

Abstract
Ciguatoxins (CTXs) are known to bind to receptor site 5 of the voltage-dependent Na channel, but the toxin's physiological effects are poorly understood. In this study, we investigated the effects of a ciguatoxin congener (CTX3C) on three different Na-channel isoforms, rNav1.2, rNav1.4, and rNav1.5, which were transiently expressed in HEK293 cells.
The toxin (1.0 μmol l−1) shifted the activation potential (V1/2 of activation curve) in the negative direction by 4–9 mV and increased the slope factor (k) from 8 mV to between 9 and 12 mV (indicative of decreased steepness of the activation curve), thereby resulting in a hyperpolarizing shift of the threshold potential by 30 mV for all Na channel isoforms.
The toxin (1.0 μmol l−1) significantly accelerated the time-to-peak current from 0.62 to 0.52 ms in isoform rNav1.2. Higher doses of the toxin (3–10 μmol l−1) additionally decreased time-to-peak current in rNav1.4 and rNav1.5.
A toxin effect on decay of INa at −20 mV was either absent or marginal even at relatively high doses of CTX3C.
The toxin (1 μmol l−1) shifted the inactivation potential (V1/2 of inactivation curve) in the negative direction by 15–18 mV in all isoforms.
INa maxima of the I–V curve (at −20 mV) were suppressed by application of 1.0 μmol l−1 CTX3C to a similar extent (80–85% of the control) in all the three isoforms. Higher doses of CTX3C up to 10 μmol l−1 further suppressed INa to 61–72% of the control.
Recovery from slow inactivation induced by a depolarizing prepulse of intermediate duration (500 ms) was dramatically delayed in the presence of 1.0 μmol l−1 CTX3C, as time constants describing the monoexponential recovery were increased from 38±8 to 588±151 ms (n=5), 53±6 to 338±85 ms (n=4), and 23±3 to 232±117 ms (n=3) in rNav1.2, rNav1.4, and rNav1.5, respectively.
CTX3C exerted multimodal effects on sodium channels, with simultaneous stimulatory and inhibitory aspects, probably due to the large molecular size (3 nm in length) and lipophilicity of this membrane-spanning toxin.
Keywords: Ciguatoxin, brevetoxin, Nav1.2, Nav1.4, Nav1.5, slow inactivation
Introduction
The ciguatoxins (CTXs) are polycyclic ethers, synthesized by the dinoflagellate Gambierdiscus toxicus (Bagnis et al., 1980; Yasumoto, 2001). These toxins, which can accumulate in tropical fish, are the causative agents in sea-food poisoning (Watters, 1995). CTX3C (Figure 1) can be isolated as a natural product from cultured G. toxicus (Satake et al., 1993). Brevetoxins (PbTxs), another member of the polycylic ethers, occur in a different species of dinoflagellate, Ptychodiscus brevis, responsible for toxic red tides (Davis, 1947). The structural formula for one of the PbTxs, PbTx-3, is given in Figure 1. Both toxins were reported to bind to receptor site 5 of the voltage-dependent sodium channel, and exert two biological actions: (1) shifting of the activation potential in the hyperpolarizing direction (Huang et al., 1984; Sheridan & Adler, 1989; Jeglitsch et al., 1998; Purkerson et al., 1999; Strachan et al., 1999), and (2) inhibition of sodium inactivation, leading to persistent channel activation (Huang et al., 1984; Jeglitsch et al., 1998). Photoaffinity labeling of purified Na channel protein by p-AB [3H]PbTx-3 located site 5 within the interface of domain I segment 6 and domain IV segment 5 of sodium channels (Trainer et al., 1991; 1994). CTX potently displaced PbTx binding, indicating that PbTx and CTX share a common binding site (Lombet et al., 1987). These pharmacologic data support the designation of CTX as a site-5 toxin. Unfortunately, the scarcity of these toxins hampers quantitative evaluation of their modulatory effects on the target protein and a more precise determination of the toxin-binding sites. In this regard, we (Hirama et al., 2001; Inoue et al., 2002) have succeeded in developing a methodology for synthesis of the ciguatoxin congener CTX3C (see Figure 1) in sufficient quantities for use in patch-clamp studies. As an initial step toward the determination of the binding sites for CTX3C, we have investigated herein possible differences in the CTX3C sensitivity of the neuronal, skeletal-muscle, and cardiac isoforms (rNav1.2, rNav 1.4, and rNav 1.5) of the voltage-dependent sodium channel. We had reason to believe that these toxins are more potent in modifying neuronal Na channels than cardiac Na channels, because the positive inotropic action of low doses of the CTXs and PbTxs was primarily due to catecholamine release from the sympathetic endings innervating cardiac muscle, and higher doses of CTXs were required to produce a direct action on cardiac Na channels (Lewis & Endean, 1986; Seino et al., 1988; Lewis et al., 1992; Lewis & Hoy, 1993; Sauviat et al., 2002). The finding of a significant sensitivity difference among the Na channel isoforms could lead to a narrowing of possible channel-binding sites for CTX3C. In the course of these studies, we uncovered novel kinetic effects of CTX3C on the channels, indicating the enhancement of slow inactivation and acceleration of channel activation.
Figure 1.

Structure of CTX3C, a representative congener of the ciguatoxins and one of the B-type brevetoxins, PbTx-3.
Methods
Electrophysiological recording
Macroscopic INa from transfected cells (see below) was measured using the whole-cell variation of the patch-clamp method. The bath solution contained (in mmol l−1): 70 NaCl, 67 N-methyl-D-glucamine, 1 CaCl2, 1.5 MgCl2, 10 glucose, and 5 HEPES (pH 7.4). The pipette solution contained (in mmol l−1): 70 CsF, 60 CsCl, 12 NaF, 5 ethylene-bis (oxonitrilo) tetraacetic acid, and 5 HEPES (pH 7.4). Whole-cell patch pipettes with a resistance of less than 2 MΩ were used to achieve optimum voltage control. Whole-cell currents (filtered at 10 kHz) were recorded using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, U.S.A.), and more than 80% of the series resistance was compensated to minimize voltage errors. Recordings were started 10 min after establishing a whole-cell recording configuration. Whole-cell membrane currents were digitized at a sampling rate of 50–100 kHz with a 12-bit analogue-to-digital converter (DigiData 1321A interface; Axon Instruments), controlled by pClamp software (Version 8; Axon Instruments). To eliminate the effect of steady-state inactivation during experiments, the holding potential was set to −140 mV. INa was determined by subtraction of linear capacitative and leak current with the P/4 protocol (Armstrong & Bezanilla, 1974) in all experiments, except for those undertaken to measure the voltage dependency of steady-state inactivation and the time course of recovery from slow inactivation.
All experiments were conducted at room temperature (22–25°C). Data are presented as mean±s.e. along with the number of observations (n), unless otherwise stated. Statistically significant differences between group means were determined by Student's t-test for paired or unpaired observations, as appropriate. The criterion for statistical significance was P<0.05.
Perfusion system
The stock solution of CTX3C (1 mmol l−1) in dimethylsulfoxide (DMSO) was diluted with bath solution to a final concentration of 0.1–10 μmol l−1. CTX3C solution in a cuvette was siphoned into the bath under a pressure head of 30 cm H2O. Siphon and vacuum tubing were connected to the bath chamber in a Y-configuration. A magnetic valve in the vacuum line was momentarily opened to prime the siphon. The resulting fan-shaped liquid stream was visualized using a test dye. Cells under study were positioned in the center of the stream. Flow of CTX3C solution could be halted as desired by occluding the flow path. Bath solution was continuously supplied at a rate of 2 ml min−1 through a separate inlet.
Transient transfection and cell culture
cDNA clones of the α-subunit of rat heart (rNav1.5), rat brain (rNav1.2), or rat skeletal muscle (rNav1.4) Na+ channels were inserted into a mammalian expression vector pCI-neo (Promega, Madison, WI, U.S.A.) or pcDNA3.1 (Invitrogen, Carlsbad, CA, U.S.A.), and were then transiently co-transfected with CD8 cDNA into HEK 293 cells using the Effectine reagent (Qiagen, Hilden, Germany). The cells were grown to 50% confluence in DMEM (Invitrogen), containing 10% fetal bovine serum (BioWhittaker, Walkersville, MD, U.S.A.), 30 units ml−1 penicillin G (Invitrogen) and 30 μg ml−1 streptomycin (Invitrogen), under a humidified atmosphere of 5% CO2 and 95% air, at 37°C. The transfected cells were used in electrophysiological experiments for up to 4 post-transfection days. Transfection-positive cells were identified by immunobeads (CD8-Dynabeads; Dynal, Oslo, Norway).
Results
Effects of external application of CTX3C on sodium channels
External application of 1.0 μmol l−1 CTX3C affected three measures of channel function, namely, the peak value of INa, the current–voltage (I–V) relationship, and the rate of channel activation. We normally allowed a 10–15 min equilibration period after membrane rupture before applying the toxin. During this period, peak INa increased to a steady-state value (most notably at clamp potentials between −30 and −10 mV, depending on the sodium-channel isoform). Figure 2 shows, in HEK 293 cells expressing rNaV1.2, the effect of CTX3C on peak INa at a series of transmembrane potentials between −120 and +50 mV. CTX3C was applied at minute 14.7 (0.3 μmol l−1) and 21.0 (1.0 μmol l−1) following membrane rupture (zero time). The I–V curves in Figure 2a were constructed at selected time points before and during application of CTX3C at 0.3 and 1.0 μmol l−1. Application of 0.3 μmol l−1 CTX3C had no effect on the current, whereas 1.0 μmol l−1 CTX3C produced a hyperpolarizing shift in the I–V curve and suppressed the I–V maximum (Figure 2a). CTX3C (1.0 μmol l−1) had various effects on INa, depending on the membrane potential. The INa records in Figure 2b demonstrate that CTX3C (1.0 μmol l−1) induces a long-lasting, non-inactivating sodium current at potentials between −100 and −80 mV, enhances INa at potentials between −80 and −30 mV, and suppresses INa at potentials more positive than −30 mV.
Figure 2.

Effects of external application of CTX3C on rNav1.2. (a) I–V curves in the absence (closed squares) and presence of CTX3C at a concentration of 0.3 μmol l−1 (open circles) and 1.0 μmol l−1 (open triangles). Note the hyperpolarizing shift of the I–V curve produced by 1.0 μmol l−1 CTX3C. (b) Current records at nine different test potentials (−120 to + 50 mV). The top panel indicates the command potential (holding potential set to −140 mV). Superimposed current records at each potential correspond to control (no drug; solid line), 0.3 μmol l−1 CTX3C (broken line), and 1.0 μmol l−1 CTX3C (dotted line).
Effects on activation process
As shown in the previous section, CTX3C induced Na currents at very negative transmembrane potentials. This caused a prominent shift in the activation potential in the direction of hyperpolarization and a reduction in the voltage dependency of activation in all the three isoforms (left column in Figure 3). Table 1a and b summarize, for isoforms rNav1.2, rNav1.5, and rNav1.4, values of half-activation voltage (Vact1/2) and slope factor (kact) obtained by fitting the Boltzmann equation to the activation curve. Vact1/2 in the control varied depending upon the isoform, rNav1.5, giving the most hyperpolarized value (−48.8±1.5 mV, n=6) and rNav1.2, the most depolarized value (−25.8±1.2 mV, n=8). CTX3C at 1.0 μmol l−1 caused a 4–9 mV hyperpolarizing shift in Vact1/2, most prominently in isoform rNav1.2. Simultaneously, the toxin (1.0 μmol l−1) reduced the voltage dependency of activation, as indicated by an increase in the slope factor from 8 to 9–12 mV, again most prominently in rNav1.2.
Figure 3.

Activation and steady-state inactivation curves for the three sodium channel isoforms in the absence (solid lines, closed squares) and presence of 1.0 μmol l−1 CTX3C (dotted lines, open circles). Boltzmann functions (smooth lines) were fitted to each set of activation (1–1/[1+exp{(V–Vact1/2)/kact}]) or inactivation (1/[1+exp{(V–Vinact1/2)/kinact}]) data (given as mean±s.e.m.). Activation curves for rNav1.2 (n=8), rNav1.4 (n=6), and rNav1.5 (n=6) were calculated from the peak INa value obtained at each membrane potential divided by the driving force (panels a, c, and e). Vact1/2 values for rNav1.2, rNav1.4, and rNav1.5 were −26, −31, and −49 mV in control and −35, −37, and −53 mV in the presence of 1.0 μmol l−1 CTX3C; slope factors (kact) for rNav1.2, rNav1.4, and rNav1.5 were 8.5, 8.8, and 9.3 mV in control and 12.4, 11.6, and 12.3 mV in 1.0 μmol l−1 CTX3C. These values are somewhat different from those in Table 1, since the latter were obtained by an alternate procedure: the parameters for each cell were initially acquired by fitting I–V data from individual cells to the Boltzmann equation, and then mean values for the parameters were computed. Steady-state inactivation curves for rNav1.2 (n=8), rNav1.4 (n=6), and rNav1.5 (n=5) were obtained by a double-pulse protocol (panels b, d, and f). INa evoked with a test pulse to −20 mV was preconditioned by a 500-ms clamp step to a variable membrane potential (−160 to −20 mV in 20-mV increments) and normalized by the unconditioned current elicited with a test pulse from the holding potential (−140 mV) (see the top panel of Figure 4). Vinact1/2 values for rNav1.2, rNav1.4, and rNav1.5 were −70, −70, and −96 mV in control, and −85, −86, and −112 mV in the presence of 1.0 μmol l−1 CTX3C. Slope factors (kinact) for rNav1.2, rNav1.4, and rNav1.5 were 9.2, 12.6, and 7.8 mV in control and 6.6, 6.6, and 9.3 mV in the presence of 1.0 μmol l−1 CTX3C. A minor inconsistency with the values reported in Table 4 is due to a procedural difference (see preceding paragraph) in calculating the values.
Table 1.
Effect of CTX on the parameters of activation (Vact1/2 and kact) in the three Na channel isoforms studied
| n | Control | 1 | 3 | 10 | |
|---|---|---|---|---|---|
| (a) Vact1/2 (mV) under control conditions (no CTX) and at the indicated concentrations of CTX (1, 3, or 10 μmol l−1) | |||||
| rNav1.2 | 8 | −25.8±1.2 | −34.5±1.6** | ||
| 3 | −20.5±4.6 | −33.7±4.2* | |||
| 3 | −30.7±2.5 | −38.1±2.3* | |||
| rNav1.4 | 6 | −31.8±2.4 | −36.3±2.9* | ||
| 3 | −26.8±1.9 | −31.1±2.3* | |||
| 3 | −26.4±5.7 | −36.2±4.2 | |||
| rNav1.5 | 6 | −48.8±1.5 | −53.9±1.8** | ||
| 3 | −47.4±1.3 | −57.0±1.4** | |||
| 3 | −51.0±3.6 | −62.9±5.7 | |||
| (b) kact (mV) under control conditions (no CTX) and at the indicated concentrations of CTX (μmol l−1) | |||||
| rNav1.2 | 8 | 8.3±0.3 | 12.0±0.7** | ||
| 3 | 10.3±0.8 | 13.6±0.5 | |||
| 3 | 9.9±0.8 | 16.9±1.5* | |||
| rNav1.4 | 6 | 8.1±0.6 | 9.2±0.6 | ||
| 3 | 9.3±0.7 | 13.5±0.3* | |||
| 3 | 9.4±0.7 | 13.0±0.7 | |||
| rNav1.5 | 6 | 8.9±0.5 | 11.4±0.5** | ||
| 3 | 9.1±0.7 | 14.0±0.5** | |||
| 3 | 10.4±0.6 | 13.8±0.7 | |||
CTX shifted V1/2 of the activation curve (Vact1/2) in the negative direction and increased the slope factor (kact), resulting in a marked hyperpolarizing shift of the threshold potential by 30 mV in all Na Channel isoforms.
P<0.05 vs control,
P<0.01 vs control.
Acceleration of activation was manifested in two other respects, changes in time-to-peak INa and enhancement of INa by conditioning prepulses. In the current record at −20 mV displayed in Figure 2b, application of the toxin decreased the time-to-peak INa from 0.90 to 0.48 ms. Table 2 summarizes the mean data for the three isoforms. At a toxin concentration of 1.0 μmol l−1, this measure of channel activation gave a statistically significant (P<0.05) result only in isoform rNav1.2; time-to-peak INa at −20 mV was shortened by 0.1 ms. Higher toxin concentrations shortened time-to-peak INa in the other two isoforms as well.
Table 2.
Effect of CTX on activation kinetics of INa as measured by time-to-peak INa
| n | Control | 1 | 3 | 10 | |
|---|---|---|---|---|---|
| rNav1.2 | 7 | 0.62±0.06 | 0.52±0.05* | ||
| 3 | 0.61±0.08 | 0.53±0.10* | |||
| 3 | 0.41±0.02 | 0.35±0.02* | |||
| rNav1.4 | 6 | 0.56±0.04 | 0.55±0.04 | ||
| 3 | 0.39±0.04 | 0.33±0.04** | |||
| 3 | 0.49±0.07 | 0.41±0.07 | |||
| rNav1.5 | 6 | 0.71±0.09 | 0.71±0.07 | ||
| 3 | 0.46±0.02 | 0.39±0.01* | |||
| 3 | 0.51±0.05 | 0.45±0.03 |
Time-to-peak INa (ms) is given for the control (no CTX) and the indicated concentrations of CTX (μmol l−1). CTX accelerated time-to-peak INa most markedly in isoform rNav 1.2, that is, by 0.1 ms.
P<0.05 vs control,
P<0.01 vs control.
In the absence of CTX, conditioning prepulses suppressed INa. Pulse pairs, consisting of a 500-ms prepulse plus a test pulse to −20 mV, were delivered at 5-s intervals from the holding potential (−140 mV); the prepulse potential was varied between −160 and −20 mV in 20-mV steps. As prepulses became more positive, test INa was progressively suppressed, essentially following the steady-state inactivation (h∞) curve that describes the voltage dependency of fast inactivation. By contrast, with 1.0 μmol l−1 CTX3C, the −120-mV conditioning prepulse produced an anomalous increase in the amplitude of INa, accompanied by acceleration in time-to-peak INa (horizontal arrow in Figure 4). This anomalous effect of conditioning can be measured as the ratio of test INa amplitudes following prepulses to −160 or −120 mV (INa-120/INa-160). INa-120/INa-160 for rNav1.2 in 1.0 μmol l−1 CTX3C was 1.09±0.02 (n=8), that is, the greatest value among the three isoforms (Table 3). Increasing the CTX3C concentration to 10 μmol l−1 resulted in comparable or greater values of the amplitude ratio in rNav1.4 (1.14±0.1, n=3) than in rNav1.2 at 1.0 μmol l−1. In contrast to rNav1.4, the ratio for rNav1.2 decreased with an increase in the concentration of CTX3C above 1.0 μmol l−1, probably due to an augmented inhibitory effect of the toxin on INa amplitude. As for rNav1.5, a concomitant shift in the h∞ curve nullified the activating effect.
Figure 4.

Stimulatory effects upon INa of preconditioning pulses that accelerate sodium-channel activation. Representative data from rNav1.2 are shown. In the presence of 1.0 μmol l−1 CTX3C, the preconditioning pulse to −120 mV did not open the channels (no inward current observable); yet INa evoked with a subsequent depolarization to −20 mV was clearly larger than that without preconditioning (see the current record in the third panel drawn with a dotted line and marked by the horizontal arrow). Simultaneously, time-to-peak INa was clearly shortened. The persistent current indicated by the arrow in the fourth panel was elicited with a conditioning pulse to −80 mV. The current at this potential shows incomplete inactivation. See text for further details.
Table 3.
Enhancement of CTX-modified INa by a subthreshold preconditioning depolarization
| n | Control | 1 | 3 | 10 | |
|---|---|---|---|---|---|
| rNav1.2 | 8 | 1.02±0.01 | 1.09±0.01** | ||
| 3 | 1.02±0.01 | 1.10±0.09 | |||
| 3 | 1.00±0.004 | 0.92±0.02* | |||
| rNav1.4 | 6 | 1.01±0.01 | 1.02±0.01 | ||
| 3 | 1.01±0.01 | 1.08±0.024 | |||
| 3 | 1.00±0.03 | 1.14±0.06 | |||
| rNav1.5 | 5 | 0.93±0.02 | 0.75±0.05* | ||
| 3 | 0.93±0.03 | 0.66±0.06* | |||
| 3 | 0.77±0.08 | 0.43±0.07* |
Cells were treated with CTX (1.0 μM) and conditioned with prepulses to −120 or −160 mv from a holding potential of −140 mV. Ratios (INa−120/INa−160) >1.0 indicate enhancement of INa by the conditioning depolarization. See text for further details.
P<0.05 vs control,
P<0.01 vs control.
Effects on inactivation process
At a constant voltage-clamp potential, decay of INa in the three isoforms consisted of two exponential components with time constants τfast and τslow. The time course of decay was hardly affected by CTX3C except in isoforms rNav1.4 and rNav1.5 at relatively high toxin concentrations. In isoform rNav1.4, 10 μmol l−1 CTX3C significantly (P<0.05) slowed τfast at −20 mV from 0.38±0.12 to 0.42±0.13 ms (n=3). The overall time course of INa decay could theoretically be affected by changes in the contribution of either component to the total current. Thus, the relative amplitude of the fast (%A1) and slow component (%A2) was estimated. CTX3C at a concentration of 1.0 μmol l−1 did not affect the relative amplitude in any channel isoform. In isoform rNav1.5, CTX3C at a concentration of 3.0 μmol l−1 decreased %A1 from a mean value of 85.3±2.2 to 81.4±1.9% and increased %A2 from 13.2±1.7 to 17.4±1.6% (n=3, P<0.05). Thus, 1.0 μmol l−1 CTX3C did not alter the time course of INa decay. Importantly, this indicates that the acceleration of time-to-peak INa at 1.0 μmol l−1 CTX3C can be attributed solely to a change in the characteristics of channel activation.
The steady-state voltage dependency of fast inactivation was evaluated by applying a 500-ms conditioning prepulse to various potentials from a holding potential of −140 mV, followed by a brief repriming interval (500 μs) and finally a test pulse to −20 mV. INa's elicited by the test pulses were inhibited with increasing depolarization of the conditioning pulse potential. The relationship between test INa and membrane potential in the presence and absence of CTX3C was fitted by the Boltzmann equation with two adjustable parameters, Vinact1/2 (half-inactivation voltage) and slope factor (kinact). The results are summarized in the right column of Figure 3 and Table 4. Control Vinact1/2 values for rNav1.2 and rNav1.4 fell into a narrow range around −70 mV, but the corresponding value for Nav1.5 was markedly hyperpolarized to −95 mV. Irrespective of the absolute control value, 1.0 μmol l−1 CTX3C shifted Vinact1/2 in the direction of hyperpolarization by 15–18 mV. The voltage dependency of inactivation became steeper both in rNav1.2 and rNav1.4, but not in rNav1.5. Higher doses of CTX3C produced still larger shifts toward negative potentials.
Table 4.
Effect of CTX on the parameters of steady-state inactivation (Vinact1/2 and kinact) for the three Na channel isoforms studied
| (a) Vinact1/2 (mV) under control conditions (no CTX) and at the indicated concentrations of CTX (μmol l−1) | |||||
|---|---|---|---|---|---|
| n | Control | 1 | 3 | 10 | |
| rNav1.2 | 8 | −69.8±2.7 | −86.9±2.3** | ||
| 3 | −64.9±4.0 | −86.1±2.8** | |||
| 3 | −69.4±0.5 | −92.8±0.3** | |||
| rNav1.4 | 6 | −70.2±2.8 | −88.2±1.6** | ||
| 3 | −66.4±0.5 | −90.6±1.7** | |||
| 3 | −65.4±1.8 | −85.2±2.6* | |||
| rNav1.5 | 5 | −95.5±1.4 | −110.1±2.7** | ||
| 3 | −94.7±1.1 | −113.6±2.7** | |||
| 3 | −108.8±2.1 | −121.2±1.8* | |||
| (b) kinact (mV) under control conditions (no CTX) and at the indicated concentrations of CTX (μmol l−1) | |||||
| rNav1.2 | 8 | 8.8±0.9 | 6.1±0.6** | ||
| 3 | 9.5±1.6 | 5.6±2.4 | |||
| 3 | 7.0±0.5 | 9.1±0.4 | |||
| rNav1.4 | 6 | 12.3±1.2 | 6.4±0.6* | ||
| 3 | 14.7±0.6 | 5.9±0.1** | |||
| 3 | 13.1±2.6 | 7.1±0.4 | |||
| rNav1.5 | 5 | 7.5±0.4 | 8.8±0.7 | ||
| 3 | 8.2±0.9 | 10.1±0.5 | |||
| 3 | 7.0±1.8 | 8.3±2.1* | |||
CTX shifted the V1/2 of inactivation curve (Vinact1/2) in the negative direction in all isoforms.
P<0.05 vs control,
P<0.01 vs control.
Effects on amplitude of INa
Owing to the influence of CTX3C on the activation process, the threshold potential for INa was dramatically shifted to hyperpolarized potentials by ∼30 mV in all three isoforms (see I–V curves in Figure 2a and activation curves in Figure 3). Simultaneously, at membrane potentials more negative than −30 mV, CTX3C produced stimulatory effects, leading to an increase in the amplitude of INa. By contrast, at membrane potentials positive to −30 mV, CTX3C produced an inhibitory effect. The inhibitory effect of CTX3C was evaluated as percent of control INa amplitude at −20 mV for each of the three isoforms (Table 5). CTX3C (1.0 μmol l−1) suppressed INa's to 83.9±5.0 (n=8), 84.5±2.4 (n=5), and 79.3±2.5% (n=6) of control in isoforms rNav1.2, rNav1.4, and rNav1.5, respectively. Increasing the concentration of CTX3C to 3.0 μmol l−1 further suppressed INa to 74.3±3.5 (n=3, P<0.05) and 65.2±5.3% (n=3, P<0.05) of control in isoforms rNav1.4 and rNav1.5.
Table 5.
Inhibition of peak INa at−20 mV as a function of CTX concentration
| 1 | 3 | 10 | ||
|---|---|---|---|---|
| rNav1.2 | 83.9±5.0 (n=8) | 72.0±1.1 (n=3) | 72.4±1.6 (n=3) | |
| ||||
| ||||
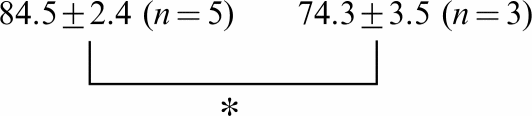
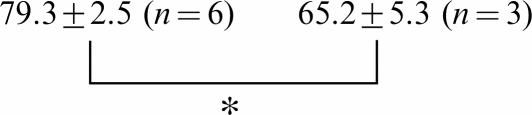
Amplitude of INa at −20 mV is given as a percentage of the control value(zero CTX) for the indicated concentrations of CTX (μmol l−1).
Significant difference between 1 and 3 μmol l−1 CTX in isoforms rNav 1.4 and rNav 1.5 (P<0.05).
CTX3C slows recovery from slow inactivation
Depolarization induces sodium inactivation that prevents the immediate re-opening of Na channels, and the channels normally require several milliseconds to fully recover their excitability. For example, in the experiments with isoform rNav1.2 shown in Figure 5, following a 10-ms depolarization to 0 mV, INa recovered with a time constant of 4.2 ms irrespective of the presence of 1.0 μmol l−1 CTX3C, and full recovery of INa was attained within 30 ms. In the absence of toxin, prolongation of the conditioning depolarization to 500 ms substantially slowed the recovery time constant to 32 ms (Figure 5). This behavior may derive from slow inactivation, as reported elsewhere (Furue et al., 1998; Vilin & Ruben, 2001). CTX3C at a concentration of 0.3 μmol l−1 did not affect the recovery time constant following a 500-ms depolarizing prepulse to 0 mV (Figure 5). However, a further increase in the concentration of CTX3C to 1.0 μmol l−1 dramatically slowed the recovery time constant to 529 ms. In the presence of CTX3C (1.0 μmol l−1), the longer conditioning depolarization (500 ms) also gave rise to a prolonged INa tail, which decayed biexponentially with time constants of 4.7 and 50.4 ms (data not shown). Thus, INa during the test depolarization was evoked on top of the immediately preceding tail current when the interval (Δt) between conditioning and test pulses was relatively brief (see lower right records in Figure 5). Table 6 summarizes the mean values of the recovery time constants obtained when sodium inactivation was induced by a 500-ms depolarizing prepulse to 0 mV. In the absence of CTX3C (‘control' in Table 6), isoforms rNav1.2, rNav1.4, and rNav1.5 recovered from inactivation with time constants of 45.2±6.5 (n=11), 64.5±5.1 (n=10), and 39.4±4.7 ms (n=9), respectively (pre-drug data were pooled from all cells tested with CTX3C). CTX3C (1.0 μmol l−1) increased the time constant for recovery from slow inactivation by 6- to 12-fold, but did not alter the sequence (Nav1.5<Nav1.2≈Nav1.4) of the three isoforms with respect to the magnitude of the recovery time constant.
Figure 5.

Comparative effects of CTX3C on time courses of INa recovery after brief or prolonged prepulses to 0 mV. Representative recovery data from rNav1.2 are shown. The pulse protocol used is displayed (inset). The duration of the conditioning depolarization (cp; first pulse in inset) was set to 10 or 500 ms. The absolute peak value of INa evoked by test depolarizations (second pulse in inset) is plotted against the interpulse interval in ms (Δt in inset). With conditioning pulses of 10 ms, recovery time course in 1.0 μmol l−1 CTX3C (inverted triangles) was identical to the control (diamonds). However, with lengthened conditioning pulses of 500 ms, application of 1.0 μmol l−1 CTX3C (triangles) markedly delayed recovery compared to the control (squares); 0.3 μmol l−1 CTX3C was without effect (circles). Sodium-current families (evoked by test depolarizations) are shown (right), depicting recovery of INa in the absence and presence of 1.0 μmol l−1 CTX (cp=500 ms). Note that CTX induced inward tail currents at the end of the preceding cp (see text for explanation).
Table 6.
Pronounced delay in recovery from slow inactivation in the presence of CTX
| n | Control | 1 | 3 | 10 | |
|---|---|---|---|---|---|
| rNav1.2 | 5 | 38.4±7.7 | 588.2±150.5* | ||
| 3 | 32.1±5.2 | 685.3±211.2* | |||
| 3 | 65.6±12.8 | 585.8±300.6 | |||
| rNav1.4 | 4 | 52.6±5.7 | 338.2±84.7* | ||
| 3 | 66.6±7.9 | 383.0±0.1** | |||
| 3 | 78.5±8.7 | 649.2±159.8 | |||
| rNav1.5 | 3 | 22.7±3.0 | 231.5±116.5 | ||
| 3 | 42.2±3.5 | 150.5±17.1* | |||
| 3 | 41.5±12.0 | 165.7±47.1 |
Time constants (ms) for recovery from slow inactivation are given for the control (no CTX) and the indicated concentrations of CTX (μmol l−1). CTX caused a marked delay in recovery from activation induced by a depolarization of intermediate duration (500 ms) to 0 mV.
P<0.05 vs control,
P<0.01 vs control.
CTX3C markedly affected the development of slow inactivation (Figure 6). The protocol employed to induce slow inactivation consisted of a conditioning prepulse to 0 mV with a variable (10–10,000 ms) duration, followed by a fixed repriming interval (500 ms) at the holding potential and finally a test pulse to −20 mV. The repriming interval chosen allows unmodified sodium channels to recover from inactivation (see Figure 5). Thus, this protocol selectively monitors CTX-modified INa. In the absence of CTX3C, prepulses up to 1000 ms did not suppress the test INa's, as expected; a 10,000-ms prepulse only slightly diminished the test INa (Figure 6). In the presence of 1.0 μmol l−1 CTX3C, however, the test INa was markedly suppressed as the duration of the conditioning prepulse was increased (40–10,000 ms). Slow inactivation, under these conditions, developed along a double-exponential time course with best-fitting time constants of 153 and 1017 ms (Figure 6).
Figure 6.

Development of slow inactivation in the absence or presence of CTX3C. Representative data from rNav1.2 are shown. The inset illustrates the pulse protocol employed. Conditioning depolarization (first pulse in inset) was variable in duration (10–10,000 ms). The interval between conditioning and test pulse (second pulse in inset) was fixed at 500 ms. Development of slow inactivation was monitored as the amplitude of test INa at −20 mV vs conditioning pulse duration. The presence of 1.0 μmol l−1 CTX3C markedly accelerated the development of slow inactivation. A 1000-ms conditioning depolarization was without effect in the absence of CTX3C, but elicited 80% of the maximally attainable slow inactivation in the presence of CTX3C.
Discussion
Extracellular application of CTX3C at a concentration of at least 1.0 μmol l−1 produced functional alterations in Na channels from three different sources, rat brain (rNav1.2), rat skeletal muscle (rNav1.4), and rat heart (rNav1.5), heterologously expressed in HEK 293 cells. The effect is multimodal, that is, stimulatory and at the same time inhibitory. The stimulatory action is mainly seen in the toxin's effects on sodium-channel activation. In this regard, CTX3C caused an alteration in the voltage dependency of activation. The threshold potential was shifted by 30 mV to the negative direction in all isoforms. Although shifts in Vact1/2 were no more than 10 mV, the simultaneous change in the slope factor made this effect significant. Thus, persistent non-inactivating sodium currents were observed at transmembrane potentials of around −80 mV. This can explain previous findings of spontaneous depolarization and oscillations of the membrane potential caused by these toxins (Wu et al., 1985; Hogg et al., 1998). The enhancing effect of CTX3C on activation could be also seen in the shortening of time-to-peak INa. A conditioning depolarization within a range of membrane potentials that neither opened sodium channels nor induced steady-state inactivation was found to effectively increase maximum peak INa. This anomalous effect of conditioning could represent the stabilizing effect of CTX3C on an electrically silent preopen state or an incompletely open substate of the modified sodium channel, as observed with PbTx-3 (Schreibmayer & Jeglitsch, 1992). Interference of CTX3C with inactivation could be also stimulatory, and such effects were previously reported (Huang et al., 1984; Jeglitsch et al., 1998). In this study, disruption of inactivation was not observed at −20 mV, except for a marginal effect detected only at a relatively high toxin concentration. Inhibitory actions of the toxin were reflected in the reduction of maximal amplitude of peak INa and promotion of slow inactivation. The former observation is confirmatory of previous findings with both CTX and PbTx (Huang et al., 1984; Strachan et al., 1999; Hidalgo et al., 2002), whereas the latter is a novel observation. In our experiments, only 500-ms conditioning prepulses substantially delayed full recovery from inactivation; recovery time constants fell below 100 ms in the absence of toxin, but were remarkably delayed to 0.5–1 s in the presence of 1.0 μmol l−1 CTX3C. Such an effect could be quite critical to cardiac function, since the long duration of cardiac action potentials (e.g., 200–500 ms) may well induce incomplete recovery from inactivation during the normal cycle of rhythmic beating.
The multimodal effect of CTX3C could arise from its large molecular dimensions (3 nm in length) and lipophilic nature; the toxin probably is membrane-spanning. In the case of PbTx, derivatives with structural modification of the K-ring retained the ability to bind to the sodium channel, but did not elicit Na+ channel openings (Purkerson-Parker et al., 2000). The increased availability of CTX3C and its congeners, owing to development of methodologies for artificial synthesis of the toxin as well as toxin derivatives with desired structural modifications, should facilitate our understanding of molecular mechanisms of Na channel gating.
Differences in sensitivity to CTX3C among sodium channel isoforms
Differences in affinity or sensitivity to either CTX3C or PbTx were previously observed by Dechraoui et al., in HEK 293 cells stably expressing Nav1.4 and Nav1.5 (Bottein Dechraoui & Ramsdell, 2003). They found that Type B brevetoxin (PbTx-3) has a reduced affinity for the sodium channel as well as a lower cytotoxicity in cells expressing Nav1.5 by comparison with those expressing Nav1.4. Effects of several CTXs have been indicated to be 10–100-fold more potent in neurons than in myocardium, in which the direct action of the toxins was assessed by monitoring contractile activity (Lewis & Hoy, 1993). Similar reports have appeared (Lewis & Endean 1986; Seino et al., 1988; Lewis et al., 1992; Sauviat et al., 2002), predicting a higher potency of either CTX or PbTx on Na channel activity in neurons as opposed to other tissues. However, a comparative study of CTXs on the activity of Na channels in a single expression system has not been reported. Our results indicated that the basic stimulatory and inhibitory actions of the toxin were similar in all the three isoforms, although there was a more marked shift in the activation curve and a more pronounced shortening of time-to-peak INa in isoform rNav1.2. Previously reported differences in CTX and PbTx action on neuronal and cardiac tissues may have been brought about by a number of indirect mechanisms, such as effects on the Na/Ca exchanger (Gaudry-Talarmain et al., 1996; Lewis et al., 2000; Sauviat et al., 2002).
Our strategy to search for the molecular site of CTX binding on the basis of differences among the three Na channel isoforms in their sensitivity to the toxin may require some refinement. The effect of 1.0 μmol l−1 CTX3C on time-to-peak INa was absent both in rNav1.4 and rNav1.5 but not in rNav1.2, thus suggesting rNav1.2 to be the most promising candidate to utilize for development of new information.
Inconsistency of effective dose with previously reported data
Polyether toxins including CTX3C have a high binding affinity for sodium channels such that these toxins inhibit [3H]-PbTx-3 binding with a Ki of less than 1.0 nmol l−1 (Dechraoui et al., 1999; Inoue et al., 2003). CTXs, including CTX3C, produced cytotoxicity, in cells pretreated with veratridine, with an ED50 of less than 0.1 nmol l−1, and PbTxs, which are less potent, had an ED50 of 5–15 nmol l−1 (Dechraoui et al., 1999). Apart from the binding properties and indirect effects of CTX and PbTx, a nanomolar range of toxin concentrations was also sufficient to provoke electrophysiological effects, such as spontaneous depolarization (Huang et al., 1984; Seino et al., 1988; Jeglitsch et al., 1998; Strachan et al., 1999; Hidalgo et al., 2002; Hogg et al., 2002; Sauviat et al., 2002). In our study, at least 1.0 μmol l−1 CTX3C was required to exert significant effects on sodium channel activity. In the only report discovered that refers to the use of higher concentrations of PbTx-3, a small shift of the I–V curve in the negative direction was exhibited at a PbTx-3 concentration of 1.2 μmol l−1, and 20 μmol l−1 PbTx-3 failed to change the mean open time of the channel (Sheridan & Adler 1989). Interestingly, the preparation studied was a cell line, that is, a neuroblastoma × glioma hybrid (NG108-15), rather than a tissue placed in primary cell culture, and was clearly different from cell types employed by others (Huang et al., 1984; Seino et al., 1988; Jeglitsch et al., 1998; Strachan et al., 1999; Hidalgo et al., 2002; Hogg et al., 2002; Sauviat et al., 2002). In our case, sodium channels were transiently expressed in HEK 293 cells. It is possible that the transduced sodium channels would have a higher sensitivity to the toxin in a unique conformation requiring the presence of other proteins that HEK-293 cells do not provide, as for example, sodium-channel β subunits. In this regard, co-expression of the sodium channel β1 subunit with the stably expressed rat brain α-subunit rBIIA (rNav1.2) increased the stimulatory effect of PbTx on the binding of [3H]-batrachotoxin to the sodium channel (Bonhaus et al., 1996). Alternatively, the toxin may have had limited access to its binding site, thereby substantially increasing the time required for the toxin to act. Accessibility of lipophilic toxins to their receptor sites on membrane-spanning proteins can be affected by several properties of the plasma membrane, including temperature, fluidity, and composition of lipids. The time of exposure to toxins in our perfusion system was limited to a few minutes, and the recording period after application of toxins was 10–20 min. A much higher sensitivity to the toxin might be observed if we incubated cells at lower toxin concentrations for a considerably longer period, as for example, 24 h.
Slow inactivation of the sodium channel and CTX
Slow inactivation in response to membrane depolarization is distinct from fast inactivation. Slow inactivation is produced only by a prolonged depolarization, on the order of tens of seconds, and has a much longer recovery time constant than fast inactivation. The molecular basis of slow inactivation is also distinct from that of fast inactivation. The IFM peptide motif in the linker between domains III and IV is responsible for fast inactivation (Vassilev et al., 1988; West et al., 1992), and this motif is considered to be the ball in the ‘ball and chain model' that occludes ion flux through the pore of the sodium channel. On the other hand, several hypotheses have been presented to explain slow inactivation, based on the following considerations: (1) rearrangement of the pore could induce slow inactivation by analogy with the known mechanism of C-type inactivation in potassium channels (Liu et al., 1996); (2) charged residues surrounding the IFM motif could affect slow inactivation (McCollum et al., 2003); (3) mid-region and C-terminal sequences in multiple domains of segment 6 (Wang & Wang, 1997; O'Reilly et al., 2001; Wang et al., 2003) could be responsible for altering slow-inactivation properties; (4) mutations of local-anesthetic binding sites modify slow inactivation (Bai et al., 2003); and (5) mutations associated with channelopathies (Vilin & Ruben, 2001) are known to affect slow inactivation. However, none of the above hypotheses is conclusive. It is likely that slow inactivation involves several domains of the channel protein acting cooperatively, so that a change in any channel region could affect slow inactivation.
CTX has been reported to accelerate recovery from inactivation induced with a 300-ms conditioning pulse to −10 mV in TTX-resistant Na channels of rat dorsal root ganglion (DRG) neurons (Strachan et al., 1999). However, there have been no reports of a toxin or drug that enhances slow inactivation, except for α-scorpion toxin that accelerated slow inactivation in human cardiac Na channels (hNav1.5) expressed in HEK-293 cells (Chen & Heinemann, 2001). In their report, α-scorpion toxin delayed recovery from slow inactivation by altering the relative amplitude of slow and fast components without changing the time constants. In addition, α-scorpion toxin inhibited fast inactivation, which may facilitate slow inactivation indirectly, since the removal of the IFM particle allows more complete slow inactivation to develop (Featherstone et al., 1996; Richmond et al., 1998). In our case, CTX3C increased the recovery time constant by more than 10-fold. Moreover, the duration of the conditioning pulses that we employed (500 ms) was rather brief compared to theirs (60 s). Thus, CTX3C actually facilitated a form of intermediate inactivation (IM) (Kambouris et al., 1998) that develops with shorter prepulses than those inducing slow inactivation (IS). In isoform rNav1.4, our time constant (45.6±7.7 ms; n=9) for recovery from IM induced by a 500-ms depolarizing pulse is comparable to their reported value (74±20 ms) using a 1-s prepulse (Kambouris et al., 1998). CTX3C at a concentration of 1 μmol l−1 increased our recovery time constant with respect to IM to 338±85 ms (n=3), which is comparable to their data for IS (in the absence of a drug treatment). Although we did not employ longer pulse duration to induce IS, the susceptibility of IM to CTX is more relevant to cardiac function, since a 500-ms pulse is similar to the duration of the cardiac action potential. Blocking of the action potential as a result of CTX-enhanced IM would cause mechanical failure of the heart.
As shown above, there are many sites on the sodium channel that affect slow inactivation. It will be useful to see if modification of any of these sites abolishes the enhancing effect of CTX3C on slow inactivation. Experimentation of this type in combination with systematic derivatization of CTX3C should permit a reasonable picture of the CTX-bound Na channel to emerge.
Pathophysiological significance of CTX effects on excitable cells
We showed in this study that CTX causes a sustained inward current at physiological resting potentials, which certainly depolarizes the membrane potential of both neuronal and cardiac cells. CTX elicits abnormal automaticity in neurons due in part to depolarization of the resting membrane; the brief duration of the nerve action potential is also contributory by preventing marked delays in channel recovery from developing in the presence of toxin. A newly discovered action of CTX, as reported herein, is its stabilizing effect on slow inactivation, a state of the sodium channel inducible by membrane depolarizations of intermediate duration (∼500 ms; see Figure 5). Cardiac sodium channels are susceptible to this aspect of CTX modification because of the long duration of the action potential in heart muscle. Enhanced slow inactivation leads to conduction block of cardiac tissue due to a decrease in action potential conduction velocity and an increase in the spatial extent of action potential refractoriness.
Acknowledgments
This work was supported by the SORST program from the Japan Science and Technology Agency (JST). We thank Dr Stephen M. Vogel (Department of Pharmacology, University of Illinois at Chicago, College of Medicine) for his critical reading of the manuscript.
Abbreviations
- CTX
ciguatoxin
- CTX3C
a ciguatoxin congener
- HEK
human embryonic kidney
- Nav
voltage-dependent sodium channel
- PbTx
brevetoxin
References
- ARMSTRONG C.M., BEZANILLA F. Charge movement associated with the opening and closing of the activation gates of the Na channels. J. Gen. Physiol. 1974;63:533–552. doi: 10.1085/jgp.63.5.533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAGNIS R., CHANTEAU S., CHUNGUE E., HURTEL J.M., YASUMOTO T., INOUE A. Origins of ciguatera fish poisoning: a new dinoflagellate, Gambierdiscus toxicus Adachi and Fukuyo, definitively involved as a causal agent. Toxicon. 1980;18:199–208. doi: 10.1016/0041-0101(80)90074-4. [DOI] [PubMed] [Google Scholar]
- BAI C.X., GLAASER I.W., SAWANOBORI T., SUNAMI A. Involvement of local anesthetic binding sites on IVS6 of sodium channels in fast and slow inactivation. Neurosci. Lett. 2003;337:41–45. doi: 10.1016/s0304-3940(02)01288-0. [DOI] [PubMed] [Google Scholar]
- BONHAUS D.W., HERMAN R.C., BROWN C.M., CAO Z., CHANG L.F., LOURY D.N., SZE P., ZHANG L., HUNTER J.C. The β1 sodium channel subunit modifies the interactions of neurotoxins and local anesthetics with the rat brain IIA alpha sodium channel in isolated membranes but not in intact cells. Neuropharmacology. 1996;35:605–613. doi: 10.1016/0028-3908(96)84631-4. [DOI] [PubMed] [Google Scholar]
- BOTTEIN DECHRAOUI M.Y., RAMSDELL J.S. Type B brevetoxins show tissue selectivity for voltage-gated sodium channels: comparison of brain, skeletal muscle and cardiac sodium channels. Toxicon. 2003;41:919–927. doi: 10.1016/s0041-0101(03)00088-6. [DOI] [PubMed] [Google Scholar]
- CHEN H., HEINEMANN S.H. Interaction of scorpion alpha-toxins with cardiac sodium channels: binding properties and enhancement of slow inactivation. J. Gen. Physiol. 2001;117:505–518. doi: 10.1085/jgp.117.6.505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DAVIS C.C. Gymnodinium breve: a cause of discolored water and animal mortality in the Gulf of Mexico. Bot Gaz. 1947;109:358–360. [Google Scholar]
- DECHRAOUI M.Y., NAAR J., PAUILLAC S., LEGRAND A.M. Ciguatoxins and brevetoxins, neurotoxic polyether compounds active on sodium channels. Toxicon. 1999;37:125–143. doi: 10.1016/s0041-0101(98)00169-x. [DOI] [PubMed] [Google Scholar]
- FEATHERSTONE D.E., RICHMOND J.E., RUBEN P.C. Interaction between fast and slow inactivation in Skm1 sodium channels. Biophys. J. 1996;71:3098–3109. doi: 10.1016/S0006-3495(96)79504-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FURUE T., YAKEHIRO M., YAMAOKA K., SUMII K., SEYAMA I. Characteristics of two slow inactivation mechanisms and their influence on the sodium channel activity of frog ventricular myocytes. Pflügers Arch. 1998;436:631–638. doi: 10.1007/s004240050682. [DOI] [PubMed] [Google Scholar]
- GAUDRY-TALARMAIN Y.M., MOLGO J., MEUNIER F.A., MOULIAN N., LEGRAND A.M. Reversed mode Na+–Ca2+ exchange activated by ciguatoxin (CTX-1b) enhances acetylcholine release from Torpedo cholinergic synaptosomes. Ann. N. Y. Acad. Sci. 1996;779:404–406. doi: 10.1111/j.1749-6632.1996.tb44814.x. [DOI] [PubMed] [Google Scholar]
- HIDALGO J., LIBERONA J.L., MOLGÓ J., JAIMOVICH E. Pacific ciguatoxin-1b effect over Na+ and K+ currents, inositol 1,4,5-triphosphate content and intracellular Ca2+ signals in cultured rat myotubes. Br. J. Pharmacol. 2002;137:1055–1062. doi: 10.1038/sj.bjp.0704980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRAMA M., OISHI T., UEHARA H., INOUE M., MARUYAMA M., OGURI H., SATAKE M. Total synthesis of ciguatoxin CTX3C. Science. 2001;294:1904–1907. doi: 10.1126/science.1065757. [DOI] [PubMed] [Google Scholar]
- HOGG R.C., LEWIS R.J., ADAMS D.J. Ciguatoxin (CTX-1) modulates single tetrodotoxin-sensitive sodium channels in rat parasympathetic neurones. Neurosci. Lett. 1998;252:103–106. doi: 10.1016/s0304-3940(98)00575-8. [DOI] [PubMed] [Google Scholar]
- HOGG R.C., LEWIS R.J., ADAMS D.J. Ciguatoxin-induced oscillations in membrane potential and action potential firing in rat parasympathetic neurons. Eur. J. Neurosci. 2002;16:242–248. doi: 10.1046/j.1460-9568.2002.02071.x. [DOI] [PubMed] [Google Scholar]
- HUANG J.M., WU C.H., BADEN D.G. Depolarizing action of a red-tide dinoflagellate brevetoxin on axonal membranes. J. Pharmacol. Exp. Ther. 1984;229:615–621. [PubMed] [Google Scholar]
- INOUE M., HIRAMA M., SATAKE M., SUGIYAMA K., YASUMOTO T. Inhibition of brevetoxin binding to the voltage-gated sodium channel by gambierol and gambieric acid-A. Toxicon. 2003;41:469–474. doi: 10.1016/s0041-0101(02)00369-0. [DOI] [PubMed] [Google Scholar]
- INOUE M., UEHARA H., MARUYAMA M., HIRAMA M. Practical total synthesis of ciguatoxin CTX3C by improved protective group strategy. Org. Lett. 2002;4:4551–4554. doi: 10.1021/ol027105m. [DOI] [PubMed] [Google Scholar]
- JEGLITSCH G., REIN K., BADEN D.G., ADAMS D.J. Brevetoxin-3 (PbTx-3) and its derivatives modulate single tetrodotoxin-sensitive sodium channels in rat sensory neurons. J. Pharmacol. Exp. Ther. 1998;284:516–525. [PubMed] [Google Scholar]
- KAMBOURIS N.G., HASTINGS L.A., STEPANOVIC S., MARBAN E., TOMASELLI G.F., BALSER J.R. Mechanistic link between lidocaine block and inactivation probed by outer pore mutations in the rat micro1 skeletal muscle sodium channel. J. Physiol. (Lond.) 1998;512:693–705. doi: 10.1111/j.1469-7793.1998.693bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEWIS R., MOLGO J., ADAMS D.J.Cigautera toxins: pharmacology of toxins involved in ciguatera and related fish poisonings Seafood and Freshwater Toxins Pharamacology, Physiology, and Detection 2000New York: Marcel-Dekker; 419–447.ed. Botana, L.M. pp [Google Scholar]
- LEWIS R.J., ENDEAN R. Direct and indirect effects of ciguatoxin on guinea-pig atria and papillary muscles. Naunyn-Schmiedebergs Arch. Pharmacol. 1986;334:313–322. doi: 10.1007/BF00508787. [DOI] [PubMed] [Google Scholar]
- LEWIS R.J., HOY A.W. Comparative action of three major ciguatoxins on guinea-pig atria and ilea. Toxicon. 1993;31:437–446. doi: 10.1016/0041-0101(93)90179-m. [DOI] [PubMed] [Google Scholar]
- LEWIS R.J., HOY A.W., MCGIFFIN D.C. Action of ciguatoxin on human atrial trabeculae. Toxicon. 1992;30:907–914. doi: 10.1016/0041-0101(92)90389-m. [DOI] [PubMed] [Google Scholar]
- LIU Y., JURMAN M.E., YELLEN G. Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron. 1996;16:859–867. doi: 10.1016/s0896-6273(00)80106-3. [DOI] [PubMed] [Google Scholar]
- LOMBET A., BIDARD J.N., LAZDUNSKI M. Ciguatoxin and brevetoxins share a common receptor site on the neuronal voltage-dependent Na+ channel. FEBS Lett. 1987;219:355–359. doi: 10.1016/0014-5793(87)80252-1. [DOI] [PubMed] [Google Scholar]
- MCCOLLUM I.J., VILIN Y.Y., SPACKMAN E., FUJIMOTO E., RUBEN P.C. Negatively charged residues adjacent to IFM motif in the DIII–DIV linker of hNaV1.4 differentially affect slow inactivation. FEBS Lett. 2003;552:163–169. doi: 10.1016/s0014-5793(03)00912-8. [DOI] [PubMed] [Google Scholar]
- O'REILLY J.P., WANG S.Y., WANG G.K. Residue-specific effects on slow inactivation at V787 in D2-S6 of Nav1.4 sodium channels. Biophys. J. 2001;81:2100–2111. doi: 10.1016/S0006-3495(01)75858-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PURKERSON-PARKER S.L., FIEBER L.A., REIN K.S., PODONA T., BADEN D.G. Brevetoxin derivatives that inhibit toxin activity. Chem. Biol. 2000;7:385–393. doi: 10.1016/s1074-5521(00)00119-8. [DOI] [PubMed] [Google Scholar]
- PURKERSON S.L., BADEN D.G., FIEBER L.A. Brevetoxin modulates neuronal sodium channels in two cell lines derived from rat brain. Neurotoxicology. 1999;20:909–920. [PubMed] [Google Scholar]
- RICHMOND J.E., FEATHERSTONE D.E., HARTMANN H.A., RUBEN P.C. Slow inactivation in human cardiac sodium channels. Biophys. J. 1998;74:2945–2952. doi: 10.1016/S0006-3495(98)78001-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SATAKE M., MURATA M., YASUMOTO T. The structure of CTX3C, a ciguatoxin congener isolated from cultured Gambierdiscus Toxicus. Tetrahedron Lett. 1993;34:1975–1978. [Google Scholar]
- SAUVIAT M.P., MARQUAIS M., VERNOUX J.P. Muscarinic effects of the Caribbean ciguatoxin C-CTX-1 on frog atrial heart muscle. Toxicon. 2002;40:1155–1163. doi: 10.1016/s0041-0101(02)00117-4. [DOI] [PubMed] [Google Scholar]
- SCHREIBMAYER W., JEGLITSCH G. The sodium channel activator Brevetoxin-3 uncovers a multiplicity of different open states of the cardiac sodium channel. Biochim. Biophys. Acta. 1992;1104:233–242. doi: 10.1016/0005-2736(92)90035-k. [DOI] [PubMed] [Google Scholar]
- SEINO A., KOBAYASHI M., MOMOSE K., YASUMOTO T., OHIZUMI Y. The mode of inotropic action of ciguatoxin on guinea-pig cardiac muscle. Br. J. Pharmacol. 1988;95:876–882. doi: 10.1111/j.1476-5381.1988.tb11717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHERIDAN R.E., ADLER M. The actions of a red tide toxin from Ptychodiscus brevis on single sodium channels in mammalian neuroblastoma cells. FEBS Lett. 1989;247:448–452. doi: 10.1016/0014-5793(89)81389-4. [DOI] [PubMed] [Google Scholar]
- STRACHAN L.C., LEWIS R.J., NICHOLSON G.M. Differential actions of pacific ciguatoxin-1 on sodium channel subtypes in mammalian sensory neurons. J. Pharmacol. Exp. Ther. 1999;288:379–388. [PubMed] [Google Scholar]
- TRAINER V.L., BADEN D.G., CATTERALL W.A. Identification of peptide components of the brevetoxin receptor site of rat brain sodium channels. J. Biol. Chem. 1994;269:19904–19909. [PubMed] [Google Scholar]
- TRAINER V.L., THOMSEN W.J., CATTERALL W.A., BADEN D.G. Photoaffinity labeling of the brevetoxin receptor on sodium channels in rat brain synaptosomes. Mol. Pharmacol. 1991;40:988–994. [PubMed] [Google Scholar]
- VASSILEV P.M., SCHEUER T., CATTERALL W.A. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- VILIN Y.Y., RUBEN P.C. Slow inactivation in voltage-gated sodium channels: molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 2001;35:171–190. doi: 10.1385/CBB:35:2:171. [DOI] [PubMed] [Google Scholar]
- WANG S.Y., BONNER K., RUSSELL C., WANG G.K. Tryptophan scanning of D1S6 and D4S6 C-termini in voltage-gated sodium channels. Biophys. J. 2003;85:911–920. doi: 10.1016/S0006-3495(03)74530-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WANG S.Y., WANG G.K. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophys. J. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WATTERS M.R. Organic neurotoxins in seafoods. Clin. Neurol. Neurosurg. 1995;97:119–124. doi: 10.1016/0303-8467(95)00015-c. [DOI] [PubMed] [Google Scholar]
- WEST J.W., PATTON D.E., SCHEUER T., WANG Y., GOLDIN A.L., CATTERALL W.A. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proc. Natl. Acad. Sci. U.S.A. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU C.H., HUANG J.M., VOGEL S.M., LUKE V.S., ATCHISON W.D., NARAHASHI T. Actions of Ptychodiscus brevis toxins on nerve and muscle membranes. Toxicon. 1985;23:481–487. doi: 10.1016/0041-0101(85)90032-7. [DOI] [PubMed] [Google Scholar]
- YASUMOTO T. The chemistry and biological function of natural marine toxins. Chem. Rec. 2001;1:228–242. doi: 10.1002/tcr.1010. [DOI] [PubMed] [Google Scholar]