

Abstract
The topoisomerase II inhibitor amsacrine is used in the treatment of acute myelogenous leukemia. Although most anticancer drugs are believed not to cause acquired long QT syndrome (LQTS), concerns have been raised by reports of QT interval prolongation, ventricular fibrillation and death associated with amsacrine treatment. Since blockade of cardiac human ether-a-go-go-related gene (HERG) potassium currents is an important cause of acquired LQTS, we investigated the acute effects of amsacrine on cloned HERG channels to determine the electrophysiological basis for its proarrhythmic potential.
HERG channels were heterologously expressed in human HEK 293 cells and Xenopus laevis oocytes, and the respective potassium currents were recorded using patch-clamp and two-microelectrode voltage-clamp electrophysiology.
Amsacrine blocked HERG currents in HEK 293 cells and Xenopus oocytes in a concentration-dependent manner, with IC50 values of 209.4 nM and 2.0 μM, respectively.
HERG channels were primarily blocked in the open and inactivated states, and no additional voltage dependence was observed. Amsacrine caused a negative shift in the voltage dependence of both activation (−7.6 mV) and inactivation (−7.6 mV). HERG current block by amsacrine was not frequency dependent.
The S6 domain mutations Y652A and F656A attenuated (Y652A) or abolished (F656A, Y652A/F656A) HERG current blockade, indicating that amsacrine binding requires a common drug receptor within the pore-S6 region.
In conclusion, these data demonstrate that the anticancer drug amsacrine is an antagonist of cloned HERG potassium channels, providing a molecular mechanism for the previously reported QTc interval prolongation during clinical administration of amsacrine.
Keywords: Acute myeloid leukemia, amsacrine, arrhythmia, DNA topoisomerase II, electrophysiology, ion channels, K+ channel, long QT syndrome, sudden cardiac death, torsade de pointes
Introduction
Amsacrine (m-AMSA) is a synthetic aminoacridine that is used in the treatment of acute myelogenous leukemia (AML), for example, as part of the induction regimen (Kantarjian et al., 1996; Arnaout et al., 2000; Rowe, 2000). Its therapeutic effectiveness has been attributed to intercalation of the compound into DNA and to inhibition of DNA topoisomerase II (Jehn & Heinemann, 1991). However, concerns have been raised by reports on QT interval prolongation, ventricular arrhythmia and death associated with amsacrine application (van Hoff et al., 1980; McLaughlin et al., 1983; Schwartz et al., 1984; Shinar & Hasin, 1984; Griffin et al., 1985; Winton et al., 1985; Dhaliwal et al., 1986; Weiss et al., 1986; Seymour, 1993), suggesting an effect of amsacrine on cardiac repolarization.
In human cardiomyocytes, repolarization is mainly carried by the rapidly and slowly activating components of the delayed rectifier potassium current, IKr and IKs (Sanguinetti & Jurkiewicz, 1990). The human ether-a-go-go-related gene (HERG) (Sanguinetti et al., 1995) encodes the α-subunit underlying IKr, and mutations in HERG account for chromosome 7-linked inherited long QT syndrome (LQT-2) (Viskin, 1999). Patients diagnosed with LQT-2 may present prolonged QT intervals in the surface electrocardiogram and have an increased risk for ventricular ‘torsade de pointes' arrhythmias and sudden cardiac death. Pharmacological inhibition of HERG potassium channels is a property of class III antiarrhythmic drugs such as amiodarone (Kiehn et al., 1999), and of a multitude of non-antiarrhythmic compounds (for a review, see Redfern et al., 2003). Block of IKr causes lengthening of the cardiac action potential, which produces a beneficial class III antiarrhythmic effect. On the other hand, excessive action potential prolongation may lead to acquired long QT syndrome and life-threatening ‘torsade de pointes' arrhythmias (Napolitano et al., 1994).
The aim of the present study was to determine the electrophysiological basis of the QTc interval prolongation associated with amsacrine treatment. Acute effects of amsacrine on HERG channels expressed heterologously in Xenopus oocytes and human embryonic kidney (HEK) 293 cells were investigated, revealing detailed insights into the biophysical mechanism of amsacrine block of HERG currents.
Methods
Molecular biology
The HERG cDNA clone was generously donated by M.T. Keating. The preparation of the HERG Y652A and HERG F656A clones has been published previously (Scholz et al., 2003). To generate the HERG Y652A/F656A clone, the respective single mutations were introduced simultaneously into the HERG wild-type cDNA template by site-directed mutagenesis using the QuikChange kit (Stratagene, La Jolla, U.S.A.) and synthetic mutant oligonucleotide primers, as described earlier in detail (Scholz et al., 2003). Procedures for in vitro transcription and oocyte injection were performed as published previously (Kiehn et al., 1999). Briefly, HERG wild type (Warmke and Ganetzky, 1994), HERG Y652A, HERG F656A and HERG Y652A/F656A cRNAs were prepared with the mMESSAGE mMACHINE kit (Ambion, Austin, U.S.A.) using SP6 RNA polymerase after linearization with EcoRI (Roche Diagnostics, Mannheim, Germany). Stage V–VI defolliculated Xenopus oocytes were injected with 46 nl of cRNA per oocyte.
The cDNA encoding the HERG potassium channel cloned in pCDNA3 was stably transfected into the human embryonic kidney cell line HEK 293 as described previously (Thomas et al., 2001). Cells were cultured in Dulbecco's modified eagle medium/nutrient mixture F-12 (D-MEM/F12, Gibco BRL, Rockville, U.S.A.) supplemented with 10% fetal bovine serum (FBS, Gibco BRL), 100 U/ml−1 penicillin G sodium, 100 μg ml−1 streptomycin sulfate and 500 μg ml−1 geneticin (G418, Gibco BRL) in an atmosphere of 95% humidified air and 5% CO2 at 37°C.
The investigation conforms to the Guide for the Care and Use of Laboratory Animals published by the US National Institute of Health (NIH Publication No. 85-23, revised 1996). All experiments followed the European Community guidelines for the use of experimental animals.
Electrophysiology
Two-microelectrode voltage-clamp recordings from Xenopus laevis oocytes were carried out as published previously (Thomas et al., 2001). In brief, recordings were performed using a Warner OC-725A amplifier (Warner Instruments, Hamden, U.S.A.) and pClamp software (Axon Instruments, Foster City, U.S.A.) for data acquisition and analysis. Microelectrodes had tip resistances ranging from 1 to 5 MΩ. The recording chamber was continually perfused. HERG current recordings from HEK 293 cells were performed by use of the whole-cell patch clamp configuration (Hamill et al., 1981) as previously reported (Thomas et al., 2001). All experiments were carried out at room temperature (22–25°C), and no leak subtraction was done during the experiments.
Solutions and drug administration
Voltage-clamp measurements of Xenopus oocytes were performed in a solution containing (in mM): 5 KCl, 100 NaCl, 1.5. CaCl2, 2 MgCl2 and 10 HEPES (pH adjusted to 7.4 with NaOH). Current and voltage electrodes were filled with 3 M KCl solution. For whole-cell patch-clamp recordings from HEK 293 cells, electrodes were filled with the following solution (in mM): 130 K-aspartate, 5.0 MgCl2, 5 EGTA, 4 ATP, 10 HEPES (pH adjusted to 7.2 with KOH). The external solution for these experiments contained (in mM): 137 NaCl, 4.0 KCl, 1.0 MgCl2, 1.8 CaCl2, 10 HEPES, 10 glucose (pH adjusted to 7.4 with NaOH). Amsacrine (Sigma) was prepared as 10 mM stock solution in DMSO and stored at −20°C. On the day of experiments, aliquots of the stock solution were diluted to the desired concentrations with the bath solution. HERG current amplitudes (recorded from Xenopus oocytes using the protocol described in Figure 1a) were not significantly altered upon application of 1% DMSO (v v−1; maximum bath concentration) for 20 min (data not shown). In addition, DMSO did not affect HERG channel currents recorded from HEK 293 cells at concentrations up to 0.3% (maximum bath concentration in this study: 0.1% DMSO) (data not shown).
Figure 1.

Inhibition of HERG channels expressed in Xenopus oocytes by amsacrine. Representative current traces recorded from the same cell under control conditions and after superfusion with amsacrine (1, 10 and 100 μM, respectively) are displayed in panel a. (b) Concentration–response relationships for the effect of amsacrine on HERG peak tail currents (n=6–7 oocytes; data are expressed as mean±s.e.m.). The IC50 yielded 2.0 μM. (c) Time course of HERG tail current inhibition by 10 μM amsacrine (n=3). For the purpose of clear presentation, not all current measurements are displayed. (d) Mean relative tail current amplitudes after application of 10 μM amsacrine (15 min) for HERG wild type (n=6), HERG Y652A (n=8), HERG F656A (n=8) and HERG Y652A/F656A currents (n=8), respectively, illustrating that the inhibitory effects of amsacrine were attenuated or completely abolished by pore region mutations (see text for voltage protocols).
Data analysis and statistics
Concentration–response relationships for drug-induced block were fit with a Hill equation of the following form: Idrug/Icontrol=1/[1+(D/IC50)n], where I indicates current, D is the drug concentration, n is the Hill coefficient, and IC50 is the concentration necessary for 50% block. Activation curves were fit with a single-power Boltzmann distribution of the form 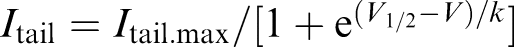 , where V is the test pulse potential V1/2 is the half-maximal activation potential, and k is the slope of the activation curve. Inactivation curves were fit to the following single-power Boltzmann equation:
, where V is the test pulse potential V1/2 is the half-maximal activation potential, and k is the slope of the activation curve. Inactivation curves were fit to the following single-power Boltzmann equation: 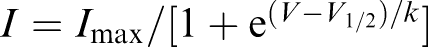 .
.
Data are expressed as mean ±s.e.m. We used paired and unpaired Student's t-tests (two-tailed tests) to compare the statistical significance of the results: P<0.05 was considered statistically significant. Multiple comparisons were performed using one-way ANOVA. If the hypothesis of equal means could be rejected at the 0.05 level, pairwise comparisons of groups were made and the probability values were adjusted for multiple comparisons using the Bonferroni correction.
Results
Amsacrine blocks HERG currents
Amsacrine blocked HERG potassium channels expressed in Xenopus laevis oocytes in a concentration-dependent manner, as displayed in Figure 1. Currents were elicited by a 2 s depolarizing step to +20 mV, followed by a repolarizing step to −40 mV for 1.6 s to produce large, slowly decaying outward tail currents which are a characteristic of HERG potassium channels (Sanguinetti et al., 1995). The holding potential was −80 mV in all experiments performed in this study, unless indicated otherwise. Pulses were applied at a frequency of 0.1 Hz during superfusion with the drug solution for 15 min. After the monitoring period, the degree of block was determined (Figure 1a). To study the concentration dependence of HERG current block by amsacrine, HERG peak tail currents in the presence of the drug were normalized to the respective control values and plotted as relative current amplitudes in Figure 1b (n=6–7 oocytes were investigated at each concentration). The half-maximal inhibition concentration (IC50) for block of tail currents yielded 2.0±0.1 μM with a Hill coefficient nH of 1.14±0.05. The time course of block is shown in Figure 1c (n=3). After a control period of 20 min, HERG channel block by 10 μM amsacrine reached steady-state conditions after 10–15 min. Upon washout (20 min), the blocking effects of amsacrine on HERG were partially reversible.
Incomplete attenuation of amsacrine block by Y652A and F656A mutations
It has been demonstrated that the aromatic residues Y652 and F656 located in the S6 domain are key determinants of drug binding to HERG channels (Mitcheson et al., 2000a). Thus, the effects of amsacrine on mutant HERG Y652A, HERG F656A and HERG Y652A/F656A channels were investigated to assess the significance of these amino-acid residues in amsacrine blockade of HERG currents. Voltage protocols were applied as described above (Figure 1a) to record currents under control conditions and after application of 10 μM amsacrine, reducing HERG wild-type currents to 20.0±2.5% (n=6; Figure 1b). As illustrated in Figure 1d, the inhibitory effect of amsacrine was significantly attenuated (HERG Y652A) or completely abolished (HERG F656A, HERG Y652A/F656A) by replacement of aromatic channel pore residues Y652 and/or F656 with alanine residues. Mean relative currents measured after amsacrine application yielded 60.5±4.6% (Y652A; n=8), 103.7±6.0% (F656A; n=8) and 105.3±4.0% (Y652A/F656A; n=8) of control currents, respectively (Figure 1d).
Effects of amsacrine on HERG current activation
The effect of amsacrine on HERG current voltage (I–V) relationship was investigated under isochronal recording conditions. Depolarizing pulses were applied for 2 s to voltages between –80 and +70 mV in 10 mV increments (0.2 Hz), and tail currents were recorded during a constant repolarizing step to –60 mV for 1.6 s. Families of current traces from one cell are shown for control conditions and after exposure to 3 μM amsacrine (15 min) in Figure 2a and b. The currents activated at potentials greater than −60 mV, reached a peak at 0 mV and then decreased at more positive potentials due to inactivation (Sanguinetti et al., 1995), giving the I–V relationship its typical bell-shaped appearance (Figure 2c and d). Figure 2e and f displays peak tail currents as a function of the preceding test pulse potential, resulting in activation curves. HERG currents at the end of the test pulse to 0 mV were reduced by 38.8±3.0%, and peak tail currents were blocked by 44.3±1.7% (n=8). The half-maximal activation voltage V1/2 was shifted by −7.6±0.8 from −12.8±0.6 mV under control conditions to −20.4±0.9 mV after amsacrine incubation (n=8). This difference was statistically significant.
Figure 2.

Effects of amsacrine on the voltage dependence of HERG activation. Control measurement (a) and the inhibitory effects of 10 μM amsacrine (15 min; b) are shown in one representative oocyte. (c and d) Resulting mean current amplitudes at the end of the test pulse as function of the preceding test pulse potential under control conditions and after incubation with prazosin (c, original current amplitudes; d, values normalized to peak step currents) (n=8). Panels e and f display activation curves, that is, peak tail current amplitudes as function of the preceding test pulse potential during the first step of the voltage protocol, recorded under isochronal conditions (e, original current amplitudes; f, values normalized to peak tail currents) (n=8). The mean half-maximal activation potential V1/2 was shifted by −7.6 mV (see text for voltage protocols).
Effects of amsacrine on HERG channel inactivation
The effects of amsacrine on HERG current inactivation were investigated using two different approaches. First, it was tested whether the time constant of inactivation was affected by the drug. Pulses were applied to 40 mV for 900 ms where channels are partially open but predominantly inactivated. A brief repolarization to –100 mV for 16 ms caused rapid recovery from inactivation without marked deactivation. During a second depolarizing pulse (150 ms) to different voltages ranging from –40 to 40 mV (increment 20 mV), large, rapidly inactivating currents were produced. Inactivating currents were recorded in the absence of the drug (Figure 3a) and after equilibration of block with 3 μM amsacrine (Figure 3b) by current monitoring for 15 min as described above. Since the inactivation time constant depends on the current amplitude, a set of control cells and matching oocytes after amsacrine application with similar mean current amplitudes at the end of the conditioning prepulse (0.49±0.08 versus 0.50±0.08 μA) were compared. Single exponential fits to the large inactivating currents yielded the time constants of inactivation at different voltages. In these experiments, no changes in the time constants for HERG channel inactivation were observed (Figure 3c; n=10 oocytes were analyzed in each series).
Figure 3.

Effects of amsacrine on HERG current inactivation. The inactivation time constant was assessed from the third step of the following voltage protocol: currents were activated by 900-ms pulses to 40 mV, followed by a brief repolarization to –100 mV (16 ms). Variable voltage steps ranging from –40 to 40 mV (150 ms; increment 20 mV) were consecutively applied to evoke inactivating currents (a, b). Current measurements recorded from different representative oocytes under control conditions (a) and after incubation with 3 μM amsacrine (b) are displayed. (c) depicts mean inactivation time constants obtained from single-exponential fits to the inactivating current traces (n=10). Panels d and e show representative single measurements of the steady-state inactivation at constant 20 mV after various potentials from –120 to 30 mV (increment 10 mV). Note that, for clarity, not all original current traces are displayed. The normalized mean inactivating current amplitudes at 20 mV are shown in panel f, giving steady-state inactivation curves. The mean half-maximal inactivation voltage was shifted by −7.6 mV towards more negative potentials (n=6) (see text for voltage protocols).
To measure steady-state inactivation relationships, channels were inactivated at a holding potential of 20 mV, before being recovered from inactivation at various potentials from –120 to 30 mV (increment 10 mV) for 20 ms. Finally, the resulting peak outward currents at constant 20 mV as a measure of steady-state inactivation were recorded. After having obtained the control measurements (Figure 3d), we applied 3 μM amsacrine to the oocytes. The holding potential was –80 mV during the current monitoring period of 15 min to avoid destruction of the cell, as it would occur when holding the cell at 20 mV. One typical recording in the presence of the drug is displayed in Figure 3e. The inactivating outward current amplitude measured at 20 mV was normalized and plotted against the test pulse potential, giving the steady-state inactivation curve (Figure 3f). Mean values for the half-maximal inactivation voltage yielded −62.9±1.8 mV for control and −70.5±1.5 mV for amsacrine measurements (n=6), displaying a relative shift of −7.6±0.8 mV. A study limitation derives from the fact that the current amplitudes at −120 mV were set as maximum values. However, since we applied the same analysis under control conditions and in the presence of amsacrine, we suggest that at least the relative shift of the half-maximal inactivation voltage may be interpreted as drug-induced effect on HERG inactivation.
The biophysical mechanism of HERG current inhibition by amsacrine
To investigate whether the channel is blocked in the closed or activated (i.e. open and/or inactivated) state, we activated currents using a protocol with a single depolarizing step to 0 mV for 7.5 s. After having obtained the control measurement, we allowed 10 μM of the drug to wash in for 15 min while holding the channels in the closed state at –80 mV. Then, measurements with amsacrine were performed (Figure 4a). The degree of inhibition (i.e. (1−current in the presence of amsacrine/control current) × 100) is displayed in Figure 4b. Analysis of the test pulse after amsacrine application revealed a time-dependent increase of block to 80% at 1250 ms in this representative cell (Figure 4b), which is consistent with block of activated HERG potassium channels. However, some degree of closed state block cannot be ruled out by this protocol, although it is rather unlikely since the initial portion of the current trace in the presence of the drug appears to be the same as the current trace recorded under control conditions. In this series of experiments, 10 μM amsacrine reduced HERG outward currents at the end of the 0 mV pulse by 77.5±2.3% (n=6).
Figure 4.

Block of activated HERG channels by amsacrine. After having recorded the control measurement, the oocyte was clamped at –80 mV for 15 min during superfusion with the drug solution (10 μM amsacrine). The control recording and the first pulse measured immediately after the incubation period are displayed (a). Panel b shows the degree of inhibition in %. Current inhibition increased time-dependently to 80% at 1250 ms in this representative experiment, indicating that mainly open and/or inactivated channels were blocked. (c) Inhibition of inactivated channels by 10 μM amsacrine. HERG channels were inactivated by a first voltage step to +80 mV, followed by channel opening at 0 mV. The corresponding relative block during the 0 mV step is displayed in panel (d) Maximum inhibition was achieved in the inactivated state during the first step, and no further time-dependent development of block occurred upon channel opening during the second voltage step (see text for voltage protocols).
To address the question whether HERG channels are also blocked by amsacrine in the inactivated state, a long test pulse to +80 mV (4 s) was applied to inactivate the channels, followed by a second voltage step (0 mV, 3.5 s) to open HERG channels (n=6). Typical current traces under control conditions and after application of 10 μM amsacrine for 15 min while holding the cell at −80 mV are displayed in Figure 4c. Figure 4d depicts the normalized relative block upon channel opening during the second voltage pulse (0 mV), illustrating that pronounced inhibition of HERG channels had already been obtained during the preceding inactivating +80 mV pulse, and no additional time-dependent block of open channels was observed during the 0 mV pulse. Currents at the end of the second voltage step (0 mV) were reduced by 78.0±2.6% (n=6). However, these results could also be interpreted as fast block of a small portion of open channels at 80 mV, followed by trapping of the amsacrine molecule at its binding site.
In summary, it is concluded from these experiments that amsacrine inhibits HERG channels predominantly in the open and inactivated state, with inhibition of closed channels being rather unlikely.
Voltage dependence of HERG channel block by amsacrine
To assess the voltage dependence of HERG channel block, we applied the following methodical approach. Since unblocking was slow, only one experiment at each potential could be carried out with one individual oocyte. Currents were elicited by 27 s depolarizing pulses ranging from –40 to 80 mV (Figure 5a), and peak inward tail currents were measured during a second step to –100 mV (400 ms). First, control currents were recorded. Then the oocyte was superfused with the drug solution (3 μM amsacrine) while holding the cell at –80 mV for 15 min where HERG channels are in the closed state. After this, measurements at the test pulse potential were performed. Relative inhibition of peak tail currents (plotted as a function of the preceding test pulse potential in Figure 5a) was not significantly different among the voltage steps applied (−40 mV: 47.8±5.2%; 0 mV: 39.7±4.5%; 40 mV: 44.3±3.9%; 80 mV: 36.9±3.2%; n=7−9 cells were studied at each potential). Thus, amsacrine reduced HERG currents in a voltage-independent manner.
Figure 5.

(a) Amsacrine block of HERG currents is not voltage-dependent. The fraction of blocked peak tail currents is plotted as function of various test pulse potentials. HERG channel block at −40, 0, 40, and 80 mV did not display significant differences (n=7–9 cells). (b) Lack of frequency-dependence of HERG channel block by amsacrine. The resulting mean relative tail current amplitudes (1 and 0.1 Hz stimulation rate) are plotted versus time (n=6 oocytes were studied at each rate). For the purpose of clear presentation, not all measurements are displayed (see text for voltage protocols).
Lack of frequency dependence of amsacrine block
The frequency dependence of block was investigated in the following set of experiments. HERG potassium channels were rapidly activated by a depolarizing step to 20 mV for 300 ms, followed by a repolarizing step to −40 mV (300 ms) to elicit outward tail currents. Pulses were applied at intervals of 1 or 10 s under control conditions and in the presence of 3 μM amsacrine, with each cell studied only at one stimulation rate. Six oocytes were used at each rate. The development of current reduction was plotted versus time (Figure 5b). The resulting level of steady-state block is a measure of the frequency dependence of block. There were no significant changes in the amount of steady-state block at both rates (35.1±5.6% (1 Hz) versus 35.3±2.7% (0.1 Hz), respectively). Thus, block was not frequency dependent.
Amsacrine blocks HERG channels in a human cell line
To demonstrate amsacrine block of HERG in human cells, we expressed HERG potassium channels heterologously in human embryonic kidney (HEK 293) cells. Channels were activated by a 2 s depolarization to +20 mV, and outward tail currents were recorded during a step to –50 mV for 2 s (Figure 6a). During the wash-in of the drug, we applied the protocol as described above (frequency 0.1 Hz), until a steady-state block was maintained for at least 30 s. HERG currents were blocked by amsacrine in a concentration-dependant manner. The IC50 value for amsacrine block of HERG tail currents was 209.4±6.0 nM with a Hill coefficient nH of 1.21±0.04 (Figure 6b; n=3 cells were studied at each concentration).
Figure 6.

Amsacrine blockade of HERG channels expressed in human HEK 293 cells. (a) Typical whole-cell patch-clamp recordings from one HEK cell under control conditions and after application of 300 nM amsacrine. Tail currents were blocked by 52.6% in this representative cell. (b) Concentration–response curve for inhibition of HERG peak tail currents in HEK 293 cells, yielding an IC50 value of 209.4 nM (n=3 cells; error bars denote s.e.m.) (see text for voltage protocols).
Discussion
Acute effects of amsacrine on HERG currents
HERG potassium channels are blocked by the anticancer drug amsacrine, a finding in line with QTc prolongation observed among AML patients treated with this drug. To our knowledge, amsacrine represents the first antineoplastic compound that inhibits HERG currents. Blockade of HERG channels expressed in Xenopus laevis oocytes displayed an IC50 value of 2.0 μM, while HERG currents in HEK 293 cells were inhibited with an IC50 value of 209.4 nM. This 9.6-fold difference corresponds well to previous reports comparing drug block of HERG currents in different expression systems. Owing to the specific properties of the Xenopus oocyte expression system (e.g. the vitelline membrane and the yolk) that reduce the actual concentration of drugs at the cell membrane, higher concentrations of drugs are necessary when applied to whole oocytes under in vitro conditions. In general, IC50 values for HERG channel block are approximately 10–20-fold higher when the drug is applied to the extracellular surface of Xenopus oocytes compared to whole-cell patch-clamp experiments using mammalian cells (Thomas et al., 2001, 2003a, 2003b). Therapeutic amsacrine plasma concentrations in humans have been reported to be 5–18 μM (Jurlina et al., 1985; Linssen et al., 1993), with approximately 97% of the drug being bound to plasma proteins (Paxton et al., 1986). Thus, it is reasonable to assume that HERG current inhibition by amsacrine is physiologically relevant.
It has been suggested that co-assembly of the regulatory β-subunit MiRP1 with HERG reconstitutes native IKr (Abbott et al., 1999). This hypothesis has been investigated by Weerapura et al. (2002) in detail, revealing that co-expression of HERG with wild-type MiRP1 (in contrast to long QT syndrome-linked mutant MiRP1; Abbott et al., 1999) does not alter its sensitivity to HERG-blocking drugs. Experiments using MiRP1 were not performed in this study since it is still controversial whether it is part of native IKr channels.
The biophysical mechanism of HERG current reduction by amsacrine
In the present study, the heterologous Xenopus oocyte expression system was used to determine the biophysical mechanism of HERG current inhibition by amsacrine in detail. One important finding of this study is that amsacrine blocks HERG channels predominantly in the open and inactivated state, as demonstrated using specially designed voltage protocols to discriminate between the different states. But still, the voltage protocols do not completely exclude partial contamination by channel states not primarily assessed by the respective protocol. In particular, recovery from inactivation and deactivation may contribute to the currents measured in Figure 3. Block at negative membrane potentials (i.e. −40 mV; Figure 5a) supports the hypothesis that amsacrine binding to closed channels might occur as well. Amsacrine application caused a −7.6 mV shift in the HERG activation curve and a −7.6 mV shift in the half-maximal inactivation voltage, which may cause a net increase of current if other biophysical parameters remained unchanged. However, due to pronounced pharmacological HERG channel block, current increase could not be detected in our study. Unblocking upon repolarization, which allows HERG channels to become available for opening, occurred rather slowly, and a complete washout could not be achieved. The lack of frequency dependence can be interpreted as the result of slow unblocking kinetics, possibly due to a trapping mechanism of the drug at its binding site (Mitcheson et al., 2000b). In summary, the biophysical mechanism of HERG current block by amsacrine shares large similarities with mechanisms observed among other drugs known to induce acquired long QT syndrome through inhibition of HERG channels (for summary and comparison, see Table 1).
Table 1.
Comparison of the biophysical parameters of HERG channel block by drugs causing acquired long QT syndrome (XO, Xenopus oocytes; MC, mammalian cells; n.i.; not investigated)
| Drug (reference) | IC50 | Blocked channel states | Voltage dependence | Frequency-dependence | Attenuation of block by mutation of aromatic pore residues |
|---|---|---|---|---|---|
| Amsacrine (this study) | 2.0 μM (XO), 209 nM (MC) | Open, inactivated | No | No | Yes (F656A; partial attenuation by Y652A) |
| Budipine (Scholz et al., 2003) | 10.2 μM (XO) | Open, inactivated | No | No | yes (F656A; partial attenuation by Y652A) |
| Chlorpromazine (Thomas et al., 2003a) | 21.6 μM (XO) | Closed, open, inactivated | Yes | reverse | n.i. |
| Fluvoxamine (Milnes et al., 2003) | 3.8 μM (MC) | (Closed), open, inactivated | Yes | n.i. | Yes (partial attenuation by Y652A and F656A) |
| Imipramine (Teschemacher et al., 1999) | 3.4 μM (MC) | Closed, open | (Yes) | n.i. | n.i. |
| Tamoxifen (Thomas et al., 2003b) | 45.3 μM (XO) | Open, (inactivated) | Reverse | No | n.i. |
The structural requirements for the drug-binding site in HERG as a basis for the unusual susceptibility of this potassium channel to block by structurally diverse drugs have recently been studied in detail. It has been demonstrated that the aromatic rings of Y652 and particularly F656 located in the S6 domain are key determinants of drug binding (Mitcheson et al., 2000a), since mutation of these residues to alanine dramatically reduced the potency of most drugs tested to date (Mitcheson, 2003). The lack of HERG F656A and HERG Y652A/F656A current inhibition clearly shows that amsacrine predominantly binds to a drug receptor within the pore-S6 region. This example supports the observation that the aromatic pore residue Y652 is less important for drug binding compared to F656, since mutation of Y652 produces only a partial attenuation of block by several drugs tested to date (including amsacrine; Table 1), compared with F656A which attenuates drug block 100-fold (with the exception of fluvoxamine, Milnes et al., 2003).
Clinical significance of HERG channel inhibition by amsacrine
Amsacrine has been shown to induce pronounced QT interval prolongation, ventricular tachycardia, ventricular fibrillation and death (van Hoff et al., 1980; McLaughlin et al., 1983; Schwartz et al., 1984; Shinar & Hasin, 1984; Griffin et al., 1985; Winton et al., 1985; Dhaliwal et al., 1986; Weiss et al., 1986; Seymour, 1993). Thus, HERG current block by amsacrine is of particular importance, since the induction regimen in AML and treatment after relapses usually include an anthracycline compound (including amsacrine) (Kantarjian et al., 1996; Arnaout et al., 2000; Rowe, 2000). Specific HERG channel inhibition by amsacrine may be of high clinical significance, since to date anticancer drugs have been thought to exert rather unspecific cardiotoxic and arrhythmogenic action. If during chemotherapy amsacrine is administered in combination with other compounds that inhibit HERG channels (e.g. antiemetic drugs such as odansetron; Kuryshev et al., 2000), the knowledge that amsacrine blocks HERG channels may help preventing fatal arrhythmias due to even more pronounced HERG block in these cases. Whether other antineoplastic drugs (in particular anthracyclines) inhibit HERG currents as well will require further studies.
Conclusions
Our results demonstrate that amsacrine is an antagonist of cloned HERG potassium channels, providing a molecular explanation for the proarrhythmic potential of this drug. Further studies need to be conducted to determine additional cardiac electrophysiological properties of amsacrine.
Acknowledgments
The excellent technical assistance of S. Lueck and R. Bloehs is gratefully acknowledged. This work was supported in part by grants from the Deutsche Forschungsgemeinschaft (project KI 663/1-1 to J.K.; project KA 1714/1-1 to C.A.K.). D.T. was supported by the University of Heidelberg (Young Investigator Award and Hans Dengler Research Scholarship), and by the Foundation Cardiology 2000 (Forssmann Scholarship).
Abbreviations
- AML
acute myelogenous leukemia
- HEK
human embryonic kidney
- HERG
human ether-a-go-go-related gene
- IK
delayed rectifier potassium current
- IKr
rapidly activating component of the delayed rectifier potassium current
- IKs
slowly activating component of the delayed rectifier potassium current
References
- ABBOTT G.W., SESTI F., SPLAWSKI I., BUCK M.E., LEHMANN M.H., TIMOTHY K.W., KEATING M.T., GOLDSTEIN S.A. MiRP1 forms IKr potassium channels with HERG and is associated with cardiac arrhythmia. Cell. 1999;97:175–187. doi: 10.1016/s0092-8674(00)80728-x. [DOI] [PubMed] [Google Scholar]
- ARNAOUT M.K., RADOMSKI K.M., SRIVASTAVA D.K., TONG X., BELT J.R., RAIMONDI S.C., BEHM F.G., SANTANA V.M., CROM W.R., MIRRO J., JR, RIBEIRO R.C. Treatment of childhood acute myelogenous leukaemia with an intensive regimen (AML-87) that individualizes etoposide and cytarabine dosages: short- and long-term effects. Leukemia. 2000;14:1736–1742. doi: 10.1038/sj.leu.2401906. [DOI] [PubMed] [Google Scholar]
- DHALIWAL H.S., SHANNON M.S., BARNETT M.J., PRENTICE H.G., BRAGMAN K., MALPAS J.S., LISTER T.A. Treatment of acute leukaemia with m-AMSA in combination with cytosine arabinoside. Cancer Chemother. Pharmacol. 1986;18:59–62. doi: 10.1007/BF00253066. [DOI] [PubMed] [Google Scholar]
- GRIFFIN J.D., MAGUIRE M.E., MAYER R.J. Amsacrine in refractory acute leukemia. Cancer Treat. Rep. 1985;69:787–789. [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMAN B., SIGWORTH F.J. Improved patch clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflug. Arch. Eur. J. Physiol. 1981;391:58–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- JEHN U., HEINEMANN V. New drugs in the treatment of acute and chronic leukemia with some emphasis on m-AMSA. Anticancer Res. 1991;11:705–711. [PubMed] [Google Scholar]
- JURLINA J.L., VARCOE A.R., PAXTON J.W. Pharmacokinetics of amsacrine in patients receiving combined chemotherapy for treatment of acute myelogenous leukemia. Cancer Chemother. Pharmacol. 1985;14:21–25. doi: 10.1007/BF00552719. [DOI] [PubMed] [Google Scholar]
- KANTARJIAN H.M., ESTEY E.H., KEATING M.A. New chemotherapeutic agents in acute myeloid leukemia. Leukemia. 1996;10 Suppl. 1:S4–S6. [PubMed] [Google Scholar]
- KIEHN J., THOMAS D., KARLE C.A., SCHÖLS W., KÜBLER W. Inhibitory effects of the class III antiarrhythmic drug amiodarone on cloned HERG potassium channels. Naunyn-Schmiedeberg's Arch. Pharmacol. 1999;359:212–219. doi: 10.1007/pl00005344. [DOI] [PubMed] [Google Scholar]
- KURYSHEV Y.A., BROWN A.M., WANG L., BENEDICT C.R., RAMPE D. Interactions of the 5-hydroxytryptamine 3 antagonist class of antiemetic drugs with human cardiac ion channels. J. Pharmacol. Exp. Ther. 2000;295:614–620. [PubMed] [Google Scholar]
- LINSSEN P., BRONS P., KNOPS G., WESSELS H., DE WITTE T. Plasma and cellular pharmacokinetics of m-AMSA related to in vitro toxicity towards normal and leukemic clonogenic bone marrow cells (CFU-GM, CFU-L) Eur. J. Haematol. 1993;50:149–154. doi: 10.1111/j.1600-0609.1993.tb00083.x. [DOI] [PubMed] [Google Scholar]
- MCLAUGHLIN P., SALVADOR P.G., CABANILLAS F., LEGHA S.S. Ventricular fibrillation following AMSA. Uncomplicated retreatment following correction of hypokalemia. Cancer. 1983;52:557–558. doi: 10.1002/1097-0142(19830801)52:3<557::aid-cncr2820520329>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- MILNES J.T., CROCIANI O., ARCANGELI A., HANCOX J.C., WITCHEL H.J. Blockade of HERG potassium currents by fluvoxamine: incomplete attenuation by S6 mutations at F656 or Y652. Br. J. Pharmacol. 2003;139:887–898. doi: 10.1038/sj.bjp.0705335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHESON J.S., CHEN J., LIN M., CULBERSON C., SANGUINETTI M.C. A structural basis for drug-induced long QT syndrome. Proc. Natl. Acad. Sci. U.S.A. 2000a;97:12329–12333. doi: 10.1073/pnas.210244497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHESON J.S., CHEN J., SANGUINETTI M.C. Trapping of a methanesulfonanilide by closure of the HERG potassium channel activation gate. J. Gen. Physiol. 2000b;115:229–240. doi: 10.1085/jgp.115.3.229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MITCHESON J.S. Drug binding to HERG channels: evidence for a ‘non-aromatic' binding site for fluvoxamine. Br. J. Pharmacol. 2003;139:883–884. doi: 10.1038/sj.bjp.0705336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NAPOLITANO C., PRIORI S., SCHWARTZ P.J. Torsade de pointes: mechanism and management. Drugs. 1994;47:51–65. doi: 10.2165/00003495-199447010-00004. [DOI] [PubMed] [Google Scholar]
- PAXTON J.W., JURLINA J.L., FOOTE S.E. The binding of ams-acrine to human plasma proteins. J. Pharm. Pharmacol. 1986;38:432–438. doi: 10.1111/j.2042-7158.1986.tb04606.x. [DOI] [PubMed] [Google Scholar]
- REDFERN W.S., CARLSSON L., DAVIS A.S., LYNCH W.G., MACKENZIE I., PALETHORPE S., SIEGL P.K., STRANG I., SULLIVAN A.T., WALLIS R., CAMM A.J., HAMMOND T.G. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- ROWE J.M. Treatment of acute myelogenous leukemia in older adults. Leukemia. 2000;14:480–487. doi: 10.1038/sj.leu.2401539. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., CHAGAN J., CURRAN M.E., KEATING M.T. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:1–20. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- SANGUINETTI M.C., JURKIEWICZ N.K. Two components of the delayed rectifier K+ current: differential sensitivity to block by class III antiarrhythmic agents. J. Gen. Physiol. 1990;96:195–215. doi: 10.1085/jgp.96.1.195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHOLZ E.P., ZITRON E., KIESECKER C., LUECK S., KATHÖFER S., THOMAS D., WERETKA S., PETH S., KREYE V.A.W., SCHOELS W., KATUS H.A., KIEHN J., KARLE C.A. Drug binding to aromatic residues in the HERG channel pore cavity as possible explanation for acquired long QT syndrome by antiparkinsonian drug budipine. Naunyn-Schmiedeberg's Arch. Pharmacol. 2003;368:404–414. doi: 10.1007/s00210-003-0805-5. [DOI] [PubMed] [Google Scholar]
- SCHWARTZ C.L., BENDER K.S., BURKE P.J., KAN J.S., CIVIN C.I. QT interval prolongation and cardiac dysrhythmia in a patient receiving amsacrine. Cancer Treat. Rep. 1984;68:1043–1044. [PubMed] [Google Scholar]
- SEYMOUR J.F. Induction of hypomagnesemia during amsacrine treatment. Am. J. Hematol. 1993;42:262–267. doi: 10.1002/ajh.2830420305. [DOI] [PubMed] [Google Scholar]
- SHINAR E., HASIN Y. Acute electrocardiographic changes induced by amsacrine. Cancer Treat. Rep. 1984;68:1169–1172. [PubMed] [Google Scholar]
- TESCHEMACHER A.G., SEWARD E.P., HANCOX J.C., WITCHEL H.J. Inhibition of the current of heterologously expressed HERG potassium channels by imipramine and amitriptyline. Br. J. Pharmacol. 1999;128:479–485. doi: 10.1038/sj.bjp.0702800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D., WENDT-NORDAHL G., RÖCKL K., FICKER E., BROWN A.M., KIEHN J. High-affinity blockade of HERG human cardiac potassium channels by the novel antiarrhythmic drug BRL-32872. J. Pharmacol. Exp. Ther. 2001;297:753–761. [PubMed] [Google Scholar]
- THOMAS D., WU K., KATHÖFER S., KATUS H.A., SCHOELS W., KIEHN J., KARLE C.A. The antipsychotic drug chlorpromazine inhibits HERG potassium channels. Br. J. Pharmacol. 2003a;139:567–574. doi: 10.1038/sj.bjp.0705283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- THOMAS D., GUT B., KARSAI S., WIMMER A.B., WU K., WENDT-NORDAHL G., ZHANG W., KATHOFER S., SCHOELS W., KATUS H.A., KIEHN J., KARLE C.A. Inhibition of cloned HERG potassium channels by the antiestrogen tamoxifen. Naunyn-Schmiedeberg's Arch. Pharmacol. 2003b;368:41–48. doi: 10.1007/s00210-003-0766-8. [DOI] [PubMed] [Google Scholar]
- VAN HOFF D.D., ELSON D., POLK G., COLTMAN C., JR Acute ventricular fibrillation and death during infusion of 4′-(9-acridinylamino)methanesulfon-m-anisidide (AMSA) Cancer Treat. Rep. 1980;64:356–358. [PubMed] [Google Scholar]
- VISKIN S. Long QT syndromes and torsade de pointes. Lancet. 1999;354:1625–1633. doi: 10.1016/S0140-6736(99)02107-8. [DOI] [PubMed] [Google Scholar]
- WARMKE J.W., GANETZKY B. A family of potassium channel genes related to eag in Drosophila and mammals. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3438–3442. doi: 10.1073/pnas.91.8.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEERAPURA M., NATTEL S., CHARTIER D., CABALLERO R., HEBERT T.E. A comparison of currents carried by HERG, with and without coexpression of MiRP1, and the native rapid delayed rectifier current. Is MiRP1 the missing link. J. Physiol. 2002;540:15–27. doi: 10.1113/jphysiol.2001.013296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WEISS R.B., GRILLO-LOPEZ A.J., MARSONI S., POSADA J.G., JR, HESS F., ROSS B.J. Amsacrine-associated cardiotoxicity: an analysis of 82 cases. J. Clin. Oncol. 1986;4:918–928. doi: 10.1200/JCO.1986.4.6.918. [DOI] [PubMed] [Google Scholar]
- WINTON E.F., HEARN E.B., MARTELO O., PRESANT C.A., ADLER S., VOGLER W.R., RANEY M., LOGAN T., SILBERMAN H.M., OMURA G.A. Sequentially administered 5-azacitidine and amsacrine in refractory adult acute leukemia: a phase I-II trial of the Southeastern Cancer Study Group. Cancer Treat. Rep. 1985;69:807–811. [PubMed] [Google Scholar]