationale of using 5 μM R-citalopram was to demonstrate that the increased recovery observed with the concomitant presence of R-citalopram was due to a specific interaction of the two enantiomers with the hSERT.
Figure 4.

Recovery of 5-HT currents inhibited by S-citalopram and fluoxetine alone or with R-citalopram. Following a stable response to 10 μM 5-HT (application 1), the oocytes were pretreated for 5 min with R- or S-citalopram alone, or combinations thereof. Immediately thereafter, the R- and/or S-citalopram was coapplied with 10 μM 5-HT (application 2) until a steady current was obtained. Subsequently, 5-HT was reapplied three times allowing a 6 min washout period between each application (applications 3–5). The recovery response to 5-HT, relative to the initial response was calculated. (a) Effect of 0.45 μM R- and 0.15 μM S-citalopram alone plus their combination. (b) Effect of 5 μM R- and 0.15 μM S-citalopram plus their combination. (c) Effect of 5 μM R-citalopram and 1 μM fluoxetine plus their combination. Values are mean values±s.e.m. obtained from three to seven oocytes from at least two different batches of oocytes. *P<0.05, **P<0.01, ***P<0.001 (one-way analysis of variance followed by post hoc comparisons of means by t-test).
The corresponding control experiments with fluoxetine show basically the opposite result: inhibition of 5-HT-elicited currents by R-citalopram and fluoxetine appeared to be additive (Figure 4c).
In a similar experimental paradigm, the determination of on- and off-sets of action for each of the citalopram enantiomers was carried out in the presence of 10 μM 5-HT. These studies (see representative traces in Figure 5) showed that the observed on-set of action for 10 μM R-citalopram is approximately three times faster than for 1 μM S-citalopram (τR: 8.5±1.3 vs τS: 22±1.6 s) (n=4–5). The slower on-set of action of S-citalopram may be due to its 10-fold lower concentration compared to R-citalopram, and may therefore merely reflect differences in the diffusion rates. However, as stated above, Ki values for the inhibition of 5-HT-elicited currents are 5 nM (for S-citalopram) and 330 nM (for R-citalopram), meaning that the concentrations used can be at least considered ‘saturating'. Furthermore, the off-set of action was significantly faster for R-citalopram than that for S-citalopram (τR:24±3.9 s vs τS: 820±300 s)(n=5). The corresponding values for fluoxetine are τon=69±19 s (n=4) and τoff=133±38 s (n=5) (data not shown). Taken together, these data suggest that S-citalopram binding to SERT results in a long-lasting inhibition of the carrier. However, in the presence of both enantiomers, increasing concentrations of R-citalopram or 5-HT attenuate the S-citalopram-induced inhibition of SERT.
Figure 5.

Representative traces from oocytes expressing hSERT illustrating on- and off-set of R- and S-citalopram action in the presence of 10 μM 5-HT. (a) Upper panel: association of 1 μM S-citalopram in the presence of 5-HT; lower panel: association of 10 μM R-citalopram in the presence of 10 μM 5-HT. (b) Upper panel: dissociation of 1 μM S-citalopram in the presence of 10 μM 5-HT. Lower panel: dissociation of 10 μM R-citalopram in the presence of 10 μM 5-HT. (c) Fitted parameters, as described in the text, illustrating the differences in association and dissociation of R- and S-citalopram from hSERT in the presence of 10 μM 5-HT. The outward-going current observed when cells are treated with R- and S-citalopram alone can be ascribed to the inhibition of the leakage current mediated by the hSERT, as described in the introduction. However, its magnitude varies significantly between oocytes.
Discussion
Why should the removal of the supposedly inactive R-citalopram from RS-citalopram improve its clinical efficacy? Published animal studies and clinical studies have indicated an earlier symptom relief and an improved efficacy for the enantiomerically pure S-citalopram, and thus an important pharmacological difference between RS- and S-citalopram (Reines, 2002; Lepola et al., 2003; Sánchez, 2003). Furthermore, in vivo microdialysis studies of 5-HT levels in the frontal cortex of freely moving rats have shown that R-citalopram counteracts the action of S-citalopram, although this study did not address the underlying mechanism (Mørk et al., 2003).
In the present study, we have used the combined approach of behavioural studies and electrophysiological recordings from Xenopus oocytes expressing hSERT. Although behavioural studies can be regarded in many ways as black box models, this model has demonstrated a direct correlation between typical serotonergic symptoms and an increased input via dosing with 5-HT precursor and an increased uptake inhibition by SSRIs. The model is therefore a relatively simple method to characterise functional consequences of changes in levels of 5-HT in vivo.
In vivo binding studies clearly demonstrate that although the affinity of R-citalopram for the [3H] MADAM labelled SERT is low, at the concentrations used in the behavioural experiment, a significant fraction of the transporter proteins will be occupied by R-citalopram. Our findings are therefore most likely the result of an in vivo interaction of R-citalopram with S-citalopram at the SERT.
The finding that S-citalopram and fluoxetine increase the behavioural score in a dose-dependent manner demonstrates that the model is working within the dynamic ranges used. Surprisingly, the coadministration of comparable, increasing amounts of R-citalopram to either S-citalopram or fluoxetine results in different effects on the 5-HTP-mediated behaviour. While R-citalopram antagonises the behavioural potentiation of S-citalopram, it appears to act additively (maybe even synergistically) with fluoxetine. The reason for the apparent synergism is not obvious, although it may be due to the discrete scoring of the behavioural read-out. The observation that 0.5 mg kg−1 S-citalopram is apparently antagonised to a much larger extent than higher doses of S-citalopram is most likely also a reflection of the same phenomenon. In general, addition of R- to S-citalopram reduced the behavioural score by 1 and 2 points for S : R ratios of 1 : 2 and 1 : 4, respectively. This type of functional antagonism by enantiomers has previously been demonstrated for the AMPA receptor agonist APPA (Ebert et al., 1994) and for G-protein-coupled receptors (Bräuner-Osborne et al., 1996). In these cases, the functional antagonism was the consequence of one enantiomer being an agonist and the other an antagonist, leading to an apparent partial agonism at the receptor level. However, with R- and S-citalopram, the interaction takes place between two antagonists with different potencies at the same transporter. The mechanism can be addressed by using a kinetic analysis. Since the behavioural data are by nature discrete values, this could only be done with the oocyte data.
In Xenopus oocytes expressing hSERT, S-citalopram proved to be more than 60 times potent than R-citalopram under equilibrium conditions. Therefore, a detectable interaction between the two compounds would not be predicted. However, as illustrated in Figures 3 and 4, the two enantiomers interact, so that the inhibitory effect of coapplication is less than that predicted from the sum of each enantiomer alone.
Based on the assumption that the two compounds are interacting competitively with hSERT, it is possible to predict the 5-HT-induced current in the presence of both enantiomers. The calculated current was significantly less than that observed, indicating an antagonistic interaction between R- and S-citalopram at the hSERT. In addition, kinetic analysis of the recovery of 5-HT-induced currents supports the hypothesis that R-citalopram antagonises the inhibition of SERT by S-citalopram, but not by fluoxetine (Figure 4). We propose that this functional antagonism is a reflection of at least two processes. First, the association/dissociation kinetic analysis indicates that both R- and S-citalopram have a relatively fast on-set of action, resulting in a comparable fraction of SERT occupied by R- or S-citalopram. The fast off-set of R-citalopram(s) is in contrast to the long-lasting action of S-citalopram (min) (Figure 5). Thus, hSERT activity will recover faster in the presence of R-citalopram, as a consequence of the fast dissociation of R-citalopram. Second, the long-lasting action of S-citalopram may not only be due to its high-affinity binding to hSERT, but ligand binding may also induce conformational changes that will lead to the long-lasting inhibition of SERT. Therefore, it seems difficult to ascribe the antagonistic interaction of R- and S-citalopram at SERT solely to thermodynamic processes, like the competition for a common binding site. Rather, the complex recovery kinetics of the 5-HT-elicited currents (Figure 4) following R- and S-citalopram or fluoxetine application prompt us to describe this phenomenon as a kinetic interaction of R- and S-citalopram with SERT. Furthermore, the long-lasting inhibition of SERT, as seen following S-citalopram binding may result in phosphorylation, homo/hetero-oligomerisation events or ligand-induced internalisation. In fact, several studies have shown that substrates maintain or even upregulate SERT activity (or the activity of related transporters) in transfected cells as well as in primary neuronal cultures, and that this effect can be blocked by nontransported inhibitors (Quick, 2002; Whitworth et al., 2002; Ramamoorthy & Blakely, 2003). In this context, it is also intriguing to speculate whether R-citalopram binding to SERT may stabilise or induce different conformational states than its S-enantiomer. In addition, it is known from in vivo pharmacokinetic studies that the steady-state serum levels of R-citalopram and its metabolites, after chronic RS-citalopram treatment, are higher than levels of S-citalopram, indicating a faster metabolism for the S-enantiomers (Kugelberg et al., 2001), thereby further increasing the ‘protective' occupancy of newly synthesised SERT by R-citalopram. According to our hypothesis, R-citalopram counteracts the S-citalopram-mediated 5-HT reuptake inhibition, resulting in lower maximal 5-HT levels and therefore a lower activation of 5-HT receptors in vivo. A persistent increase in 5-HT levels may be essential for SSRI efficacy. It could be argued that peak 5-HT concentrations may be of equal importance. However, the present data do not favour the latter interpretation, since, as illustrated in Figure 4, the combined initial effect of R- and S-citalopram is at least as great as that produced by S-citalopram alone.
Recovery of the 5-HT-elicited current as shown in Figure 4 is not monophasic. Even after the application of R-citalopram alone, the recovery is not complete, suggesting that SERT inhibition by nontransported inhibitors, including R-citalopram, has a longer-lasting component.
There are difficulties in extrapolating the antagonistic effects of R-citalopram, which were observed on a second-to-minute time scale in the in vitro and in vivo assays, to the improved clinical efficacy of S-citalopram observed over days or weeks. However, it is intriguing that R-citalopram antagonism of the effect of S-citalopram is reproduced in very different experimental paradigms, such as voltage-clamp recordings in oocytes expressing hSERT, in vivo microdialysis in freely moving rats (Mørk et al., 2003) and 5-HTP potentiation in a behavioural mouse model, but does not extend to fluoxetine.
In conclusion, the data indicate that R- and S-citalopram interact with hSERT in an antagonistic manner, resulting in a reduction in the efficacy of S-citalopram. This antagonistic behaviour can, in part, be explained by the kinetic interaction of the two enantiomers with the SERT. In addition, we suggest that S-citalopram induces a long-lasting inhibited state of the transporter, which is attenuated by R-citalopram binding to SERT. These preclinical data are, therefore, in agreement with clinical results, which indicates an increased efficacy of S-citalopram compared with an equivalent dose of RS-citalopram. Although the concentrations used in this study are within the clinically relevant concentration range, it remains to be established if this phenomenon can adequately explain the clinical results.
Acknowledgments
Jan Egebjerg, Peter Høngaard Andersen, David J. Simpson and Marianne Faber are gratefully acknowledged for their suggestions and discussions during the course of the study. Nikki J. Damsgaard, Dorit Skov and Christian Spang Pedersen for their technical assistance.
Abbreviations
- (h)SERT
(human) serotonin transporter
- 5-HTP
5-hydroxytryptophan
- MADAM
N,N-dimethyl-2-(2-amino-4-methylphenyl thio)benzylamine
- SSRI
selective serotonin reuptake inhibitor
- 5-HT
serotonin
References
- AMARA S.G., KUHAR M.J. Neurotransmitter transporters: recent progress. Annu. Rev. Neurosci. 1993;16:73–93. doi: 10.1146/annurev.ne.16.030193.000445. [DOI] [PubMed] [Google Scholar]
- ANDERSEN P.H., JANSEN J.A., NIELSEN E.B. [3H]GBR 12935 binding in vivo in mouse brain: labelling of a piperazine acceptor site. Eur. J. Pharmacol. 1987;144:1–6. doi: 10.1016/0014-2999(87)90002-1. [DOI] [PubMed] [Google Scholar]
- ARTIGAS F., PEREZ V., ALVAREZ E. Pindolol induces a rapid improvement of depressed patients treated with serotonin reuptake inhibitors. Arch. Gen. Psychiatry. 1994;51:248–251. doi: 10.1001/archpsyc.1994.03950030084009. [DOI] [PubMed] [Google Scholar]
- ARTIGAS F., ROMERO L., DE MONTIGNY C., BLIER P. Acceleration of the effect of selected antidepressant drugs in major depression by 5-HT1A antagonists. Trends Neurosci. 1996;19:378–383. doi: 10.1016/S0166-2236(96)10037-0. [DOI] [PubMed] [Google Scholar]
- BLIER P., BERGERON R., DE MONTIGNY C. Selective activation of postsynaptic 5-HT1A receptors induces rapid antidepressant response. Neuropsychopharmacology. 1997;16:333–338. doi: 10.1016/S0893-133X(96)00242-4. [DOI] [PubMed] [Google Scholar]
- BLIER P., DE MONTIGNY C. Current advances and trends in the treatment of depression. Trends Pharmacol. Sci. 1994;15:220–226. doi: 10.1016/0165-6147(94)90315-8. [DOI] [PubMed] [Google Scholar]
- BRÄUNER-OSBORNE H., EBERT B., BRANN M.R., FALCH E., KROGSGAARD-LARSEN P. Functional partial agonism at cloned human muscarinic acetylcholine receptors. Eur. J. Pharmacol. 1996;313:145–150. doi: 10.1016/0014-2999(96)00501-8. [DOI] [PubMed] [Google Scholar]
- CHALON S., TARKIAINEN J., GARREAU L., HALL H., EMOND P., VERCOUILLIE J., FARDE L., DASSE P., VARNAS K., BESNARD J.C., HALLDIN C., GUILLOTEAU D. Pharmacological characterization of N,N-dimethyl-2-(2-amino-4-methylphenyl thio)benzylamine as a ligand of the serotonin transporter with high affinity and selectivity. J. Pharmacol. Exp. Ther. 2003;304:81–87. doi: 10.1124/jpet.102.042226. [DOI] [PubMed] [Google Scholar]
- EBERT B., MADSEN U., LUND T.M., LENZ S.M., KROGSGAARD-LARSEN P. Molecular pharmacology of the AMPA agonist, (S)-2-amino-3-(3-hydroxy-5- phenyl-4-isoxazolyl)propionic acid [(S)-APPA] and the AMPA antagonist, (R)-APPA. Neurochem. Int. 1994;24:507–515. doi: 10.1016/0197-0186(94)90001-9. [DOI] [PubMed] [Google Scholar]
- GADDUM J.H. The quantitative effects of antagonistic drugs. J. Physiol. (Lond.) 1937;89:7P–9P. [Google Scholar]
- HYTTEL J., BØGESØ K.P., PERREGAARD J., SÁNCHEZ C. The pharmacological effect of citalopram residues in the (S)-(+)- enantiomer. J. Neural Transm. Gen. Sect. 1992;88:157–160. doi: 10.1007/BF01244820. [DOI] [PubMed] [Google Scholar]
- KUGELBERG F.C., APELQVIST G., CARLSSON B., AHLNER J., BENGTSSON F. In vivo steady-state pharmacokinetic outcome following clinical and toxic doses of racemic citalopram to rats. Br. J. Pharmacol. 2001;132:1683–1690. doi: 10.1038/sj.bjp.0704015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LARSEN A.K., BRENNUM L.T., EGEBJERG J., SÁNCHEZ C., HALLDIN C., ANDERSEN P.H.Selectivity of 3H-MADAM binding to 5-hydroxytryptamine transporters in vitro and in vivo in mice; correlation with behavioural data Br. J. Pharmacol. 2004(in press) [DOI] [PMC free article] [PubMed]
- LEPOLA U.M., LOFT H., REINES E.H. Escitalopram (10–20 mg/day) is effective and well tolerated in a placebo-controlled study in depression in primary care. Int. Clin. Psychopharmacol. 2003;18:211–217. doi: 10.1097/00004850-200307000-00003. [DOI] [PubMed] [Google Scholar]
- LIMAN E.R., TYTGAT J., HESS P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- MAGER S., MIN C., HENRY D.J., CHAVKIN C., HOFFMAN B.J., DAVIDSON N., LESTER H.A. Conducting states of a mammalian serotonin transporter. Neuron. 1994;12:845–859. doi: 10.1016/0896-6273(94)90337-9. [DOI] [PubMed] [Google Scholar]
- MONTGOMERY S.A., LOFT H., SÁNCHEZ C., REINES E.H., PAPP M. Escitalopram (S-enantiomer of citalopram): clinical efficacy and onset of action predicted from a rat model. Pharmacol. Toxicol. 2001;88:282–286. doi: 10.1034/j.1600-0773.2001.d01-118.x. [DOI] [PubMed] [Google Scholar]
- MORTENSEN O.V., KRISTENSEN A.S., RUDNICK G., WIBORG O. Molecular cloning, expression and characterization of a bovine serotonin transporter. Brain Res. Mol. Brain Res. 1999;71:120–126. doi: 10.1016/s0169-328x(99)00178-3. [DOI] [PubMed] [Google Scholar]
- MØRK A., KREILGAARD M., SÁNCHEZ C. The R-enantiomer of citalopram counteracts escitalopram-induced increase in extracellular 5-HT in the frontal cortex of freely moving rats. Neuropharmacology. 2003;45:167–173. doi: 10.1016/s0028-3908(03)00138-2. [DOI] [PubMed] [Google Scholar]
- QUICK M.W. Substrates regulate gamma-aminobutyric acid transporters in a syntaxin 1A-dependent manner. Proc. Natl. Acad. Sci. U.S.A. 2002;99:5686–5691. doi: 10.1073/pnas.082712899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAMAMOORTHY S., BLAKELY R.D. Phosphorylation and sequestration of serotonin transporters differentially modulated by psychostimulants. Science. 2003;285:763–766. doi: 10.1126/science.285.5428.763. [DOI] [PubMed] [Google Scholar]
- REINES E.H. Escitalopram is efficacious and well tolerated in the treatment of depression in primary care. Eur. Neuropsychopharmcol. 2002;12 Suppl 3:S254. [Google Scholar]
- SÁNCHEZ C. R-citalopram attenuates anxiolytic effects of escitalopram in a rat ultrasonic vocalisation model. Eur. J. Pharmacol. 2003;464:155–158. doi: 10.1016/s0014-2999(03)01376-1. [DOI] [PubMed] [Google Scholar]
- SÁNCHEZ C., BERGQVIST P.B.F., BRENNUM L.T., GUPTA S., HOGG S., LARSEN A., WIBORG O. Escitalopram, the (S)-(+)-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with potent effects in animal models predictive antidepressant and anxiolytical activities. Psychopharmacology. 2003;167:353–362. doi: 10.1007/s00213-002-1364-z. [DOI] [PubMed] [Google Scholar]
- SMITH P.K., KROHN R.I., HERMANSON G.T., MALLIA A.K., GARTNER F.H., PROVENZANO M.D., FUJIMOTO E.K., GOEKE N.M., OLSON B.J., KLENK D.C. Measurement of protein using bicinchoninic acid. Anal. Biochem. 1985;150:76–85. doi: 10.1016/0003-2697(85)90442-7. [DOI] [PubMed] [Google Scholar]
- SONDERS M.S., AMARA S.G. Channels in transporters. Curr. Opin. Neurobiol. 1996;6:294–302. doi: 10.1016/s0959-4388(96)80111-5. [DOI] [PubMed] [Google Scholar]
- WHITWORTH T.L., HERNDON L.C., QUICK M.W. Psychostimulants differentially regulate serotonin transporter expression in thalamocortical neurons. J. Neurosci. 2002;22:RC192. doi: 10.1523/JNEUROSCI.22-01-j0003.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]