

Abstract
In this study, intracellular Ca2+ was measured as the Fura-2 ratio (R) of fluorescence excited at 340 and 380 nm (F340/F380) in nonpressurized rat mesenteric small arterioles (Ø (lumen diameter) 10–25 μm). The response to depolarization using 75 mM KCl was an increase in R from a baseline of 0.96±0.01 ([Ca2+]i ∼74 nM) to 1.04±0.01 (∼128 nM) (n=80).
The response to 75 mM K+ was reversibly abolished in Ca2+-free physiological saline solution, whereas phentolamine (10 μM) or tetrodotoxin (1 μM) had no effects. LaCl3 (200 μM) inhibited 61±9% of the response.
A [K+]-response curve indicated that the Ca2+ response was activated between 15 and 25 mM K+. The data suggest that the Ca2+ response was caused by the activation of voltage-dependent Ca2+ channels.
Mibefradil use dependently inhibited the Ca2+ response to 75 mM K+ by 29±2% (100 nM), 73±7% (1 μM) or 89±7% (10 μM). Pimozide (500 nM) use dependently inhibited the Ca2+ response by 85±1%.
Nifedipine (1 μM) inhibited the Ca2+ response to 75 mM K+ by 41±12%. The response was not inhibited by calciseptine (500 nM), ω-agatoxin IVA (100 nM), ω-conotoxin MVIIA (500 nM), or SNX-482 (100 nM).
Using reverse transcriptase–polymerase chain reaction, it was shown that neither CaV2.1a (P-type) nor CaV2.1b (Q-type) voltage-dependent Ca2+ channels were expressed in mesenteric arterioles, whereas the CaV3.1 (T-type) channel was expressed. Furthermore, no amplification products were detected when using specific primers for the β1b, β2, or β3 auxiliary subunits of high-voltage-activated Ca2+ channels.
The results suggest that the voltage-dependent Ca2+ channel activated by sustained depolarization in mesenteric arterioles does not classify as any of the high-voltage-activated channels (L-, P/Q-, N-, or R-type), but is likely to be a T-type channel. The possibility that the sustained Ca2+ influx observed was the result of a T-type window current is discussed.
Keywords: Microcirculation, resistance vessel, arteriole, vascular smooth muscle cell, voltage-dependent Ca2+ channel, conducted vascular response, mibefradil, pimozide, calciseptine, use dependence
Introduction
Voltage-dependent Ca2+ channels are crucial for excitation–contraction coupling in muscle cells from the heart, skeletal muscle and various types of smooth muscle including vascular smooth muscle cells (VSMC). These channels have been roughly divided into high-voltage-activated (L-, P/Q-, N-, R-type) vs low-voltage-activated calcium channels (T-type) (Hofmann et al., 1999), referring to the fact that the former requires larger depolarization for activation than the latter.
In the majority of VSMC, excitation–contraction coupling is maintained by L-type Ca2+ channels pharmacologically distinguished by their sensitivity to dihydropyridines such as nifedipine (McDonald et al., 1994; Perez-Reyes, 2003). However, in microvascular resistance vessels other types of voltage-dependent Ca2+ channels have emerged that were previously believed to function only in the heart or brain tissue. In rat cremaster muscle arterioles, T-type Ca2+ channels are involved in the maintenance of vascular tone (VanBavel et al., 2002), and in renal afferent arterioles from rabbit or rat, both T-type (Hansen et al., 2001) and P/Q-type (Hansen et al., 2000) channels appear to have a functional role in depolarization-induced vasoconstriction and Ca2+ influx. Furthermore, in a recent study, we demonstrated that mRNA coding for the pore-forming α1C-subunit (CaV1.2) of the L-type Ca2+ channel was absent in rat mesenteric arterioles, whereas that of α1G- (CaV3.1) and α1H- (CaV3.2) subunits of the T-type Ca2+ channel were expressed (Gustafsson et al., 2001). In addition, vasoconstriction in rat mesenteric arterioles (Ø (lumen diameter) <40 μm) in vivo appeared to rely on mibefradil-sensitive and dihydropyridine-insensitive Ca2+ channels (Gustafsson et al., 2001).
In agreement with the above findings, isolated smooth muscle cells from rat and guinea-pig mesenteric artery terminal branches (Ø 50–100 μm) displayed nifedipine-insensitive, voltage-activated Ca2+ currents with T-channel-like pharmacological properties (Morita et al., 2002). However, the biophysical properties of this channel were different from ‘classical' low voltage-activated T-type channels (Morita et al., 1999). The major difference to T-type characteristics were activation at more depolarized potentials (threshold at −50 to −40 mV and peak current at −15 to −10 mV) than typical T-type currents with activation threshold at −70 to −60 mV and peak current at −40 to −30 mV. Moreover, physical ion pore properties (Ba2+/Ca2+ permeability; Cd2+/Ni2+ blocking efficacy) suggested that this channel belongs to the class of high-voltage activated Ca2+ channels (e.g. R-type), rather than low-voltage activated (T-type) channels (Morita et al., 1999; 2002). These authors therefore suggested that this channel is either a novel type of voltage-activated Ca2+ channel with mixed properties known from R- and T-type channels or an alternative splice variant of the T-type channel.
Owing to the fast inactivating nature of native T-type channels (τinact 5−50 ms), they have been thought to play little role in Ca2+ influx during sustained depolarization. However, small steady-state Ca2+ currents occurring in the voltage ‘window' between overlapping steady-state activation and inactivation curves (window currents) may account for sustained Ca2+ influx into cells (McDonald et al., 1994; Morita et al., 1999; Perez-Reyes, 2003). A physiological role for T-type window currents has recently been demonstrated in human myoblast terminal differentiation where they mediated the sustained Ca2+ entry required for myoblast fusion (Bijlenga et al., 2000).
In the present study, we have characterized the pharmacological properties of depolarization-induced Ca2+ influx in isolated rat mesenteric small arterioles (Ø 10–25 μm) stimulated by elevated bath K+ concentration. The induced Ca2+ increases are hypothesized to rely on window currents through Ca2+ channels that display fast voltage-dependent inactivation (T-type). Using reverse transcriptase–polymerase chain reaction (RT–PCR), we have also investigated the mRNA expression profile of a selection of known α1- (pore-forming) and β-(auxiliary) subunits of voltage-dependent Ca2+ channels.
Methods
Animals and microdissection of mesenteric arterioles
A total of 51 male Wistar rats (age 7–10 weeks; weight 261±24 g) were anesthetized by inhalation of 2−5% halothane or sevofluran in a mixture of 35% O2 with 65% N2. A catheter was inserted in the left jugular vein for infusion and a tracheal catheter was used to secure free airways. At this point, halothane/sevofluran anesthesia was stopped and a continuous intravenous infusion of pentobarbital sodium (154 μg min−1) initiated.
A median laparotomy was performed and a loop of the small intestine with adjacent mesentery was pinned onto Sylgard® covering the bottom of a Petri dish filled with isotonic NaCl kept at room temperature. The mesenteric microcirculation was examined with a Leica Wild M10 stereomicroscope (× 8−63 magnification). The arterioles (resistance vessels) of interest derive from first- or second-order branches of the superior mesenteric artery and give rise to capillary networks supplying the mesentery. The criteria for selection of arterioles (Ø 10−25 μm) branching from an artery into the transparent part of the mesentery were observation of a fast pulsating blood flow and that they gave rise to smaller capillaries downstream of the point of observation. Arterioles with surrounding tissue were isolated and transferred onto a glass beaker containing ice-cold physiological saline solution (PSS) with 1% bovine serum albumin (BSA). With the aid of a microscope (× 95), microdissection was carried out in a glass dish placed in an ice-cold metal block. Each arteriole was dissected free from fat and as much of the surrounding tissue as possible while avoiding stretching or damaging the vessel.
RNA isolation, RT–PCR and Southern blotting
A total of 4–6 mm arteriole (Ø 10–25 μm) per rat (n=2) was microdissected and transferred onto an Eppendorf tube (on ice) containing 400 μl RLT buffer (Qiagen, Germany) and freshly added β-mercaptoethanol (1%).
Arterioles were added directly to guanidinium thiocyanate (4 mol l−1). For homogenization, the fragments were repeatedly triturated through syringes with decreasing dimension ending with 25 G. Yeast tRNA (12 μg) was added as carrier. Total RNA was extracted by phenol–chloroform extraction, precipitated with isopropanol and repeatedly washed with 70% ethanol (Andreasen et al., 2000). RNA pellets were suspended in diethyl pyrocarbonate water and used for cDNA synthesis as previously described in detail (Andreasen et al., 2000).
PCRs were performed with DNA oligonucleotides (Invitrogen) for rat CaV2.1, CaV3.1, smooth muscle auxiliary β3-subunit, cardiac β2-subunit, or neuronal β1b-subunit. β-actin primers and reaction components were as described previously (Andreasen et al., 2000). As negative control, reverse transcription of total RNA was carried out in the absence of reverse transcriptase and subsequently amplified by PCR. As positive control for PCR, cDNA (50 ng) synthesized from various rat organs (cerebral cortex, left cardiac ventricle, and renal inner medulla) was used.
A dual set of PCR primers was designed to distinguish between CaV2.1a (P-type) and CaV2.1b (Q-type) at the splice site determining the ω-agatoxin IVA affinity, namely, the N1605-P1606 insertion. One forward primer was designed to hybridize upstream from the splice site. Two reverse primers were designed to overlap the splice site in such a way that each reverse primer would hybridize only to the chosen splice variant. All PCR products were verified by sequencing. Sequences were read on an ABI Prism genetic analyzer using ready-reaction mix from ABI, and templates were PCR products or on subcloned PCR products (plasmid pSP73, Promega).
CaV2.1a sense primer: 5′-gcc ctt cga gtg ttc aac-3′; antisense CaV2.1a: 5′-ctc agg ttg atg aag tta ttc c-3′, covering bases 4642–4826, 185 bp.
CaV2.1b sense primer: 5′-cag gtt gat gaa gtt att cgg-3′ (covering bases 4642–4824+6 bp), 189 bp. GenBank accession no. M64373 (Bourinet et al., 1999).
CaV3.1 sense primer: 5′-gaa cgt gag gcc aag agt-3′; antisense: 5′-gct tgt atg cgt tcc cct-3′, covering bases 3910–4130, 221 bp. GenBank accession no. AF027984 (Andreasen et al., 2000).
β1b (neuronal) sense primer: 5′-tgt caa act gga cag c; antisense: 5′-aag aag tca aac aac gcc, 282 bp. GenBank accession no. X61394.
β2 (cardiac) sense primer: 5′-gga gga aat tca tca tcc; antisense: 5′-ttc cgc taa gct cga cc, 459 bp. GenBank accession no. 0M80545.
β3 (smooth muscle) sense primer: 5′-caa aca gga aca gaa gg; antisense: 5′-gat ggt cct ctt gcc, 324 bp. GenBank accession no. M88751.
RT–PCR was performed with annealing temperature between 50 and 60°C for 32–33 cycles (Eppendorf, Mastercycler).
Ca2+ indicator loading and intracellular Ca2+ measurements
Microdissected arterioles were transferred onto a glass dish containing PSS-based loading buffer to which Fura-2/AM had been added, and loaded for 120 min at room temperature in the dark. Alternatively, vessels were glued onto the cover slide bottom of the recording/perfusion chamber (Warner Instrument, Inc., CT, U.S.A.) using Cell-Tak (Becton Dickinson Labware, MA, U.S.A.), and incubated with Fura-2 herein under similar conditions. Following loading, the vessels were rinsed with fresh PSS, mounted between two suction pipettes (tip o.d. ∼20 μm) if not already immobilized using Cell-Tak, and rested for 20–30 min before performing measurements. There was no difference between results obtained with arterioles immobilized using Cell-Tak or suction pipettes.
The recording chamber (∼0.1 ml capacity) was mounted onto the stage of an inverted fluorescence microscope (Olympus IX50). Solutions were added via a continuous superfusion flow (∼1.7 ml min−1) and excess solution was removed by suction. A quartz oil-immersion objective (Olympus × 40/1.35) was used for Ca2+ measurements and the nonpressurized arterioles were visualized (see Figure 1) on a PC monitor using a digital video camera (SensiCam) and the Image Workbench (Axon) software package. Using a monochromator controlled by the software, the preparation was excited with fluorescent light of alternating wavelengths (340 vs 380 nm). A region of interest (ROI) for the measurement of intracellular calcium concentration ([Ca2+]i) was selected using a software routine. The fluorescence emitted in the ROI was detected by the digital camera and stored to a PC hard disk. Results are expressed as the Fura-2 ratio (R=F340/F380), which was calculated after a constant background signal had been subtracted from each wavelength. [Ca2+]i was calculated using the Grynkiewics equation (Grynkiewicz et al., 1985):
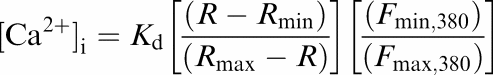 |
Figure 1.

Micrograph of Fura-2-loaded arteriole in isolated rat mesentery. The tissue was excited at 340 nm and emission collected at 510 nm, giving rise to the bright fluorescence seen in the arteriolar wall. For the purpose of illustration, a dimmed transmission light was used to distinguish the arteriolar lumen and wall from the mesenteric tissue. The few bright spots seen in the mesentery probably correspond to fibroblasts loaded with Fura-2. The freshly isolated tissue was glued to a cover slide and subsequently loaded with Fura-2/AM in the recording chamber. In some cases, excess mesenteric tissue was carefully removed by microdissection prior to Fura loading and arterioles mounted between two suction pipettes during recordings.
Here, Kd is the Fura-2 dissociation constant for Ca2+, R is the ratio of emitted fluorescence (F) at 340 over 380 nm, min and max denotes values obtained with Ca2+-free solution or a solution with saturating [Ca2+], respectively. The Kd value used was 227 nM and Kd, Rmin, Rmax, Fmin, 380, and Fmax, 380 were determined in vitro using commercially available calibration buffers and Fura-2 pentapotassium salt (Molecular Probes, Leiden, The Netherlands).
Prior to experimentation, arterioles were rejected if the background-subtracted fluorescence of the adjacent tissue was >50% of that observed in arterioles or if changes in flow rate or solution level caused excess noise. Less than 5% of arterioles were rejected due to the lack of a Ca2+ response to 75 mM K+. In acceptable preparations, a stable baseline measurement of 1–2 min was recorded before any stimulus was applied.
In most of the experiments, arterioles were superfused two to four times with high-K+ solution for 40–100 s and with 2 min intervals. Subsequently, arterioles were exposed to an experimental drug dissolved in normal PSS for 1–5 min before the high-K+ solution was applied two to four times in the presence of the same drug. A washout and resting period of 2–3 min was followed by two to four times high-K+ stimulation in the absence of any drugs. In neurotoxin experiments, solutions were added by pipette (>20 times the bath volume) instead of by superfusion to reduce the amount of toxin needed. There was no difference between basal high-K+ responses obtained with the protocol for neurotoxin experiments compared to that used in the remainder of the study.
Solutions and chemicals
PSS contained (in mM): 140 NaCl, 6 KCl, 1 MgCl2, 1.8 CaCl2, 5 N-(2-hydroxyethyl)piperazine-N′-(2-ethanesulfonic acid; 4-(2-hydroxyethyl)piperazine-1-ethanesulfonic acid (HEPES), and 5 glucose, with pH adjusted to 7.4. In solutions with elevated [K+] (high-K+ PSS), NaCl was substituted with equimolar amounts of KCl. In calcium-free PSS, CaCl2 was substituted with 1 mM ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA). In Na+-free PSS, NaCl was substituted mole for mole with N-Methyl-D-glucosamine chloride (NMDG-Cl). The loading buffer consisted of 5 μM Fura-2/AM (Molecular Probes), 1% BSA, 0.5% DMSO, 0.02% Pluronic F127, 0.06% Cremophore EL, and 2.5 mM Probenecid (all from Sigma-Aldrich, Steinheim, Germany) dissolved in PSS.Mibefradil (F. Hoffmann-La Roche Ltd, Basel, Switzerland), LaCl3, phentolamine (both Sigma-Aldrich, Steinheim, Germany), calciseptine, ω-agatoxin IVA, ω-conotoxin MVIIA, SNX-482, and tetrodotoxin (TTX) (all Alomone Labs, Jerusalem, Israel) were added from stock solutions prepared from water. Nifedipine and pimozide (both Sigma-Aldrich, Steinheim, Germany) were dissolved in DMSO, which was finally diluted to 0.001–0.005% in PSS. In vehicle control experiments, there were no effects of 0.005% DMSO alone (n=4).
Data presentation and statistics
Baseline Ca2+ measurements were reported as crude ratios or means±s.e. of the ratio, and in addition global mean values were reported as approximate [Ca2+]i values. Calcium responses to high K+ were summarized as % of baseline R(100 × Rstim/Rbas), and calcium responses during drug application were summarized as % inhibition (100−(100 × ΔRdrug/ΔRctr)). Statistical tests were paired Student's t-tests (unless otherwise stated), two-sided, and performed at the 5% level of significance. In cases where use of a nonparametric test is indicated, the data could not pass the test for normality.
Results
Ca2+ measurements under resting and stimulated conditions (high [K+])
In a total of 80 arterioles from 51 rats the baseline Fura-2 ratio was 0.96±0.01, corresponding to a global mean resting [Ca2+]i of ∼74 nM. Superfusing arterioles with PSS containing 75 mM K+ instead of 6 mM resulted in an increase in the ratio from the resting level to 1.04±0.01 (P<0.001, signed-rank test) or 108±1% of the baseline value (see Figure 2a and b). The response to 75 mM K+ corresponds to a [Ca2+]i increase of approximately 54 nM from ∼74 to ∼128 nM.
Figure 2.

(a) Original recording of Fura-2 ratio (F340/F380) in mesenteric arteriole. Sustained depolarization induced by 75 mM K+ (K) resulted in an increase of intracellular [Ca2+] (F340/F380) from the baseline level. The Ca2+ response was reversibly abolished in the presence of Ca2+-free PSS with Ca2+ chelator (EGTA). (b) Summary of Ca2+ responses to 75 mM K+ shown as % of baseline Fura-2 ratio. Compared to the control stimulation (K75 Control), the Ca2+ response was not altered by treatment with phentolamine (10 μM) or addition of TTX (1 μM) on top of phentolamine. However, the response was partially blocked in the presence of 200 μM La3+. The number of arterioles used is shown within parenthesis. *P<0.05, **P<0.01; t-test vs control stimulation.
Ca2+ entry vs mobilization
The calcium response to high K+ could in principle be induced by depolarization of nerve terminals, thereby causing release of neurotransmitters such as noradrenaline, which would lead to receptor-mediated calcium mobilization in arterioles. To verify that this was not the case, the effect of the nonspecific α-adrenergic antagonist phentolamine and the neuronal voltage-dependent Na+ channel blocker TTX was investigated. There was no significant difference between the high-K+ response in the absence or presence of 10 μM phentolamine or with 1 μM TTX added to the phentolamine (Figure 2b).
To investigate the Ca2+ dependence of the response, arterioles were superfused with a Ca2+-free PSS including a Ca2+ chelator (EGTA). As opposed to the control response in the presence of extracellular Ca2+, the high-K+ response was completely and reversibly abolished in Ca2+-free PSS, indicating that the response was strictly dependent on calcium entry (see Figure 2a and b).
Trivalent metal ions (e.g. La3+) have been used to block currents through transiently expressed voltage-activated Ca2+ channels (Beedle et al., 2002). In the present study, we used lanthanum (LaCl3) to test whether the calcium influx was sensitive to trivalent metal ions. The response to high K+ was reversibly reduced in the presence of 200 μM La3+, corresponding to an inhibition of 61±9%.
A dose−peak response relationship was obtained for elevated extracellular K+ concentrations of 15, 25, 50, 75, and 110 mM (Figure 3). The calcium response was activated between 15 and 25 mM K+ and it was maximal at 75–110 mM K+. The above data suggest that the high-K+-induced Ca2+ response was mediated by voltage-dependent Ca2+ channels in the arteriolar wall.
Figure 3.

In a number of unpaired experiments, arterioles were subjected to depolarization with varying extracellular [K+]. The concentration−peak response curve indicates activation of Ca2+ entry between 15 and 25 mM extracellular K+ and the response is maximal at approximately 75 mM K+. The number of arterioles used is shown within parenthesis.
Pharmacological characterization of the influx pathway
Effect of L-type blockers
Our previous investigation (Gustafsson et al., 2001) revealed that rat mesenteric arterioles do not possess mRNA coding for the CaV1.2 α1-subunit of the L-type Ca2+ channel, and in addition vasoconstriction in vivo was insensitive to dihydropyridines (10 μM nifedipine or nimodipine). To confirm these results, we tested the effect of nifedipine (1 μM) on the Ca2+ response to high-K+ stimulation. Nifedipine induced a variable block from 0 to 70% inhibition (Figure 4). As this could be a nonspecific effect of nifedipine, we also tested the venom neurotoxin calciseptine reported to be a potent blocker of L-type Ca2+ channels with little or no effect on N- or T-type voltage-dependent Ca2+ channels (de Weille et al., 1991). In five mesenteric arterioles, 500 nM calciseptine did not inhibit the Ca2+ response to high K+ (Figure 4), indicating that L-type channels do not play a role in this response and that the effect of nifedipine was nonspecific. To verify the efficacy of calciseptine, we tested its effect in freshly isolated rat renal afferent arterioles known to contain L-type Ca2+ channels (Salomonsson & Arendshorst, 1999; Loutzenhiser & Loutzenhiser, 2000). Using the same protocol as for mesenteric arterioles, 500 nM calciseptine induced a 47±4% inhibition of the Ca2+ response to 75 mM K+ in afferent arterioles (P<0.01, n=4).
Figure 4.

Effects of various Ca2+ channel blockers shown as % inhibition of the Ca2+ response to 75 mM K+. The L-type Ca2+ channel blocker nifedipine (1 μM) inhibited 40% of the response, whereas there was no effect of a highly specific L-type blocker, the neurotoxin calciseptine (500 nM). Highly specific neurotoxin blockers of P/Q-type (ω-agatoxin IVA, 100 nM), N-type (ω-conotoxin MVIIA, 500 nM), or R-type Ca2+ channels (SNX-482, 100 nM) did not inhibit the response to high K+. There was a minor increase of the Ca2+ response in the presence of ω-agatoxin IVA. The number of arterioles is within parenthesis. *P<0.05; t-test vs untreated control experiments.
Effect of P/Q-, N- and R-type blockers
Functional evidence for P/Q-type Ca2+ channels and mRNA expression of the CaV2.1 α1-subunit of this channel has recently been described in afferent arterioles from rat kidney (Hansen et al., 2000). We therefore tested the effect of the venom neurotoxin ω-agatoxin IVA, which is a specific blocker of P/Q-type Ca2+ channels (Mintz et al., 1992). In a total of four experiments, 100 nM ω-agatoxin IVA did not inhibit high-K+-induced Ca2+ response in rat mesenteric arterioles, but instead induced a small, reversible, and significant increase (Figure 4). Specific neurotoxin blockers also exist for N-type (Feng et al., 2003) and R-type Ca2+ channels (Newcomb et al., 1998). There were no effects of the N-type blocker ω-conotoxin MVIIA (500 nM) or the R-type blocker SNX-482 (100 nM) (Figure 4).
Effect of mibefradil
A recent study on rat mesenteric arterioles indicated a role for dihydropyridine-insensitive, mibefradil-sensitive Ca2+ channels (Gustafsson et al., 2001). We therefore tested the effects of mibefradil, which is known to inhibit T-type Ca2+ channels at submicromolar concentrations, whereas L-type channels are affected at micromolar concentrations. To exploit the use-dependent effects of mibefradil, we applied a protocol in which four brief (40 s) stimulations with 75 mM K+ were followed by four stimulations in the presence of the drug to facilitate binding to channels in the open or inactivated state. Mibefradil did not change the baseline Fura-2 ratio, but it inhibited the response to high-K+ (Figure 5a). Throughout the concentration range from 100 nM to 10 μM, there was a difference between the initial and fourth high-K+ response in the presence of drug, indicating use dependence of mibefradil block (Figure 5b). This effect was time independent since there was no difference between responses obtained with 1, 3, or 5 min preincubation with mibefradil (data not shown). Using one-way repeated measures ANOVA with Student–Newman–Keuls post hoc test, there was a significant difference between the first vs the third and fourth high-K+ stimulation in the presence of mibefradil at both 100 nM (P<0.001, n=4) and 1 μM (P<0.05, n=5), but not at 10 μM (n=4). This difference was not detected in the untreated controls (n=4). A summary of % inhibition by dose level is presented in Figure 5b. Importantly, the use-dependent block induced by 100 nM mibefradil, a concentration believed to be specific for T-type channels, was statistically significant.
Figure 5.

Effects of the T-type Ca2+ channel blockers mibefradil and pimozide. (a) In this original trace, an arteriole was stimulated repeatedly (12 stimulations of 40 s duration) with 75 mM K+ (K). After four stimulations mibefradil (1 μM) was applied for a total period of ∼10 min. The inhibition induced by mibefradil was maximal after three to four stimulations, and this time-independent effect is referred to as use-dependent block. (b) Open columns depict % inhibition of the first stimulation (initial block) and hatched columns depict inhibition of the last stimulation (use-dependent block) in the presence of mibefradil at all concentrations tested (n=4−5). The untreated control experiments are shown here as 0.0 μM mibefradil (n=4). (c) Ca2+ responses to 75 mM K+ (K) in an arteriole exposed to 500 nMof the antipsychotic pimozide, clearly showing a use-dependent effect of the drug. A small reversible decrease of the baseline ratio is seen after the addition of pimozide. (d) Summary of responses obtained in arterioles exposed to pimozide (n=4) and in vehicle control experiments (n=4). *P<0.05, **P<0.01, and *** P<0.001; t-test vs untreated or vehicle control experiments.
Effect of pimozide
The neuroleptic agent pimozide is, apart from being a D2 dopamine receptor antagonist, an effective blocker of voltage-dependent calcium channels (Enyeart et al., 1990; 1993; Sah & Bean, 1994). In analogy with mibefradil, block of calcium channels by pimozide appears to be use dependent (Arnoult et al., 1998). The use-dependent effect of pimozide was therefore investigated using the same protocol as for mibefradil (Figure 5c). In four arterioles, pimozide (500 nM) efficiently and use dependently blocked 85% of the calcium response to 75 mM K+, whereas a slight inhibition (10%) was observed in the vehicle control experiments (Figure 5d). It should be noted that the baseline Fura-2 ratio was reversibly decreased from 0.965±0.009 to 0.959±0.010 after adding 500 nM pimozide. In vehicle control experiments, there was no change of the baseline ratio before compared to 1 min after adding 0.005% DMSO to the perfusate.
Activation of reverse mode Na+/Ca2+ exchanger
With Na+ as the major cation in the superfusate, the only way to increase bath [K+] at a constant osmolarity is to substitute equimolar amounts of Na+ in the bath. Depolarization of the cell and a concomitant lowering of the bath [Na+] might reverse the electrogenic Na+/Ca2+ exchanger that functions as a Ca2+ extrusion mechanism under control conditions. We investigated the effect of mibefradil on the Ca2+ response to bath Na+ substitution with NMDG, as such an effect might compromise our conclusions with regard to mibefradil's effect on voltage-dependent Ca2+ channels. When bath Na+ was substituted, a slowly activating (t1/2=41.8±10.5 s) sustained increase of the Fura-2 ratio was observed from a baseline of 1.01±0.04 to 1.36±0.15 (P=0.051, n=5), corresponding to an approximate increase in [Ca2+]i from ∼106 to ∼352 nM. There was no effect of 1 μM mibefradil on this response (4.7±10.5% inhibition, n=5).
Expression of α1 and β calcium channel subunits
RNA was extracted from mesenteric arterioles of two rats and analyzed along with RNA obtained from various rat organs as positive controls. Amplification products for P- and Q-type calcium channels CaV2.1a and CaV2.1b were not detected in mesenteric arterioles with template cDNA from 0.5 mm vessel length and after amplification for up to 34 cycles. Amplification products were readily obtained for both P- and Q-type channels with 50 ng cerebral cortex cDNA as template (Figure 6). Sequencing of amplification products revealed 100% homology to the respective splice variant sequences for P- and Q-type calcium channel α1-subunits. Previously reported expression in mesenteric arterioles of T-type channel α1-subunit CaV3.1 (Gustafsson et al., 2001) was confirmed on the same cDNA batches using the same amount of starting cDNA (Figure 6).
Figure 6.

Agarose-gel electrophoresis showing PCR products obtained when using specific primers for selected α1- and β-subunits of voltage-dependent Ca2+ channels and cDNA from mesenteric arterioles. The CaV2.1-P and CaV2.1-Q are alternative splice variants of the same gene and corresponds to the P-type and Q-type α1-subunits, respectively. CaV3.1-T corresponds to the α1G-subunit of T-type channels. β-2 (cardiac), β-3 (smooth muscle), and β-B (β1b, neuronal) are known β-subunits of high-voltage-activated Ca2+ channels. Specific primer for β-actin was also included in the reactions. Pooled cDNA from mesenteric arterioles in each of two rats were used (Prep 1, Prep 2). Appropriate controls are shown in which cDNA was omitted (neg. control) or 50 ng template cDNA isolated from various rat organs (cerebral cortex, left cardiac ventricle, or renal inner medulla) was included in the PCR (pos. control).
We did not detect products for any of the tested β-subunits: neuronal β1b, cardiac β2, or neuronal/smooth muscle β3 using cDNA corresponding to 0.5 mm vessel length as starting amount and 33 cycles of amplification (Figure 6). In each of the PCRs, significant amplification products were obtained in the positive controls (cerebral cortex, left cardiac ventricle, or renal inner medulla). When arteriolar cDNA was amplified for β-actin with intron-spanning primers, significant products were obtained at the correct size, indicating intact template and a cDNA origin of the amplification products. Amplification products were obtained only in the presence of cDNA in each of the RT–PCRs.
Discussion
We have measured intracellular Ca2+ in mesenteric small arterioles and provide evidence of a depolarization-activated influx pathway dominated by mibefradil-sensitive Ca2+ channels. The magnitude of the Ca2+ response to 75 mM extracellular K+ was an increase to 108% of the baseline Fura-2 ratio (Figure 2b), corresponding to an increase in [Ca2+]i from ∼70 nMat rest to ∼130 nMduring sustained depolarization.
Basal characteristics of the KCl-induced Ca2+ response
The Ca2+ response to high-K+ was strictly dependent on extracellular Ca2+, arguing in favor of Ca2+ entry via plasma membrane channels. The ∼60% inhibition of the response by LaCl3 is another indication of the involvement of Ca2+ channels, since La3+ and other trivalent metal ions are known to inhibit voltage-dependent as well as voltage-independent Ca2+ entry pathways (Beedle et al., 2002; Jung et al., 2002).
To rule out that the preparation contained functional nerve endings we tested the effect of TTX, an inhibitor of voltage-dependent Na+ channels, and phentolamine, a nonspecific α-adrenoceptor antagonist. As none of these substances had an effect on the response to 75 mM K+ (Figure 2b), we concluded that the activation of α-adrenergic receptors via local neurotransmitter release did not play a role in the responses.
Since mibefradil inhibits both vasoconstriction of mesenteric arterioles in vivo (Gustafsson et al., 2001) and the nifedipine-insensitive Ca2+ current in isolated mesenteric VSMC (Morita et al., 2002), we conclude that Ca2+ channels are expressed in the arteriolar smooth muscle. However, based on our Ca2+ response data, we cannot exclude that the endothelial cells might also express mibefradil-sensitive Ca2+ channels. The current view is that endothelial cells do not express voltage-dependent Ca2+ channels (Domenighetti et al., 1998; Nilius & Droogmans, 2001), although the opposite has also been suggested (Vinet & Vargas, 1999; Wu et al., 2003).
Channel pharmacology and inhibitor specificity
In rat mesenteric arterioles (<40 μm), a role for dihydropyridine-insensitive channels in both local and conducted vasomotor responses was suggested because 10 μM nifedipine or nimodipine had no effects, and because the L-type CaV1.2 α1-subunit was not expressed at the mRNA level (Gustafsson et al., 2001). Surprisingly, we found an effect of 1 μM nifedipine on the Ca2+ response to 75 mM K+. However, there was no effect of 500 nMcalciseptine on the Ca2+ response in mesenteric arterioles (Figure 4). The efficacy of 500 nMcalciseptine was confirmed using rat renal afferent arterioles known to express L-type channels (Salomonsson & Arendshorst, 1999; Loutzenhiser & Loutzenhiser, 2000). Together with our previous finding that L-type channel message is not expressed in mesenteric arterioles (Gustafsson et al., 2001), these data suggest that the observed inhibition by nifedipine is a nonspecific effect.
At present, we cannot explain the discrepancy between the dihydropyridine insensitivity of vasoconstriction in mesenteric arterioles in vivo (Gustafsson et al., 2001) and our in vitro data, but there are several possible explanations. A 40% inhibition of the Ca2+ increase in vivo by nifedipine could be insufficient to block vasoconstriction. Furthermore, we cannot directly compare the results obtained in vivo (37°C) with those obtained in vitro (room temp.), especially because the stimulation mode was different in the two studies: in the previous study vasoconstriction was induced by locally applied current pulses, whereas in the present study Ca2+ influx was measured during sustained depolarization using high KCl.
The P- and Q-type inhibitor ω-agatoxin IVA (10 nM) inhibited the KCl-induced Ca2+ increase in rabbit afferent arterioles (Hansen et al., 2000) and has low interaction with N- or L-type Ca2+ channels when applied in nanomolar concentrations (Mintz et al., 1992; McDonough et al., 2002). Since we observed no inhibition at 100 nM (Figure 4), it is safe to conclude that the P/Q-type channel is absent in rat mesenteric arterioles. This is in line with the RT–PCR, which did not show amplification of message for the two splice variants of CaV2.1 (α1A-subunit) corresponding to the P- or Q-type channel (Figure 6).
In nanomolar concentrations, the peptide neurotoxin ω-conotoxin MVIIA is a specific inhibitor of transiently expressed CaV2.2 (α1B) N-type channels (Feng et al., 2001; 2003). Another peptide neurotoxin, SNX-482, inhibited CaV2.3 channels (α1E, R-type) stably expressed in a mammalian cell line with an EC50 of 15–30 nM. At high concentration (500 nM), SNX-482 had no effects on L- or T-type currents, but some effect on N-type currents (Newcomb et al., 1998). Compared to the reported EC50 values, the concentrations of ω-conotoxin MVIIA (500 nM) or SNX-482 (100 nM) employed in the present study were high, and the lack of any effects (Figure 4) indicates that N- and R-type Ca2+ channels are not expressed in this microvessel.
The above results suggested that inhibitors of the T-type voltage-dependent Ca2+ channels might be effective. Mibefradil has been shown to block T-type channels with an IC50 of 100−300 nM(Mishra & Hermsmeyer, 1994; Martin et al., 2000; Hansen et al., 2001; Morita et al., 2002). In rat mesenteric small arterioles, mibefradil was indeed effective at all the concentrations tested when a special protocol designed for use-dependent drugs was applied (Figures 5a and b). In comparison with the IC50 values mentioned above, the use-dependent inhibition of 29% seen with 100 nMmibefradil (Figure 5b) indicates binding to a channel with T-type pharmacology.
Recent studies show use- or voltage-dependent block by mibefradil for both high- and low-voltage-activated Ca2+ channels (Bezprozvanny & Tsien, 1995; Arnoult et al., 1998; McDonough & Bean, 1998; Jimenez et al., 2000; Morita et al., 2002). We designed a stimulation protocol to be able to show use dependence of mibefradil. The concentration-dependent effect of mibefradil increased proportionally with the number of prior high-K+ stimulations and was maximal at the fourth consecutive stimulation (Figure 5a). The effect was independent of the length of the preincubation period prior to stimulation.
Pimozide is another drug with use-dependent effects (Arnoult et al., 1998). Cloned T-type channels were blocked with EC50 values around 30–50 nM(Santi et al., 2002), whereas native channels were blocked with EC50∼500 nM(Enyeart et al., 1993; Arnoult et al., 1998). In the present study, 500 nMpimozide inhibited 85% of the Ca2+ response to 75 mM K+ (Figure 5c and d). Pimozide was previously shown to block L-type channels with an EC50 of 75 nM(Enyeart et al., 1990) and N- and P-type Ca2+ channels with an EC50 in the micromolar range (Sah & Bean, 1994). Although pimozide is a potent blocker of L-type channels, these channels are not expressed in mesenteric arterioles, indicating that it blocked a channel with T-type pharmacology.
Identification of T-type subunits in mesenteric arterioles
In a previous study, we detected RT–PCR amplification of the T-type CaV3.1 and CaV3.2 α1-subunits in both mesenteric arteries and arterioles from the rat (Gustafsson et al., 2001), and in the present study CaV3.1 expression in mesenteric arterioles was confirmed (Figure 6). Owing to careful dissection of the arterioles used in RT–PCR experiments, it is unlikely that RNA of nonvascular origin, for example, from fibroblasts, have contaminated the RNA extracted. However, we cannot rule out that endothelial RNA might have contributed to the RT–PCR products, and it is therefore a possibility that CaV3.1 may be expressed in endothelial cells.
The T-type channel has not been purified and the exact subunit composition is therefore unknown (Hofmann et al., 1999; Perez-Reyes, 2003). It has been shown that α1–β interactions within the high-voltage-activated Ca2+ channel complex are promiscuous with respect to subunit isoforms (Walker & De Waard, 1998), suggesting that several α1–β combinations would be possible in mesenteric arterioles expressing two T-type α1-subunits (CaV3.1 and CaV3.2) and an unknown diversity of β-subunits. In rat mesenteric arteries, Ca2+ channels with T-type pharmacology display higher 50% activation and inactivation voltages (V0.5) compared to classical T-type channels (Morita et al., 2002). We were therefore interested in testing whether known β-subunits were coexpressed with the α1-subunits and, thus, could modify channel properties. However, our RT–PCR showed no amplification of the neuronal β1b, cardiac β2, or neuronal/smooth muscle β3 subunits (Figure 6). Mapping the expression of known β- or α2δ-subunits would give a better understanding of possible subunit interactions in mesenteric arterioles and is an interesting subject for future investigation.
Biophysical characteristics of non-L-type channels in mesenteric resistance vessels
In VSMC from rat or guinea-pig mesenteric artery, nifedipine-insensitive Ca2+ currents with T-type pharmacology activated at −50 to −40 mV and peaked at −10 to 0 mV (Morita et al., 1999; 2002). In contrast, the three cloned T-type α1-subunits (CaV3.1, CaV3.2, and CaV3.3) all have an activation threshold at −70 mV and peak of I–V curve at −30 mV at physiological [Ca2+] (Perez-Reyes, 2003). It has therefore been proposed that the high-voltage-activated nifedipine-insensitive Ca2+ channels in mesenteric arteries, although they display T-type pharmacology, do not belong to the T-type channels. However, native T-type currents in some peripheral tissues, including smooth muscle, display activation threshold and peak current at −50 and −10 mV, respectively (Perez-Reyes, 2003), and with our demonstration of channels with T-type pharmacology and mRNA expression profile (Gustafsson et al., 2001); we think that it will be important to investigate the expression profile of auxiliary subunits capable of modifying the function of Ca2+ channel α1-subunits.
Both mesenteric nifedipine-insensitive Ca2+ channels (Morita et al., 2002) and cloned CaV3.1 and CaV3.2 T-type channels (Perez-Reyes, 2003) display fast inactivation kinetics (τinact=10−30 ms). This raises the question as to how mesenteric Ca2+ channels are able to sustain prolonged Ca2+ influx during high K+. One possibility is through small window currents in the voltage range where steady-state activation and inactivation curves overlap (McDonald et al., 1994; Perez-Reyes, 2003). It has been reported that a T-type window current was responsible for a physiological, steady-state Ca2+ entry in human myoblasts (Bijlenga et al., 2000), and window currents in the Vm range from −50 to −20 mV were suggested to account for high-K+-induced, nifedipine-insensitive Ca2+ entry and vasoconstriction in rat mesenteric artery (Morita et al., 1999; 2002; Itonaga et al., 2002; Inoue & Mori, 2003). In pressurized small arterioles (hamster cheek pouch) resting Vm measured in vivo was −35 mV (Welsh & Segal, 1998). Under physiological pressure, it is therefore likely that a window type Ca2+ current in mesenteric arterioles is either present or can be activated by small changes in Vm.
Different effects of high KCl in vitro and in vivo
The dihydropyridine-insensitive voltage-dependent Ca2+ channels involved in conducted vasoconstriction (Gustafsson et al., 2001) most likely correspond to the mibefradil-sensitive channels we characterize in the present study. However, while 75 mM K+ induced a sustained Ca2+ increase in vitro in the present study, superfusing the mesentery with 75 mM K+ did not induce vasoconstriction in vivo (Gustafsson et al., 2001). One explanation might be that in vivo the blood perfusion maintained [K+] in the vicinity of VSMC below 75 mM, whereby the induced depolarization was not in the membrane potential range where a sufficient window current can be reached. Another possibility is that KCl induced a counter regulatory effect in vivo. A recent report states that exposing the perfused mesenteric bed of the rat to 70 mM K+ leads to an intraluminal release of nitric oxide (NO) via a NO synthase-independent pathway (Mendizabal et al., 2000). Release of NO might counteract the Ca2+ increase in VSMC with the net effect being no diameter change.
In summary, a voltage-dependent Ca2+ channel with T-type pharmacology is responsible for depolarization-induced Ca2+ influx in mesenteric arterioles where the L-type Ca2+ channel is not expressed. The sustained Ca2+ entry through this channel is most likely due to a window current in a narrow membrane voltage range in depolarized VSMC.
Acknowledgments
Ms. Anni Salomonsson and Mr. Ian Godfrey are acknowledged for their skillful technical assistance. This project received financial support from The Danish Heart Association (Grant Nos. 22938 and 22977), the Danish Medical Research Council (Grant Nos. 22-01-0459 and 22-02-0263), and the Novo Nordisk Foundation (Grant No. 2003-11-28). Mibefradil was a kind gift from F. Hoffmann-La Roche Ltd, Basel, Switzerland.
Abbreviations
- EGTA
ethylene glycol-bis(2-aminoethylether)-N,N,N′,N′-tetraacetic acid
- NMDG
N-Methyl-D-glucosamine
- NO
nitric oxide
- PSS
physiological saline solution
- TTX
tetrodotoxin
- Vm
membrane potential
- VSMC
vascular smooth muscle cell
- Ø
lumen diameter
References
- ANDREASEN D., JENSEN B.L., HANSEN P.B., KWON T.H., NIELSEN S., SKØTT O. The α1G-subunit of a voltage-dependent Ca2+ channel is localized in rat distal nephron and collecting duct. Am. J. Physiol. Renal Physiol. 2000;279:F997–1005. doi: 10.1152/ajprenal.2000.279.6.F997. [DOI] [PubMed] [Google Scholar]
- ARNOULT C., VILLAZ M., FLORMAN H.M. Pharmacological properties of the T-type Ca2+ current of mouse spermatogenic cells. Mol. Pharmacol. 1998;53:1104–1111. [PubMed] [Google Scholar]
- BEEDLE A.M., HAMID J., ZAMPONI G.W. Inhibition of transiently expressed low- and high-voltage-activated calcium channels by trivalent metal cations. J. Membr. Biol. 2002;187:225–238. doi: 10.1007/s00232-001-0166-2. [DOI] [PubMed] [Google Scholar]
- BEZPROZVANNY I., TSIEN R.W. Voltage-dependent blockade of diverse types of voltage-gated Ca2+ channels expressed in Xenopus oocytes by the Ca2+ channel antagonist mibefradil (Ro 40-5967) Mol. Pharmacol. 1995;48:540–549. [PubMed] [Google Scholar]
- BIJLENGA P., LIU J.H., ESPINOS E., HAENGGELI C.A., FISCHER-LOUGHEED J., BADER C.R., BERNHEIM L. T-type α1H Ca2+ channels are involved in Ca2+ signaling during terminal differentiation (fusion) of human myoblasts. Proc. Natl. Acad. Sci. U.S.A. 2000;97:7627–7632. doi: 10.1073/pnas.97.13.7627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOURINET E., SOONG T.W., SUTTON K., SLAYMAKER S., MATHEWS E., MONTEIL A., ZAMPONI G.W., NARGEOT J., SNUTCH T.P. Splicing of α1A subunit gene generates phenotypic variants of P- and Q-type calcium channels. Nat. Neurosci. 1999;2:407–415. doi: 10.1038/8070. [DOI] [PubMed] [Google Scholar]
- DE WEILLE J.R., SCHWEITZ H., MAES P., TARTAR A., LAZDUNSKI M. Calciseptine, a peptide isolated from black mamba venom, is a specific blocker of the L-type calcium channel. Proc. Natl. Acad. Sci. U.S.A. 1991;88:2437–2440. doi: 10.1073/pnas.88.6.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOMENIGHETTI A.A., BENY J.L., CHABAUD F., FRIEDEN M.An intercellular regenerative calcium wave in porcine coronary artery endothelial cells in primary culture J. Physiol. 1998513103–116.(Part 1) [DOI] [PMC free article] [PubMed] [Google Scholar]
- ENYEART J.J., DIRKSEN R.T., SHARMA V.K., WILLIFORD D.J., SHEU S.S. Antipsychotic pimozide is a potent Ca2+ channel blocker in heart. Mol. Pharmacol. 1990;37:752–757. [PubMed] [Google Scholar]
- ENYEART J.J., MLINAR B., ENYEART J.A. T-type Ca2+ channels are required for adrenocorticotropin-stimulated cortisol production by bovine adrenal zona fasciculata cells. Mol. Endocrinol. 1993;7:1031–1040. doi: 10.1210/mend.7.8.8232302. [DOI] [PubMed] [Google Scholar]
- FENG Z.P., DOERING C.J., WINKFEIN R.J., BEEDLE A.M., SPAFFORD J.D., ZAMPONI G.W. Determinants of inhibition of transiently expressed voltage-gated calcium channels by ω-conotoxins GVIA and MVIIA. J. Biol. Chem. 2003;278:20171–20178. doi: 10.1074/jbc.M300581200. [DOI] [PubMed] [Google Scholar]
- FENG Z.P., HAMID J., DOERING C., BOSEY G.M., SNUTCH T.P., ZAMPONI G.W. Residue Gly1326 of the N-type calcium channel α1B subunit controls reversibility of ω-conotoxin GVIA and MVIIA block. J. Biol. Chem. 2001;276:15728–15735. doi: 10.1074/jbc.M100406200. [DOI] [PubMed] [Google Scholar]
- GRYNKIEWICZ G., POENIE M., TSIEN R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- GUSTAFSSON F., ANDREASEN D., SALOMONSSON M., JENSEN B.L., HOLSTEIN-RATHLOU N. Conducted vasoconstriction in rat mesenteric arterioles: role for dihydropyridine-insensitive Ca2+ channels. Am. J. Physiol. Heart Circ. Physiol. 2001;280:H582–H590. doi: 10.1152/ajpheart.2001.280.2.H582. [DOI] [PubMed] [Google Scholar]
- HANSEN P.B., JENSEN B.L., ANDREASEN D., FRIIS U.G., SKØTT O. Vascular smooth muscle cells express the α1A subunit of a P-/Q- type voltage-dependent Ca2+ Channel, and it is functionally important in renal afferent arterioles. Circ. Res. 2000;87:896–902. doi: 10.1161/01.res.87.10.896. [DOI] [PubMed] [Google Scholar]
- HANSEN P.B., JENSEN B.L., ANDREASEN D., SKØTT O. Differential expression of T- and L-type voltage-dependent calcium channels in renal resistance vessels. Circ. Res. 2001;89:630–638. doi: 10.1161/hh1901.097126. [DOI] [PubMed] [Google Scholar]
- HOFMANN F., LACINOVA L., KLUGBAUER N. Voltage-dependent calcium channels: from structure to function. Rev. Physiol. Biochem. Pharmacol. 1999;139:33–87. doi: 10.1007/BFb0033648. [DOI] [PubMed] [Google Scholar]
- INOUE R., MORI Y. New target molecules in the drug control of blood pressure and circulation. Curr. Drug Targets Cardiovasc. Haematol. Disord. 2003;3:59–72. doi: 10.2174/1568006033337348. [DOI] [PubMed] [Google Scholar]
- ITONAGA Y., NAKAJIMA T., MORITA H., HANANO T., MIYAUCHI Y., ITO Y., INOUE R. Contribution of nifedipine-insensitive voltage-dependent Ca2+ channel to diameter regulation in rabbit mesenteric artery. Life Sci. 2002;72:487–500. doi: 10.1016/s0024-3205(02)02286-5. [DOI] [PubMed] [Google Scholar]
- JIMENEZ C., BOURINET E., LEURANGUER V., RICHARD S., SNUTCH T.P., NARGEOT J. Determinants of voltage-dependent inactivation affect mibefradil block of calcium channels. Neuropharmacology. 2000;39:1–10. doi: 10.1016/s0028-3908(99)00153-7. [DOI] [PubMed] [Google Scholar]
- JUNG S., STROTMANN R., SCHULTZ G., PLANT T.D. TRPC6 is a candidate channel involved in receptor-stimulated cation currents in A7r5 smooth muscle cells. Am. J. Physiol. Cell Physiol. 2002;282:C347–C359. doi: 10.1152/ajpcell.00283.2001. [DOI] [PubMed] [Google Scholar]
- LOUTZENHISER K., LOUTZENHISER R. Angiotensin II-induced Ca2+ influx in renal afferent and efferent arterioles: differing roles of voltage-gated and store-operated Ca2+ entry. Circ. Res. 2000;87:551–557. doi: 10.1161/01.res.87.7.551. [DOI] [PubMed] [Google Scholar]
- MARTIN R.L., LEE J.H., CRIBBS L.L., PEREZ-REYES E., HANCK D.A. Mibefradil block of cloned T-type calcium channels. J. Pharmacol. Exp. Ther. 2000;295:302–308. [PubMed] [Google Scholar]
- MCDONALD T.F., PELZER S., TRAUTWEIN W., PELZER D.J. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiol Rev. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- MCDONOUGH S.I., BEAN B.P. Mibefradil inhibition of T-type calcium channels in cerebellar purkinje neurons. Mol. Pharmacol. 1998;54:1080–1087. doi: 10.1124/mol.54.6.1080. [DOI] [PubMed] [Google Scholar]
- MCDONOUGH S.I., BOLAND L.M., MINTZ I.M., BEAN B.P. Interactions among toxins that inhibit N-type and P-type calcium channels. J. Gen. Physiol. 2002;119:313–328. doi: 10.1085/jgp.20028560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MENDIZABAL V.E., POBLETE I., LOMNICZI A., RETTORI V., HUIDOBRO-TORO J.P., ADLER-GRASCHINSKY E. Nitric oxide synthase-independent release of nitric oxide induced by KCl in the perfused mesenteric bed of the rat. Eur. J. Pharmacol. 2000;409:85–91. doi: 10.1016/s0014-2999(00)00789-5. [DOI] [PubMed] [Google Scholar]
- MINTZ I.M., VENEMA V.J., SWIDEREK K.M., LEE T.D., BEAN B.P., ADAMS M.E. P-type calcium channels blocked by the spider toxin ω-Aga-IVA. Nature. 1992;355:827–829. doi: 10.1038/355827a0. [DOI] [PubMed] [Google Scholar]
- MISHRA S.K., HERMSMEYER K. Selective inhibition of T-type Ca2+ channels by Ro 40-5967. Circ. Res. 1994;75:144–148. doi: 10.1161/01.res.75.1.144. [DOI] [PubMed] [Google Scholar]
- MORITA H., COUSINS H., ONOUE H., ITO Y., INOUE R. Predominant distribution of nifedipine-insensitive, high voltage-activated Ca2+ channels in the terminal mesenteric artery of guinea pig. Circ. Res. 1999;85:596–605. doi: 10.1161/01.res.85.7.596. [DOI] [PubMed] [Google Scholar]
- MORITA H., SHI J., ITO Y., INOUE R. T-channel-like pharmacological properties of high voltage-activated, nifedipine-insensitive Ca2+ currents in the rat terminal mesenteric artery. Br. J. Pharmacol. 2002;137:467–476. doi: 10.1038/sj.bjp.0704892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEWCOMB R., SZOKE B., PALMA A., WANG G., CHEN X., HOPKINS W., CONG R., MILLER J., URGE L., TARCZY-HORNOCH K., LOO J.A., DOOLEY D.J., NADASDI L., TSIEN R.W., LEMOS J., MILJANICH G. Selective peptide antagonist of the class E calcium channel from the venom of the tarantula Hysterocrates gigas. Biochemistry. 1998;37:15353–15362. doi: 10.1021/bi981255g. [DOI] [PubMed] [Google Scholar]
- NILIUS B., DROOGMANS G. Ion channels and their functional role in vascular endothelium. Physiol. Rev. 2001;81:1415–1459. doi: 10.1152/physrev.2001.81.4.1415. [DOI] [PubMed] [Google Scholar]
- PEREZ-REYES E. Molecular physiology of low-voltage-activated T-type calcium channels. Physiol. Rev. 2003;83:117–161. doi: 10.1152/physrev.00018.2002. [DOI] [PubMed] [Google Scholar]
- SAH D.W., BEAN B.P. Inhibition of P-type and N-type calcium channels by dopamine receptor antagonists. Mol. Pharmacol. 1994;45:84–92. [PubMed] [Google Scholar]
- SALOMONSSON M., ARENDSHORST W.J. Calcium recruitment in renal vasculature: NE effects on blood flow and cytosolic calcium concentration. Am. J. Physiol. 1999;276:F700–F710. doi: 10.1152/ajprenal.1999.276.5.F700. [DOI] [PubMed] [Google Scholar]
- SANTI C.M., CAYABYAB F.S., SUTTON K.G., MCRORY J.E., MEZEYOVA J., HAMMING K.S., PARKER D., STEA A., SNUTCH T.P. Differential inhibition of T-type calcium channels by neuroleptics. J. Neurosci. 2002;22:396–403. doi: 10.1523/JNEUROSCI.22-02-00396.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- VANBAVEL E., SOROP O., ANDREASEN D., PFAFFENDORF M., JENSEN B.L. Role of T-type calcium channels in myogenic tone of skeletal muscle resistance arteries. Am. J. Physiol. Heart Circ. Physiol. 2002;283:H2239–H2243. doi: 10.1152/ajpheart.00531.2002. [DOI] [PubMed] [Google Scholar]
- VINET R., VARGAS F.F. L- and T-type voltage-gated Ca2+ currents in adrenal medulla endothelial cells. Am. J. Physiol. 1999;276:H1313–H1322. doi: 10.1152/ajpheart.1999.276.4.H1313. [DOI] [PubMed] [Google Scholar]
- WALKER D., DE WAARD M. Subunit interaction sites in voltage-dependent Ca2+ channels: role in channel function. Trends Neurosci. 1998;21:148–154. doi: 10.1016/s0166-2236(97)01200-9. [DOI] [PubMed] [Google Scholar]
- WELSH D.G., SEGAL S.S. Endothelial and smooth muscle cell conduction in arterioles controlling blood flow. Am. J. Physiol. 1998;274:H178–H186. doi: 10.1152/ajpheart.1998.274.1.H178. [DOI] [PubMed] [Google Scholar]
- WU S., HAYNES J, Jr, TAYLOR J.T., OBIAKO B.O., STUBBS J.R., LI M., STEVENS T. CaV3.1 (α1G) T-type Ca2+ channels mediate vaso-occlusion of sickled erythrocytes in lung microcirculation. Circ. Res. 2003;93:346–353. doi: 10.1161/01.RES.0000087148.75363.8F. [DOI] [PubMed] [Google Scholar]