

Abstract
The objective of this study was to investigate if there is a synergistic effect of a combination of P2Y12 and P2Y1 inhibition and P2Y12 and thrombin inhibition, on ADP- and thrombin-induced platelet activation, respectively. The rationale being that these combinations will cause a concurrent inhibition of both Gαq and Gαi signalling.
Blood from healthy volunteers was preincubated with AR-C69931MX, a reversible P2Y12 antagonist; MRS2179, a reversible P2Y1 antagonist; or melagatran, a direct reversible thrombin inhibitor; alone or in various combinations prior to activation with ADP or thrombin. Platelet function in whole blood was assessed by flow cytometry using the antibody PAC-1 to estimate the expression of active αIIbβ3 (the fibrinogen receptor GPIIb/IIIa). A synergistic effect was evaluated by comparing the concentrations in the different combinations with those of corresponding equipotent concentrations of each single inhibitor alone. The equipotent single concentrations were experimentally obtained from concentration response curves performed in parallel.
A synergistic effect regarding inhibition of ADP-induced platelet activation (10 μM) was obtained with different combinations of AR-C69931MX and MRS2179.
Inhibition of thrombin-induced platelet activation (2 nM) with combinations of AR-C69931MX and the thrombin inhibitor melagatran did also result in a strong synergistic effect.
To our knowledge, this is the first time that data supporting a synergistic effect has been published for the inhibitor combinations described.
Whether this synergistic effect in vitro also results in an improved antithrombotic effect in vivo with or without an increased risk of bleeding remains to be studied in well-conducted clinical studies.
Keywords: P2Y12, P2Y1, flow cytometry, αIIbβ3, platelets, thrombin, melagatran, AR-C69931MX, ADP
Introduction
ADP and thrombin are two important platelet agonists that both act through seven transmembrane G-protein-coupled receptors (GPCRs) expressed on the platelet cell surface. Thrombin induces a unique and specific proteolysis of a subclass of GPCRs designated protease-activated receptors (PARs). Proteolysis of PARs by thrombin exposes a tethered ligand, at the extracellular N-terminus of the receptor, resulting in intramolecular activation (Vu et al., 1991). Two of the four known PARs, PAR1 and PAR4, are expressed on human platelets (Kahn et al., 1999). PAR1 has a higher affinity for thrombin than PAR4 (Kahn et al., 1999; Covic et al., 2000; Ayala et al., 2001) and a sequential activation mechanism by thrombin has been hypothesised (Covic et al., 2000). Thus, PAR1 is first cleaved at low thrombin concentrations; then, as the thrombin concentration increases, the low-affinity PAR4 is cleaved.
ADP, which is a weaker agonist than thrombin, is stored in the dense granules of the platelet and is released in response to primary agonists like thrombin. Thus, part of the thrombin response is via an autocrine, paracrine effect by secreted ADP. ADP also activates two different GPCRs on the platelet surface, P2Y1 (Jin & Kunapuli, 1998) and P2Y12 (Hollopeter et al., 2001; Zhang et al., 2001). Similar to PAR1 and PAR4, P2Y1 signals via Gαq, whereas P2Y12 signals via Gαi. ADP-induced P2Y1 signalling has been shown to activate phospholipase C and to be responsible for elevation of cytosolic Ca2+ and shape change. P2Y12 signalling inhibits adenylyl cyclase resulting in a reduced concentration of cAMP, which acts as an inhibitor of platelet activation. In addition to inhibition of adenylyl cyclase, P2Y12 also potentiates the release reaction (Dangelmaier et al., 2001) and can directly activate αIIbβ3 via phosphoinositide-3 kinase (Kauffenstein et al., 2001). It is known that both a P2Y1 and a P2Y12 antagonist alone can potently inhibit ADP-induced platelet activation (Jin et al., 1998; Eckly et al., 2001). In addition, the aggregatory response to ADP is markedly impaired in both P2Y1 (Leon et al., 1999) and P2Y12–null mice (Foster et al., 2001). Although an additive effect of combining blockade of P2Y12 and P2Y1 has been described (Lenain et al., 2003), data supporting a synergistic effect have not been published. Hence, in this study, we have investigated if there is a synergistic effect when a P2Y12 antagonist, AR-C69931MX (MX stands for tetrasodium salt) (Humphries et al., 1994), is combined with a P2Y1 antagonist, MRS2179 (Baurand et al., 2001) on ADP-induced platelet activation. The term synergism is often used casually, or even erroneously, and its use has been critically and extensively reviewed by Berenbaum (1977), who's stringent criteria for the term synergy has been used in this study.
It has been shown that the only Gαi signal resulting from thrombin activation is via secreted ADP and activation of P2Y12 (Kim et al., 2002). Since there is also strong evidence for the importance of both a Gαq and Gαi signal (Jin & Kunapuli, 1998) to obtain complete platelet activation, it is likely that P2Y12 is of greater importance than P2Y1 in the thrombin response. Indeed, this finding has also been confirmed in an earlier study in our laboratory (Nylander et al., 2003). On the basis of these findings, we decided to investigate the possibility of a synergistic effect by combining a thrombin inhibitor with a P2Y12 antagonist since this would inhibit thrombin-induced platelet activation via two different mechanisms. Our idea was that a direct thrombin inhibitor would decrease the active thrombin concentration and thus indirectly inhibit the PAR4 and PAR1 Gαq signals, whereas a P2Y12 antagonist would inhibit the Gαi signal from thrombin's secondary response, that is, the action of released ADP via P2Y12. The antithrombotic effect of a combination of a synthetic hexadecasaccharide (SanOrg123781) with antithrombin activity and clopidogrel, which is an indirect irreversible P2Y12 antagonist requiring hepatic metabolism, has also been shown to be more effective than the two compounds alone in a mouse model of electrically induced, carotid artery injury (Lorrain et al., 2002). Hence, in this study, we have also investigated a possible synergistic effect on thrombin-induced platelet activation when a direct-acting reversible P2Y12 antagonist, AR-C69931MX, is combined with a direct-acting reversible thrombin inhibitor, melagatran (Gustafsson et al., 2001), using the same stringent criteria for the definition of synergy as for inhibition of ADP-induced platelet activation. In order to verify that these criteria for synergy could distinguish synergy from a mere additive effect, we also tested a drug combination that could only result in additive effect, namely a combination of two different P2Y1 antagonists, MRS2179 and A3P5P (Boyer et al., 1996).
Methods
Antibodies
The anti-CD42a-peridinin chlorophyll protein (PerCP) (clone Beb1) and anti-αIIbβ3-fluorescein isothiocyanate (FITC) (clone PAC-1) antibodies were purchased from BD Pharmingen (San Diego, CA, U.S.A.).
Reagents
ADP, MRS2179, Adenosine 3′,5′-diphosphate (A3P5P), and bovine serum albumin (BSA) were purchased from Sigma Chemical Company (St Louis, MO, U.S.A.). Human α-thrombin was purchased from Haematologic Technologies Incorporated (Essex Junction, VT, U.S.A.). The fibrin polymerisation inhibitor Pefablock FG (GPRP) was purchased from Pentapharm (Basel, Switzerland). AR-C69931MX was produced by AstraZeneca (Charnwood, U.K.) and melagatran was produced by AstraZeneca (Mölndal, Sweden).
Blood collection
This study was conducted using blood samples obtained from healthy adult volunteers. Informed consent was obtained from all participants in the study, which was performed in accordance with local ethical regulations. Blood was collected by venipuncture through a 17-gauge butterfly needle, without a tourniquet to minimize platelet activation. The first 2 ml of blood was discarded before collecting 4.5 ml aliquots in tubes containing 0.5 ml of 0.129 M sodium citrate. Within 1 min of collection, the blood was diluted 1 : 10 with modified Tyrodes buffer (TB; 137 mM NaCl, 2.8 mM KCl, 1 mM MgCl2, 12 mM NaHCO3, 0.4 mM Na2HPO4, 0.35% BSA, 10 mM HEPES, 5.5 mM glucose, pH 7.4). Blood samples were used within 15 min of collection.
Platelet activation
Platelet activity was measured by means of a whole-blood flow cytometry assay. CD42a, also known as GPIX, is a membrane glycoprotein, which is part of a noncovalent complex, GPIb-IX-V, found on both resting and activated platelets. It was, therefore, used as a general platelet marker. The fibrinogen receptor αIIbβ3 is normally expressed in an inactive conformation, which is transformed into an active form after platelet activation. There is also an intracellular pool of αIIbβ3 stored in the α-granules, which is only expressed on the outer membrane after platelet activation, leading to granule secretion, see review by Phillips et al. (1988). The αIIbβ3 antibody PAC-1 specifically recognises the active conformation of αIIbβ3 (Shattil et al., 1985; 1987) and was therefore used as a marker of platelet activation. Activation of αIIbβ3 is an absolute prerequisite for platelet aggregation, and by measuring this activation, the PAC-1 assay evaluates the aggregation potential of each individual platelet. GPRP, a fibrin polymerisation inhibitor, was used to prevent fibrin polymerisation when thrombin was used as agonist. GPRP does not inhibit either platelet activation or thrombin activity (Michelson, 1994).
All incubations were performed at room temperature in the dark. AR-C69931MX, MRS2179, A3P5P, melagatran, thrombin, and ADP were diluted in TB, and 5 μl of each diluted antagonist in the combination or TB was added to each tube and preincubated for 5 min with a mix constituting the following reagents: 19.5 μl human diluted whole blood, 1.5 μl mouse antihuman CD42a-PerCP, 2.5 μl PAC-1-FITC, and 1.5 μl of TB or 66 mM GPRP (when thrombin was used as agonist). Agonist or TB (5 μl) was added to each tube and the samples were incubated for 15 min. The reaction was stopped by fixing the cells for 30 min with 40 μl of 1.5% formaldehyde in TB. The samples were then diluted with 2 ml TB prior to analysis on a flow cytometer. In order to avoid nonagonist-induced platelet activation, the procedure did not involve any washing, centrifugation, or vortex mixing.
Flow cytometry
Samples were analysed with a FACSCalibur using CellQuest software (Becton Dickinson, Palo Alto, CA, U.S.A.) within 2 h of fixing. The threshold was set at fluorescence 3 (FL3; CD42a-PerCP), and platelets were defined as CD42a positive and within the platelet cluster in a log forward-scatter (FSC) versus log CD42a-PerCP dot plot. Data on 5000 platelets were acquired from each sample. The data were analysed using WinList 5.0 software (Verity Software House, Topsham, ME, U.S.A.), and the platelet population was analysed with respect to PAC-1 mean fluorescence intensity (MFI).
Data analysis
Inhibition of ADP- and thrombin-induced platelet activation was assessed as the downregulation of PAC-1 MFI in the platelet population and expressed as a percentage of the PAC-1 MFI in the absence of inhibitor. The latter was assigned an arbitrary activity of 100%. All data were corrected for background, which was defined as the MFI in the absence of agonist. The percentage of inhibition was calculated for platelet activation as 100−((PAC-1 MFIagonist+inhibitor/PAC-1 MFIagonist) × 100). Per cent inhibition was plotted versus the antagonist concentration (log10 transformed) and fitted to sigmoidal concentration−response curves using Grafit 4.10 (Erytacus Software, London, U.K.). The antagonist concentrations that gave half-maximum inhibition (IC50) were calculated according to the equation, y=a/(1+(x/IC50)s), where y=percentage inhibition (range 0–100); a=maximum range, that is, 100 in the absence of inhibitor; s=the slope of the concentration−response curve; x=inhibitor concentration; and IC50=inhibitor concentration that inhibits upregulation of PAC-1 by 50%.
All values were expressed as mean±standard error of the mean (s.e.m.).
Calculation of synergy
A possible synergistic effect was tested according to the criteria described by Berenbaum (1977), and the experiments were designed as follows. A concentration–response curve covering a wide concentration range was constructed for each individual antagonist. Combinations of different concentrations of the antagonists were prepared in parallel, using blood from the same subject. Percent inhibitions of ADP- (10 μM) and thrombin-induced (2 nM) platelet activation were then determined for each of these combinations in parallel with the concentration–response for each single antagonist. The values obtained for the combinations were fitted to the equation for each antagonist's concentration–response curve in order to calculate which concentration of the single antagonists that would give the same response as the different combinations (equipotent concentrations). These equipotent concentrations were then applied to the equation used by Berenbaum as follows:
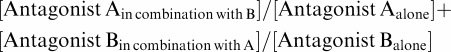 |
The value calculated from this formula is referred to as the synergy factor and a value less than 1 indicates true mathematical synergy. A value equal to 1 indicates additive effect, and more than 1 antagonistic effect. The formula can be clarified by the use of the following example: combining the same dose (e.g. 50 nM) of one and the same antagonist (e.g. A) should of course only give an additative effect. This is also the result from using the formula for this example: (50 nM/100 nM)+(50 nM/100 nM)=1. From this example it is obvious that in order to have a synergistic effect, the sum of the two divisions needs to be less than 1.
Results
Potential synergy between P2Y12 and P2Y1 receptor antagonists on inhibition of ADP-induced platelet activation
Both AR-C69931MX and MRS2179 inhibited ADP-induced (10 μM) platelet activation in a concentration-dependent manner with IC50 values of 3.1±0.7 nM (n=5) and 5300±900 nM (n=5), respectively (Figure 1).
Figure 1.

Inhibition of 10 μM ADP-induced platelet activation by AR-C69931MX in (a) and MRS2179 in (b) compared with different combinations of AR-C69931MX and MRS2179, expressed as a percentage of PAC-1 MFI in the absence of antagonist. Data represents mean values of five separate experiments.
Three concentrations of AR-C69931MX were combined in a matrix with three concentrations of MRS2179. Except for combinations with the lowest concentration of MRS2179, there was a stronger effect for all combinations compared with the effect obtained using the same concentration of each individual inhibitor. The corresponding equipotent concentrations for each single antagonist compared to the different dose combinations were calculated from the respective antagonist dose–response curve (Figure 2). The actual increase in per cent inhibition compared with a concentration response of AR-C69931MX and MRS2179 is shown in Figure 1.
Figure 2.

Calculated equipotent single doses of AR-C69931MX and MRS2179 compared to the different tested doses in the combinations. Data from five separate experiments expressed as mean±s.e.m.
Whether the increased effect of the combinations is synergistic in nature or simply an additive effect was tested according to Berenbaum's model. A synergistic effect was evident in most cases as the synergy factor was less than 1 in 36 of 45 tests performed in blood from five different volunteers. The mean synergy factor for all 45 tests was 0.70±0.05. The combinations that were not synergistic were in all cases combinations with the lowest concentration of MRS2179, which by itself gave 19±3% inhibition. Expressing the results from five experiments, all of which used blood obtained from different subjects, as mean±95% confidence interval (CI) show that there is a true synergistic effect as the 95% CI is less than 1 for all but those combinations with the lowest concentration of MRS2179 (Figure 3). If these combinations are excluded, the mean synergy factor for the remaining six combinations is 0.49±0.02.
Figure 3.

Synergy factor for the different combinations of AR-C69931MX and MRS2179 on inhibition of 10 μM ADP-induced platelet activation calculated as described by Berenbaum (1977). Data expressed as mean±95% CI from five separate experiments.
Also the P2Y1 antagonist A3P5P inhibited ADP-induced (10 μM) platelet activation, with an IC50 value of 105,000±11,000 nM (n=4). Three concentrations of A3P5P (37,500, 75,000, 150,000 nM) were combined in a matrix with the same three concentrations of MRS2179 used together with AR-C69931MX. Berenbaum's model was used as in the previous drug combinations and in all tested combinations of MRS2179 and A3P5P, the synergy factor±95% CI was not separated from 1 and thus only additive effect was present (Figure 4). The mean synergy factor for all 36 tests was 1.11±0.21.
Figure 4.

Synergy factor for the different combinations of MRS2179 and A3P5P on inhibition of 10 μM ADP-induced platelet activation calculated as described by Berenbaum (1977). Data expressed as mean±95% CI from four separate experiments.
Potential synergy between a thrombin inhibitor and a P2Y12 receptor antagonist on inhibition of thrombin-induced platelet activation
Both melagatran and AR-C69931MX inhibited thrombin-induced (2 nM) platelet activation, with IC50 values of 10.5±1.8 nM (n=5) and 2.3±0.3 nM (n=5), respectively (Figure 5). While melagatran was able to completely block the thrombin-induced response, AR-C69931MX reached a plateau of approximately 70% of maximal inhibition at concentrations greater than 15 nM. The IC50 value for AR-C69931MX was, therefore, calculated with 70% inhibition as the maximum. As in the previous experiments, three concentrations of melagatran were combined in a matrix with three concentrations of AR-C69931MX. In all individuals and at all tested combinations, the single concentration had to be increased compared with the same concentration in combination with the other antagonist to achieve the same inhibitory effect. The equipotent concentrations of melagatran and AR-C69931MX that alone yielded the same inhibition as each combination were then calculated from the individual concentration–responses as described for the P2Y1 and P2Y12 antagonists above (Figure 6). The actual increase in potency (per cent inhibition) for the combinations compared with the concentration–response curves for melagatran and AR-C69931MX is shown in Figure 5. Synergy was evident as the synergy factor was less than 1 in 36 out of 36 tests performed in blood from four different subjects. However in 24 of these 36 tests, the equipotent concentration of AR-C69931MX alone could not be accurately calculated, as the effect of the combination was greater than the maximal effect possibly achieved by AR-C69931MX alone, that is, 70%. In these cases, a stringent approximation of the equipotent concentration was made and set equal to the lowest AR-C69931MX concentration that gave the maximal inhibitory effect; that is, in these cases, the synergy factors will be overestimated and therefore, synergy will be underestimated. Despite this, there was a significant synergistic effect, that is, 95% CI less than 1, for all the combinations but one, 1 nM AR-C69931MX and 8 nM melagatran (Figure 7). The mean synergy factor for all 36 tests was 0.58±0.02.
Figure 5.

Inhibition of 2 nM thrombin-induced platelet activation by melagatran in (a) and AR-C69931MX in (b) compared with different combinations of melagatran and AR-C69931MX, expressed as a percentage of PAC-1 MFI in the absence of antagonist. Data represents mean values of four to five separate experiments.
Figure 6.

Calculated equipotent single doses of melagatran and AR-C69931MX compared to the different tested doses in the combinations. Data from four to five separate experiments expressed as mean±s.e.m. *Equipotent dose could not be calculated, the absolute minimal concentration needed is expressed.
Figure 7.

Synergy factor for the different combinations of melagatran and AR-C69931MX on inhibition of 2 nM thrombin-induced platelet activation calculated as described by Berenbaum (1977). Data expressed as mean±95% CI from four separate experiments.
Discussion
Both a Gαq and a Gαi signal are needed for potent platelet activation as shown by (Jin & Kunapuli, 1998). ADP can, by itself, activate both Gαq and Gαi signalling via P2Y1 and P2Y12, respectively. A concurrent inhibition of P2Y1 and P2Y12 may, therefore, result in a synergistic response, which was tested for in this study.
Unlike ADP, thrombin cannot, by itself, activate both Gαq and Gαi signalling. Thrombin needs the Gαi signal from P2Y12 and secreted ADP to complement the Gαq signal mainly from PAR1 but also, at high thrombin concentrations, from PAR4. Results from earlier studies (Nylander & Mattsson 2003; Nylander et al., 2003) indicate the possibility of a synergistic effect by a combination of a P2Y12 antagonist and a direct thrombin inhibitor on thrombin-induced platelet activation. We have, therefore, in the present investigation constructed concentration–response curves for all antagonists that cover a wide concentration range and, in parallel, have determined inhibition of ADP- or thrombin-induced platelet activation with various combinations of the antagonists. The effect of the combinations (percent inhibition) can then be transferred to the concentration–response equation for each single antagonist, and the concentration of the single antagonist that gives the same effect as the combination can thus be determined. By using this approach, we found a significant synergistic inhibition of ADP-induced platelet activation by combining a P2Y1 and a P2Y12 receptor antagonist, as evidenced by a mean synergy factor well below 1 (0.49±0.02). However, the efficiency of the combinations was dependent on the concentrations of MRS2179, as all combinations with the lowest concentration failed to show a significant synergistic effect (Figure 3). Another way to express the power of synergy data is to calculate the dose-reduction index (DRI) defined as folds of dose reduction allowed for each drug in the combination compared with each drug alone at a given effect (Chou, 1998). For instance, a combination of 1.5 nM AR-C69931MX and 2500 nM MRS2179 gave 64% inhibition and a synergy factor of 0.51. The same effect (64% inhibition) was obtained with a single dose of 6.0 nM AR-C69931MX or 9600 nM MRS2179. This gives a DRI of 4.0 and 3.8 for AR-C69931MX and MRS2179, respectively. Thus, at this specific combination, a synergy factor of 0.51 (close to the mean synergy factor) indicates that the dose can be reduced ≈4-fold without loss of efficacy regardless of the single drug used.
A significant synergistic effect was also evident for a combination of a P2Y12 antagonist and a thrombin inhibitor when thrombin was used as agonist (Figure 7). The mean synergy factor was well below 1 (0.58±0.02) and, thus, comparable with that found for the combined P2Y12/P2Y1 blockade when ADP was used as agonist. A combination of 2.5 nM AR-C69931MX and 8 nM melagatran resulted in 87% inhibition with a synergy factor of 0.58. The single doses required for the same effect were, (⩾) 22 and 28 nM, for AR-C69931MX and melagatran, respectively. This gives a DRI of 8.8 and 3.5 for AR-C69931MX and melagatran, respectively. Thus, at this specific combination, a synergy factor of 0.58 (equal to the mean synergy factor) indicates that the dose can be reduced ≈9- and four-fold without loss of efficacy depending on the single drug used.
When combining two different P2Y1 antagonists, MRS2179 and A3P5P, no synergy was present as the mean synergy factor±95% CI was not separated from 1 (Figure 4), and thus shows only additive effect. This shows that the criteria for synergy as described by Berenbaum could distinguish synergy from a mere additive effect.
A mechanism that could explain these synergistic effects is that the two compounds in each combination are acting (directly or indirectly) on two different signalling pathways, Gαq and Gαi, which are both essential for complete platelet activation (Jin & Kunapuli, 1998). Combination of a P2Y12 and a P2Y1 antagonist will directly block Gαi and Gαq, respectively, and thus inhibit ADP-induced platelet activation. At thrombin concentrations high enough to activate both PAR1 and PAR4, inhibition of thrombin-induced platelet activation with a reversible thrombin inhibitor, such as melagatran, will indirectly prevent Gαq signalling via cleavage and activation mainly of the low-affinity PAR4 by decreasing the active thrombin concentration (Nylander & Mattsson 2003). However, at a thrombin concentration of 2 nM, which was used in these experiments, there is only a limited cleavage of PAR4. Therefore, melagatran will inhibit this limited PAR4 Gαq signal but have its main effect via partial inhibition of the PAR1 Gαq signal. A P2Y12 antagonist, conversely, will indirectly inhibit thrombin-induced platelet activation by blockade of the only Gαi signal that is generated by thrombin. P2Y12 blockade is most potent at low thrombin concentrations, which is sufficient to cleave the high-affinity PAR1 and, thereby, induce complete ADP release (Nylander et al., 2003). Thus, the combination of a reversible thrombin inhibitor and a P2Y12 antagonist also cause a concurrent inhibition of both Gαq and Gαi signalling. The third possible combination of the tested inhibitors would be a mixture of a P2Y1 antagonist and a thrombin inhibitor. This would be a combination that inhibits two separate Gαq receptors, PAR1 and P2Y1, and an indirect inhibition of Gαi via P2Y12 due to inhibition of degranulation and ADP release. However, this combination was not tested in the present study since our previous results (Nylander et al., 2003) showed that P2Y12, but not P2Y1, has a role in thrombin-induced αIIbβ3 activation and thereby excludes that such a combination could have a synergistic effect.
As we have hypothesized that the demonstrated synergistic effect is due to a concurrent inhibition of a Gαq and a Gαi signal, it would be desirable to demonstrate that no synergistic effect can be achieved via a concurrent inhibition of two separate Gαq or Gαi signals. This however would require a very complex experimental design since, as soon as platelets are degranulated, ADP will give both a Gαq and a Gαi signal. This would also require tools, which are not commercially available, for example, potent antagonists against PAR1 and PAR4. Unfortunately we do not have these tools and therefore cannot perform this control experiment. Thus, whether the reason for the shown synergy is the proposed concurrent inhibition of a Gαq and a Gαi signal remains to be proven.
Thrombin formation and platelet activation are two major mechanisms causing acute coronary events after plaque rupture. Use of a synergistic combination of two drugs that inhibit both these events may allow the use of lower doses in order to reduce bleeding events with maintained efficacy, as shown by the results from three recent studies. In the ASPECT-2 study involving patients with acute coronary events (van Es et al., 2002), the combination of moderate-intensity oral anticoagulation (international normalized ratio (INR) 2.0–2.5) and low-dose aspirin therapy was as effective as high-intensity oral anticoagulation (INR 3–4) alone in the reduction of subsequent cardiovascular events and death and was more effective than aspirin therapy alone. There was no significant difference in incidence of major bleeding. In the large, randomized multicenter trial WARIS-2, which addressed secondary prevention after myocardial infarction, treatment with both high-intensity warfarin (INR 2.8–4.2) and moderate-intensity warfarin (INR 2.0–2.5) combined with low-dose aspirin therapy was better than aspirin therapy alone. There was no statistical significant difference in effect or major bleeding between the two groups receiving warfarin (Hurlen et al., 2002). The combined use of two platelet inhibitors with different modes of action has been shown to be better than a single agent, for example, in the CURE trial. Patients with acute coronary syndrome taking the combination of clopidogrel and aspirin had a 20% relative risk reduction in the combined end point of cardiovascular death, nonfatal myocardial infarction, and stroke in comparison with those taking aspirin alone, but were at increased risk for major bleeding, 38%, with an absolute risk of 1% (Yusuf et al., 2001).
Whether a synergistic effect in vitro of combinations of direct and reversible inhibitors of platelet activation and thrombin opens the possibility for the use of low-concentration combinations with maintained efficacy but reduced bleeding problems remains, however, to be seen and needs to be evaluated in large clinical studies.
In conclusion, the results of this study show that a synergistic inhibition of ADP-induced platelet activation can be achieved by combining inhibition of P2Y12 and P2Y1. In addition, true synergy is also shown for inhibition of thrombin-induced platelet activation by a combination of thrombin and P2Y12 inhibition. Together, these results indicate a possible clinical benefit for combining these inhibitors, provided that bleeding problems do not outweigh this benefit. This finding suggests the need for well-conducted clinical studies to determine whether these synergistic effects in vitro also results in an improved antithrombotic effect in vivo, with or without an increased risk of bleeding.
Acknowledgments
This study was supported by the Swedish Research Council project K2004-71X-15060-01A and grants from the County Council of Östergötland.
Abbreviations
- A3P5P
adenosine 3′,5′-diphosphate
- CI
confidence interval
- DRI
dose-reduction index
- FITC
fluorescein isothiocyanate
- FL
fluorescence
- GPCR
G-protein-coupled receptor
- MFI
mean fluorescence intensity
- PAR
protease-activated receptor
- PerCP
peridinin chlorophyll protein
- SEM
standard error of the mean
- TB
Tyrodes buffer
References
- AYALA Y.M., CANTWELL A.M., ROSE T., BUSH L.A., AROSIO D., DI CERA E. Molecular mapping of thrombin-receptor interactions. Proteins: Struct. Funct. Genet. 2001;45:107–116. doi: 10.1002/prot.1130. [DOI] [PubMed] [Google Scholar]
- BAURAND A., RABOISSON P., FREUND M., LEON C., CAZENAVE J.P., BOURGUIGNON J.J., GACHET C. Inhibition of platelet function by administration of MRS2179, a P2Y1 receptor antagonist. Eur. J. Pharmacol. 2001;412:213–221. doi: 10.1016/s0014-2999(01)00733-6. [DOI] [PubMed] [Google Scholar]
- BERENBAUM M.C. Synergy, additivism and antagonism in immunosuppression. A critical review. Clin. Exp. Immunol. 1977;28:1–18. [PMC free article] [PubMed] [Google Scholar]
- BOYER J.L., ROMERO-AVILA T., SCHACHTER J.B., HARDEN T.K. Identification of competitive antagonists of the P2Y1 receptor. Mol. Pharmacol. 1996;50:1323–1329. [PubMed] [Google Scholar]
- CHOU T.C. Drug combinations: from laboratory to practice. J. Lab. Clin. Med. 1998;132:6–8. doi: 10.1016/s0022-2143(98)90018-x. [DOI] [PubMed] [Google Scholar]
- COVIC L., GRESSER A.L., KULIOPULOS A. Biphasic kinetics of activation and signaling for PAR1 and PAR4 thrombin receptors in platelets. Biochemistry. 2000;39:5458–5467. doi: 10.1021/bi9927078. [DOI] [PubMed] [Google Scholar]
- DANGELMAIER C., JIN J., SMITH J.B., KUNAPULI S.P. Potentiation of thromboxane A2-induced platelet secretion by Gi signaling through the phosphoinositide-3 kinase pathway. Thromb. Haemost. 2001;85:341–348. [PubMed] [Google Scholar]
- ECKLY A., GENDRAULT J.L., HECHLER B., CAZENAVE J.P., GACHET C. Differential involvement of the P2Y(1) and P2Y(T) receptors in the morphological changes of platelet aggregation. Thromb. Haemost. 2001;85:694–701. [PubMed] [Google Scholar]
- FOSTER C.J., PROSSER D.M., AGANS J.M., ZHAI Y., SMITH M.D., LACHOWICZ J.E., ZHANG F.L., GUSTAFSON E., MONSMA F.J., JR, WIEKOWSKI M.T., ABBONDANZO S.J., COOK D.N., BAYNE M.L., LIRA S.A., CHINTALA M.S. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J. Clin. Invest. 2001;107:1591–1598. doi: 10.1172/JCI12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GUSTAFSSON D., NYSTROM J., CARLSSON S., BREDBERG U., ERIKSSON U., GYZANDER E., ELG M., ANTONSSON T., HOFFMANN K., UNGELL A., SORENSEN H., NAGARD S., ABRAHAMSSON A., BYLUND R. The direct thrombin inhibitor melagatran and its oral prodrug H 376/95: intestinal absorption properties, biochemical and pharmacodynamic effects. Thromb. Res. 2001;101:171–181. doi: 10.1016/s0049-3848(00)00399-6. [DOI] [PubMed] [Google Scholar]
- HOLLOPETER G., JANTZEN H.M., VINCENT D., LI G., ENGLAND L., RAMAKRISHNAN V., YANG R.B., NURDEN P., NURDEN A., JULIUS D., CONLEY P.B. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409:202–207. doi: 10.1038/35051599. [DOI] [PubMed] [Google Scholar]
- HUMPHRIES R.G., TOMLINSON W., INGALL A.H., CAGE P.A., LEFF P. FPL 66096: a novel, highly potent and selective antagonist at human platelet P(2T)-purinoceptors. Br. J. Pharmacol. 1994;113:1057–1063. doi: 10.1111/j.1476-5381.1994.tb17100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HURLEN M., ABDELNOOR M., SMITH P., ERIKSSEN J., ARNESEN H. Warfarin, aspirin, or both after myocardial infarction. N. Engl. J. Med. 2002;347:969–974. doi: 10.1056/NEJMoa020496. [DOI] [PubMed] [Google Scholar]
- JIN J., DANIEL J.L., KUNAPULI S.P. Molecular basis for ADP-induced platelet activation. II. The P2Y1 receptor mediates ADP-induced intracellular calcium mobilization and shape change in platelets. J. Biol. Chem. 1998;273:2030–2034. doi: 10.1074/jbc.273.4.2030. [DOI] [PubMed] [Google Scholar]
- JIN J., KUNAPULI S.P. Coactivation of two different G protein-coupled receptors is essential for ADP-induced platelet aggregation. Proc. Natl. Acad. Sci. U.S.A. 1998;95:8070–8074. doi: 10.1073/pnas.95.14.8070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAHN M.L., NAKANISHI-MATSUI M., SHAPIRO M.J., ISHIHARA H., COUGHLIN S.R. Protease-activated receptors 1 and 4 mediate activation of human platelets by thrombin. J. Clin. Invest. 1999;103:879–887. doi: 10.1172/JCI6042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KAUFFENSTEIN G., BERGMEIER W., ECKLY A., OHLMANN P., LEON C., CAZENAVE J.P., NIESWANDT B., GACHET C. The P2Y(12) receptor induces platelet aggregation through weak activation of the alpha(IIb)beta(3) integrin-a phosphoinositide 3-kinase-dependent mechanism. FEBS Lett. 2001;505:281–290. doi: 10.1016/s0014-5793(01)02824-1. [DOI] [PubMed] [Google Scholar]
- KIM S., FOSTER C., LECCHI A., QUINTON T.M., PROSSER D.M., JIN J., CATTANEO M., KUNAPULI S.P. Protease-activated receptors 1 and 4 do not stimulate Gi signaling pathways in the absence of secreted ADP and cause human platelet aggregation independently of Gi signaling. Blood. 2002;99:3629–3636. doi: 10.1182/blood.v99.10.3629. [DOI] [PubMed] [Google Scholar]
- LENAIN N., FREUND M., LEON C., CAZENAVE J.-P., GACHET C. Inhibition of localized thrombosis in P2Y1-deficient mice and rodents treated with MRS2179, a P2Y1 receptor antagonist. J. Thromb. Haemost. 2003;1:1144–1149. doi: 10.1046/j.1538-7836.2003.00144.x. [DOI] [PubMed] [Google Scholar]
- LEON C., HECHLER B., FREUND M., ECKLY A., VIAL C., OHLMANN P., DIERICH A., LEMEUR M., CAZENAVE J.P., GACHET C. Defective platelet aggregation and increased resistance to thrombosis in purinergic P2Y(1) receptor-null mice. J. Clin. Invest. 1999;104:1731–1737. doi: 10.1172/JCI8399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LORRAIN J., LECHAIRE I., GAUFFENY C., MASSON R., ROOME N., O'CONNOR S., SCHAEFFER P., HERBERT J.Synergistic anti-thrombotic effect of SanOrg123781A, a synthetic heparin mimetic, in combination with the antiplatelet agent clopidogrel, in a mouse model of electrically-induced carotid artery injury Blood 2002. abstract1966 [DOI] [PubMed]
- MICHELSON A.D. Platelet activation by thrombin can be directly measured in whole blood through the use of the peptide GPRP and flow cytometry: methods and clinical applications. Blood Coag. Fibrinol. 1994;5:121–131. doi: 10.1097/00001721-199402000-00014. [DOI] [PubMed] [Google Scholar]
- NYLANDER S., MATTSSON C. Thrombin-induced platelet activation and its inhibition by anticoagulants with different modes of action. Blood Coag. Fibrinol. 2003;14:159–167. doi: 10.1097/00001721-200302000-00007. [DOI] [PubMed] [Google Scholar]
- NYLANDER S., MATTSSON C., RAMSTRÖM R., LINDAHL T.L. The relative importance of the ADP receptors, P2Y12 and P2Y1 in thrombin-induced platelet activation. Thrombo. Res. 2003;111:65–73. doi: 10.1016/j.thromres.2003.08.021. [DOI] [PubMed] [Google Scholar]
- PHILLIPS D.R., CHARO I.F., PARISE L.V., FITZGERALD L.A. The platelet membrane glycoprotein IIb–IIIa complex. Blood. 1988;71:831–843. [PubMed] [Google Scholar]
- SHATTIL S.J., CUNNINGHAM M., HOXIE J.A. Detection of activated platelets in whole blood using activation-dependent monoclonal antibodies and flow cytometry. Blood. 1987;70:307–315. [PubMed] [Google Scholar]
- SHATTIL S.J., HOXIE J.A., CUNNINGHAM M., BRASS L.F. Changes in the platelet membrane glycoprotein IIb IIIa complex during platelet activation. J. Biol. Chem. 1985;260:11107–11114. [PubMed] [Google Scholar]
- VAN ES R.F., JONKER J.J., VERHEUGT F.W., DECKERS J.W., GROBBEE D.E. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet. 2002;360:109–113. doi: 10.1016/S0140-6736(02)09409-6. [DOI] [PubMed] [Google Scholar]
- VU T.K., HUNG D.T., WHEATON V.I., COUGHLIN S.R. Molecular cloning of a functional thrombin receptor reveals a novel proteolytic mechanism of receptor activation. Cell. 1991;64:1057–1068. doi: 10.1016/0092-8674(91)90261-v. [DOI] [PubMed] [Google Scholar]
- YUSUF S., ZHAO F., MEHTA S.R., CHROLAVICIUS S., TOGNONI G., FOX K.K. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N. Engl. J. Med. 2001;345:494–502. doi: 10.1056/NEJMoa010746. [DOI] [PubMed] [Google Scholar]
- ZHANG F.L., LUO L., GUSTAFSON E., LACHOWICZ J., SMITH M., QIAO X.D., LIU Y.H., CHEN G.D., PRAMANIK B., LAZ T.M., PALMER K., BAYNE M., MONSMA F.J. ADP is the cognate ligand for the orphan G protein-coupled receptor SP1999. J. Biol. Chem. 2001;276:8608–8615. doi: 10.1074/jbc.M009718200. [DOI] [PubMed] [Google Scholar]