

Abstract
We determined the structure of the photolytic intermediate of a sperm whale myoglobin (Mb) mutant called Mb-YQR [Leu-(B10)→Tyr; His(E7)→Gln; Thr(E10)→Arg] to 1.4-Å resolution by ultra-low temperature (20 K) x-ray diffraction. Starting with the CO complex, illumination leads to photolysis of the Fe–CO bond, and migration of the photolyzed carbon monoxide (CO*) to a niche in the protein 8.1 Å from the heme iron; this cavity corresponds to that hosting an atom of Xe when the crystal is equilibrated with xenon gas at 7 atmospheres [Tilton, R. F., Jr., Kuntz, I. D. & Petsko, G. A. (1984) Biochemistry 23, 2849–2857]. The site occupied by CO* corresponds to that predicted by molecular dynamics simulations previously carried out to account for the NO geminate rebinding of Mb-YQR observed in laser photolysis experiments at room temperature. This secondary docking site differs from the primary docking site identified by previous crystallographic studies on the photolyzed intermediate of wild-type sperm whale Mb performed at cryogenic temperatures [Teng et al. (1994) Nat. Struct. Biol. 1, 701–705] and room temperature [Šrajer et al. (1996) Science 274, 1726–1729]. Our experiment shows that the pathway of a small molecule in its trajectory through a protein may be modified by site-directed mutagenesis, and that migration within the protein matrix to the active site involves a limited number of pre-existing cavities identified in the interior space of the protein.
Keywords: cryo-crystallography, heme proteins, ligand binding, function
Conformational fluctuations have been proposed to account for the observation that diffusion of water or small ligands through the protein matrix is a global phenomenon involving virtually all regions of the protein, as suggested (among others) by Lakowicz and Weber (1) to account for the efficient fluorescence quenching of internal tryptophans by dioxygen. In the crystal structure of enzymes processing large substrates, a channel involved in the capture and optimal presentation to the catalytic site is seen (2). Cavities and connecting channels identified in the interior space of a protein (3) confer flexibility and alternative packing arrangements that allow rapid transitions between structurally distinct states; in small globular proteins, however, it is not known whether these packing defects are important in controlling function by defining alternative pathways and hosting stations in the diffusion of a ligand to the active site. The ultra-low temperature x-ray diffraction experiment on the sperm whale myoglobin (Mb) mutant presented in this paper supports the viewpoint that pre-existing internal cavities play a major role in controlling the dynamics of ligand binding and can be modified by protein engineering.
The role of protein relaxation in Mb has been extensively investigated by laser photolysis because the photosensitivity of the complex of ferrous Mb with CO, O2, and NO makes it possible to populate intermediate states and to follow the structural dynamics involved in the relaxation of the protein and the migration of the ligand through the matrix. These diatomic molecules are small enough to migrate rapidly away from the metal and large enough to probe accessibility of atomic-sized cavities that are sparse in the structure and interconnected through fluctuating channels (3–5). The high photosensitivity of the MbCO adduct, with unitary quantum yield (6), makes this derivative an ideal model for time-resolved crystallography (7) using intense synchrotron x-ray sources.
Previous crystallographic studies on the photolyzed state of wild-type (wt) sperm whale Mb showed the photodissociated CO* to reside in the heme pocket near the metal but at nonbonding distance, both at room temperature in the first time-resolved nsec diffraction experiment (7) and at ultra-low temperatures (8–10). Differences in the location of CO* observed in the cryogenic x-ray diffraction experiments were attributed (partially or totally) to a variable degree of photolysis related to the experimental protocol and/or the temperature. The results indicate (see ref. 10 for a discussion) that the photolyzed CO* is located largely 3.6–3.7 Å from the Fe (which moves out of the heme plane by ≈ 0.3 Å), in agreement with optical absorption and polarized IR time-resolved studies, as well as molecular dynamics calculations (11–14). A study of the structural dynamics of Mb by x-ray diffraction may provide an answer to some interesting questions, such as: (i) How far could the niche hosting the photolyzed CO* be considered a docking site? (ii) To what extent could this niche be controlled by the nature of the side chains in the distal heme pocket? (iii) Could protein engineering allow one to modify the pathways for ligand migration through cavities and channels within the protein matrix, to and from the iron? An answer may come from a combination of time-resolved spectroscopy, molecular dynamics simulations, x-ray crystallography of intermediate states, and mutagenesis, as shown by the experiment reported below.
Our results on the Mb mutant called Mb-YQR (15) indicate that the intramolecular pathway of the photolyzed CO* can be modified by hindering the primary docking site identified in wt Mb (see above), while making another pre-existing cavity more easily available to host the ligand; thus engineering proteins with alternative dynamic pathways for ligands and substrates seems possible. The result supports the view that internal cavities and channels play a role in determining the overall reaction kinetics and thus acquire physiological value by controlling the oxygen affinities of heme proteins.
Materials and Methods
Biochemistry.
Mutant Mb-YQR was expressed in Escherichia coli and purified as described by Travaglini-Allocatelli et al. (16). Crystals were grown at 21°C by using the vapor diffusion technique as described by Phillips et al. (17), by mixing 5 μl of 1 mM protein with an equal volume of the reservoir solution consisting of 2.7 M ammonium sulfate in 20 mM Tris⋅HCl at pH 8.7 and 1 mM EDTA. Crystals grow in approximately 2 weeks and exhibit the hexagonal space group P6 (for details on data collection and cell dimensions see Table 1).
Table 1.
Summary of crystallographic data
| YQR-CO | YQR⋯CO* | |
|---|---|---|
| Data statistics | ||
| Temperature, K | 100 | 20 |
| Unit cell (Å) in P6 | a = b = 90.49, c = 45.28 | a = b = 90.61, c = 45.33 |
| Resolution, Å | 19.6–1.4 | 18.0–1.4 |
| # Reflection (total/last shell) | 143,218/15,373 | 136,182/14,395 |
| # Unique refl. (total/last shell) | 40,518/7,138 | 41,213/7,686 |
| Completeness (total/last shell), % | 96.5/91.4 | 98.5/99.1 |
| I/σ (total/last shell) | 19.8/5.8 | 19.8/5.3 |
| Rsym (total/last shell), % | 3.6/12.5 | 3.6/11.1 |
| Refinements | ||
| Resolution, Å | 19.6–1.4 | 18.0–1.4 |
| # Water molecules | 259 | 266 |
| R/Rfree,* % | 12.9/15.4 | 13.2/16.6 |
| rms bond/over all coord. [Å], ESU or ML based‡ | 0.014/0.024 | 0.015/0.025 |
| Biso overall/Biso water [Å2] | 14.0/27.0 | 14.8/28.0 |
| B CO: [Å2] | C: 8.5/O: 14.6 | 13.6/21.4† |
*The same set of 5% randomly chosen reflections was used for calculation of Rfree.
†The diffraction data do not allow assigning the carbon and oxygen atoms, respectively.
‡ESU, estimated standard uncertainty; ML, maximum likelihood
Crystallography.
Data collection.
Mb-YQR-CO crystals were obtained by soaking crystal fragments of ≈ 100 μm3 in CO-saturated mother liquor containing 8 mg/ml dithionite until a clear color change occurred. The same solution supplemented with 10% xylitol and 10% glucose was used as cryoprotectant in which the crystals were rinsed briefly before flash-freezing in liquid nitrogen.
Both data sets were collected to 1.4-Å resolution from the same crystal: first, the CO-bound complex was collected at 100 K by using an Oxford cryostream; then the photolyzed data set was collected at 20 K by using an open-flow helium cryostat. Photolysis was achieved by using the white light from a fiber-optic microscope illuminator run continuously at low power 10 min before and during data collection, as described by Schlichting et al. (8). Diffraction data were collected at beamline X12C at the Brookhaven synchrotron by using a wavelength of 0.91 Å and the Brandeis 1k × 1k charge-coupled device detector. Data reduction and scaling were done with xds and xscale, respectively (18). Data statistics are shown in Table 1.
Refinement.
The structure of the unliganded complex of Mb-YQR (15) was used as a starting model for the refinement of the photolyzed complex after omitting water and sulfate molecules. After one round of refinement with xplor [including simulated annealing (T = 2,000 K) to remove model bias], the refinement was continued with protein/refmac (19, 20). Electron density maps were inspected with the program o (21). The CO molecule had good electron density from the first round of refinement onward and was included at the last step that included anisotropic B factor refinement and a wARP run to pick water molecules (22).
The final model of the photolyzed complex (with B factors set to 15.0 Å2) was used as a starting model for the refinement of the data of the bound complex. The same set of reflections was used for calculation of Rfree in both cases, and the refinement procedure (including the simulated annealing step) was the same. The final refinement statistics are given in Table 1.
Results and Discussion
The Structure of Carbonyl Mb-YQR: L(B10)Y/H(E7)Q/T(E10)R.
The high quality of the crystals of Mb-YQR and their diffraction data allow a detailed analysis of the structural features in the ground state (CO-bound) and after photolysis. The structure of the mutant already was shown by Brunori et al. (15) to be identical to wt sperm whale Mb outside the immediate environment of the distal heme pocket for both the deoxy and the oxy derivatives (at 1.7- to 1.8-Å resolution). The new data at higher resolution (1.4 Å) confirm this conclusion for the CO derivative. It may be recalled that in wt deoxy Mb a water molecule is present in the distal pocket and stabilized by a H bond with the distal His(E7)64 (23); in contrast, deoxy Mb-YQR does not have a water molecule because Tyr(B10)29 would interfere sterically.
Examination of the CO geometry with respect to the heme (Table 2) shows the angle Fe–C–O to be within the range observed for wt and other mutants of sperm whale MbCO (23–25). The electron density maps and the derived parameters of the heme iron geometry (Table 3) show that the Fe(II) is essentially in the heme plane in the CO derivative of Mb-YQR, while being out of plane (by ≈ 0.3 Å) in the deoxy derivative.
Table 2.
Ligand geometry for wt sperm whale Mb and three mutants
Table 3.
Heme iron geometry
| Mb⋯CO*
|
Mb-CO
|
Deoxy Mb
|
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| YQR | wt† | YQR | wt‡ | wt§ | wt¶ | YQR | wt‡ | wt§ | wt¶ | |
| Fe-Npplane, Å | 0.25 | 0.19 | 0.07 | 0.001 | 0.05 | 0.015∥ | 0.30 | 0.25 | 0.32 | 0.29 |
| Fe-Heme plane, Å | 0.31 | 0.27 | 0.14 | 0.05 | 0.10 | — | 0.36 | 0.32 | 0.4 | — |
| Fe-Nɛ2[His93], Å | 2.12 | 2.25 | 2.12 | 2.30 | 2.27 | — | 2.23 | 2.2 | 2.25 | — |
| Temperature, K | 20 | 20 | 100 | 100 | 277 | ∼293 | 120 | 100 | 277 | ∼293 |
| Resolution, Å | 1.4 | 1.5 | 1.4 | 1.15 | 2.0 | 1.15 | 1.8 | 1.15 | 2.0 | 1.15 |
Binding of CO to Mb-YQR is associated with a displacement of Tyr(B10)29 that is larger (by 0.9 Å) than that associated with O2 binding; Gln(E7)64, the new distal residue in the mutant, is also in a more “open” position (Fig. 1A and ref. 15). The displacement of Gln(E7)64 induced by CO binding to the single mutant His(E7) → Gln(23) is considerably smaller than that seen in Mb-YQR (0.7 vs. 2.9 Å); this may be caused by (i) either a steric effect between the hydroxyl group of Tyr(B10)29 and the Nɛ2 of Gln(E7)64, or (ii) an effect of Arg(CD3)45 present in Mb-YQR, which may pull out Gln(E7) by H bonding (see ref. 15).
Figure 1.
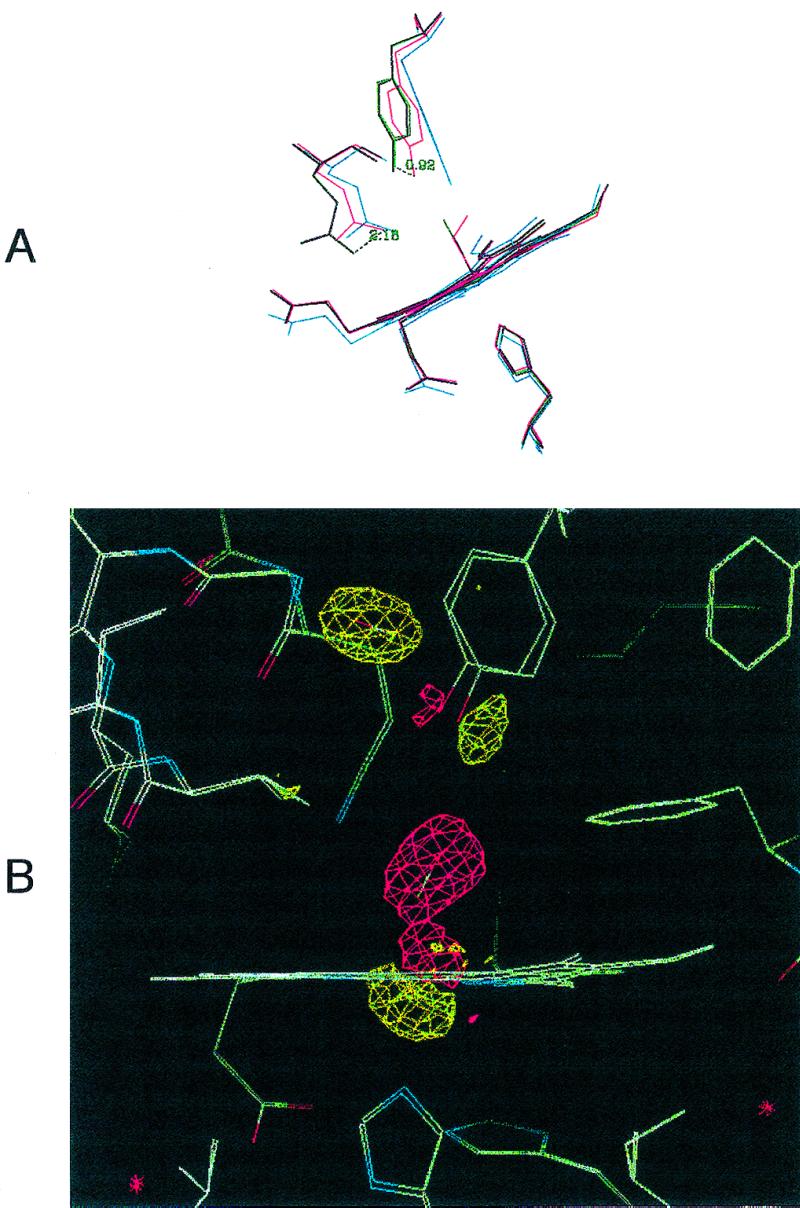
(A) Distal site pocket in Mb-YQR. The heme, its ligands (O2 and CO), and residues Tyr(B10)29, Gln(E7)64, and His(F8)93 are shown in the following ligation states: deoxygenated, blue; oxygenated, red; carbonmonoxy, purple; CO* photolyzed at 20 K, green. The displacements of the hydroxyl group of Tyr(B10)29 and the amino group of Gln(E7)64, respectively, in going from the oxygenated to the carbonmonoxy state, are given in Å. It also may be noted that the side chain of Tyr(B10)29 slightly “bounces” toward the outside of the pocket in the photolyzed state with respect to the CO-bound state. (B) Electron density maps in the heme region calculated by using the measured structure factor amplitudes for Mb-YQR⋯CO* at 20 K minus the ones from Mb-YQR-CO bound. The map is contoured at 3-σ electron density. Shown are the bound CO of Mb-YQR-CO in a negative density region (red) and CO* clearly positioned in an elongated sphere of positive density (yellow). In addition, the position of other residues in the structure of the CO bound and the photolyzed states change: the iron moves out the porphyrin plane, and the proximal His(F8)93 (Lower) and Tyr(B10)29 (Upper) shift.
The Photolytic Intermediate.
Irradiation of the Mb-YQR-CO crystal with white light by using the experimental setup described by Schlichting et al. (8) is associated with very clear changes in the electron density map. As shown in Fig. 1B, several differences are observed between the electron density maps of the CO-bound and the photolyzed state. The most prominent one is the loss of density of the CO bound to the Fe(II) and the appearance of new density away from the pocket in a site behind Tyr(B10)29. The new prominent peak seen in the photolytic intermediate can be fitted with CO (and is inconsistent with H2O), and its integrated electron density accounts for a full occupancy of CO, with a B factor comparable to the average B factors of the molecule (see Table 1). This finding implies that under the conditions of the experiment essentially all of the photolyzed ligand moves to the new position and none remains bound to the heme, nor is present in the primary docking site, where it was observed in the photolyzed state of wt Mb (7–10).
The niche hosting the photolyzed CO* (Fig. 2) is lined by residues Gly(B6)26, Ile(B9)28, Tyr(B10)29, Gly(E8)65, Val(E11)68, Leu(E12)69, and Ile(G9)107. This secondary docking site corresponds to one of the Xe-binding sites identified by Tilton et al. (26) in the crystallographic structure of wt met Mb obtained at high pressure (7 atm, 1 atm = 101.3 kPa) of Xe, and in fact is the so-called Xe[4] binding site. Upon Xe binding, a significant decrease in the thermal B factors of these side chains was reported (26).
Figure 2.

Stereo view of the docking site of the photolyzed CO* in Mb-YQR at 20 K. The site is defined as being formed by the residues having at least one atom at 5 Å or less from one of the two atoms of CO* and include: Gly(B6)25, Ile(B9)28, Tyr(B10)29, Gly(E8)65, Val(E11)68, Leu(E12)69, and Ile(G8)107. The distance of the CO* from the heme Fe is shown in Å.
The protein side chains in the photolytic intermediate of Mb-YQR undergo small displacements, which are nevertheless interesting. On the distal side, Tyr(B10)29, Gln(E7)64, and Val(E11)68 are only slightly perturbed (see Fig. 1A); the shift of Tyr(B10)29 is small (0.4 Å) compared with that seen in going to deoxy Mb-YQR (where the hydroxyl is 2.7 Å away from that seen in the CO state), but nevertheless it swings further away. On the proximal side, the metal and the proximal His(F8)93 move away from the heme plane upon photolysis (Fig. 1 A and B and Table 3), as shown also for wt Mb at cryogenic and room temperatures (7–10).
The secondary docking site hosting CO* corresponds to that predicted by molecular dynamics simulations carried out to account for the time course of the geminate recombination of Mb-YQR (15); upon laser photolysis of NO bound to reduced Mb-YQR, the quantum yield for photodissociation is unusually high (≈4% compared with 0.1% for wt Mb) and the geminate rebinding has a prominent, unusually slow kinetic component (τ ≈ 300 nsec) that almost disappears when the photolysis experiment is carried out in the presence of 10 atm of Xe. Molecular dynamics simulations (15, 27) indicated the photolyzed ligand occupies a secondary docking site that was assumed to correspond to the Xe[4] of Tilton et al. (26). This prediction is now confirmed experimentally, as shown in Figs. 1B and 2. The distance of CO* from the Fe(II) was calculated to be 8.5 Å, similar (but not identical) to that determined by diffraction, which is 8.1 Å. The laser photolysis data of geminate NO rebinding to Mb-YQR at 20°C indicate that only a fraction of the photolyzed ligand migrates to the secondary docking site, whereas the diffraction data at 20 K show that most of the CO* is located there.
Ligand Migration in the Protein.
The secondary docking site was suggested (15) to become accessible to the photolyzed ligand because of flipping of the δ-carbon of Ile(G8)107 occurring around 10 psec in the simulation, opening an access channel to the site. This same residue was reported to be along one of the exit channels for CO in simulations carried out by Elber and Karplus (4). Obviously if this was the primary access channel, fluctuations of Ile(G8)107 also occur at 20 K, suggesting a very small barrier. It is of interest that in the case of several mutants of Ile(G8)107, occupancy of a secondary docking site (presumably the same) was found to be unchanged when Ile was substituted by smaller side chains such as Ala, Val, or Leu, but was significantly reduced when substituted by Trp (28). This finding is in agreement with the hypothesis by Brunori et al. (15) that a significant role may be assigned to Phe, which in Ascaris suum Hb occupies the same topological position (G8); in this protein both the geminate kinetics (13) and the overall O2 dissociation rate constant (29) suggest that access channel to the secondary docking site is blocked by Phe.
Previous crystallographic work on the photolytic intermediate of wt Mb at cryogenic temperatures (20–40 K) and room temperature (20°C), in spite of some differences, shows that CO* resides by and large in the distal site, 3.6–3.7 Å from the metal, above the pyrrole C of the heme. The amino acid side chains confining the CO* were identified as: Phe(CD1)43, Ile(G8)107, Leu(B10)29, Val(E11)68, and His(E7)64. This primary docking site obviously is hosting the photodissociated ligand long enough to allow observation of geminate rebinding by laser photolysis. This extensively investigated phenomenon is most evident with NO because of higher intrinsic reactivity, but is also seen with O2 and (least of all) with CO (13, 30, 31).
The obvious question is why and how the new side chains in the distal pocket of the mutant Mb-YQR control the location of CO* in the protein matrix. An answer lies, in our opinion, largely in the nature of the residue at position (B10). As shown in Fig. 3, if we superimpose the structures of the photolytic intermediates of wt Mb and Mb-YQR, the substitution of Leu(B10)29 with Tyr seems sufficient to account for the observation that the preferential docking site of CO* in the two proteins is different and (by and large) an alternative. The position of CO* in the primary docking site seems incompatible with Tyr at (B10)29 because the hydroxyl is only 2.8 Å away from CO*, which may account for the very low occupancy of CO* in the primary docking site in the photolytic intermediate of Mb-YQR. On the other hand, Leu at (B10)29 in wt Mb would clash with CO* if this were to occupy the secondary docking site, being only 2.4 Å away from it (see Fig. 3); thus this niche, which hosts Xe[4], is less available for CO* in wt Mb. Other substitutions at position B10 were found to have an effect on the geminate kinetics of O2, as reported by Scott and Gibson (28). An interesting observation is that the fraction of the photolyzed ligand occupying the secondary docking site increased when Leu(B10)29 was substituted by Ala; in this mutant one expects a smaller steric interference compared with that shown in Fig. 3, and thus the observation by Scott and Gibson (28) is compatible with the interpretation given above.
Figure 3.

The position of CO* in wt Mb and Mb-YQR, relative to residue at B10. Superposition of wt Mb⋯CO* (8) (dark) and [Mb-YQR⋯CO*] (light). For both proteins the model shows the heme, the photolyzed CO*, and the residue at position B10 i.e., Leu for wt Mb and Tyr for Mb-YQR. We also show: (i) the distance (2.9 Å) expected between the hydroxyl group of Tyr(B10)29 and the photolyzed CO* if this was to occupy the primary docking site (dark), characteristically observed in wt Mb; and (ii) the distance (2.2 Å) expected between Leu(B10)29 and the photolyzed CO* in the secondary docking site (light) identified in the photolyzed intermediate of Mb-YQR (see Fig. 1B).
Analysis of the structure (see Fig. 4) suggests a possible interpretation of the events occurring in the geminate rebinding after laser photolysis of liganded Mb-YQR and allows us to propose a pathway for ligand diffusion to the metal. The peculiar and unique kinetic feature of Mb-YQR is the very slow geminate rebinding of NO (15). It may be thought that migration of the photolyzed ligand from the secondary docking site to hit the iron could be the cause; however, because Ile(G8)107 in the access channel seems not to provide enough of a barrier, a more plausible explanation may be found in the substantial conformational change of Tyr(B10)29 and Gln(E7)64 occurring upon deoxygenation of Mb-YQR (Figs. 1A and 4). This motion, which is not seen at 20 K, is likely to occur at room temperature quickly enough to impose a substantial barrier to ligand approach near and rebinding to the Fe(II). Therefore, migration of CO* from the secondary docking site may be rapid, but close approach to the iron may depend on a concerted movement of Tyr(B10)29 and Gln(E7)64 to their deoxy configuration, which imposes a substantial barrier.
Figure 4.

Motions of Tyr(B10)29 in going from the photolyzed intermediate (where CO* is in the secondary docking site shown as 2 to the deoxygenated state. The position of CO* in the primary docking site is shown as 1. The figure depicts that approach of the ligand to the metal and rebinding would demand a substantial movement of Tyr(B10)29 from the unliganded configuration. Deoxy, light; photolyzed, dark.
Concluding Remarks.
This crystallographic experiment on the intermediate obtained by laser photolysis of the CO complex of the Mb mutant called Mb-YQR has shown that at 20 K the photolyzed ligand CO* occupies a niche or docking site that is completely different from that seen for the photolytic intermediate in wt Mb (7–10). The agreement we observed between the room temperature laser photolysis data obtained with the NO adduct (15), the molecular dynamics simulations (15), and the present crystallographic data at 20 K strongly suggests that the secondary docking site hosting the ligand (Fig. 1B) is the same over this temperature range. This finding substantiates the significance of cryogenic experiments for the interpretation of biochemically relevant reactions studied at room temperature, in agreement with the conclusions drawn from the first temperature-dependent x-ray diffraction experiment carried out on Mb (32).
Previous work by molecular dynamics simulations (4, 5) and random mutagenesis (33) indicated that the photolyzed ligand can migrate through the protein via a limited number of pathways. Three predominant trajectories were identified (4), with the ligand hopping between cavities before escape to the solvent. Ile(G8)107, gating the channel to the secondary docking site, and Leu(B10)29 were identified along the escape route for the photodissociated ligand. The mutations introduced in Mb-YQR, which include B10 as well as the distal residue (E7), seem to have altered the preferential pathway of the diatomic molecule diffusing inside the protein, and thus the docking site that the dissociated ligand is going to occupy. To a first approximation, the single mutation at Leu(B10)29→Tyr dictates the docking site predominantly occupied by CO* in wt Mb and Mb-YQR. Although this experiment has been carried out with CO, the significance of dynamic events and fast fluctuations of protein side chains in controlling the overall kinetics and thus the affinity of a heme protein for O2, recently has been highlighted (34) in a short provocative paper. A possible mechanism for control of the O2 geminate rebinding rate to deoxy Mb-YQR is suggested to demand a concerted conformational change of Tyr(B10)29 and Gln(E7)64 for close approach of the ligand to the metal.
In conclusion, we have evidence that, even for a small diatomic molecule, migration occurs through a limited number of pathways and docking sites, which can be engineered. This view is consistent with the approach taken by Scott and Gibson (28), who described the geminate rebinding time course of a photodissociated ligand with two exponential events, reflecting migration to the iron from two distinct docking sites. In the case of the NO geminate rebinding to Mb-YQR (15), this was clearly sufficient to account for the observed time course, and because in this mutant the quantum yield for NO photodissociation is unusually large, analysis was very reliable. Thus, there was no need to resort to an analysis based on a stretched exponential equation to describe ligand rebinding coupled to protein relaxation. The picture emerging from the observation that migration of the diatomic molecule occurs through a small number of specific docking sites may seem at odds with the hyerarchical model proposed by Frauenfelder and coworkers (35) to account for the dynamics of ligand binding to proteins. Further work by protein engineering and time-resolved crystallography may lead to a better definition of the role of cavities and channels in allowing the diffusion of a ligand from the solvent to the active site through specific pathways, thereby controlling the biochemistry and functional dynamics of proteins.
Acknowledgments
We are very grateful to Prof. D. Tsernoglou for his crucial role in establishing the Protein Crystallography Unit at the University of Rome “La Sapienza,” and to Prof. Q.H. Gibson (Rice University, Houston) for reading and commenting the manuscript. The work was generously supported by the Human Frontiers Science Program (I.S. and J.B.), the Richard Anne-Liese Gielen-Leyendecker-Stiftung (I.S.), the Agenzia Spaziale Italiana and the Ministero dell' Università e della Ricerca Scientifica e Tecnologica (Structural biology and dynamics of redox proteins, 1999) (M.B.). Beamline X12C is supported by the U.S. Department of Energy Offices of Health and Environmental Research and Basic Energy Sciences and by the National Science Foundation.
Abbreviations
- Mb
myoglobin
- wt
wild type
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Data deposition: The atomic coordinates and structure factors have been deposited in the Protein Data Bank, www.rcsb.org (PDB ID codes 1dxd and 1dxc).
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040459697.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040459697
References
- 1.Lakowicz J R, Weber G. Biochemistry. 1983;12:4171–4179. doi: 10.1021/bi00745a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miles E W, Rhee S, Davies D R. J Biol Chem. 1999;274:12193–12196. doi: 10.1074/jbc.274.18.12193. [DOI] [PubMed] [Google Scholar]
- 3.Richards F M. Annu Rev Biophys Bioeng. 1977;6:151–176. doi: 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- 4.Elber R, Karplus M. J Am Chem Soc. 1990;112:9161–9175. [Google Scholar]
- 5.Carlson M L, Regan R M, Gibson Q H. Biochemistry. 1996;35:1125–1136. doi: 10.1021/bi951767k. [DOI] [PubMed] [Google Scholar]
- 6.Antonini E, Brunori M. Hemoglobin and Myoglobin in Their Reactions with Ligands. Amsterdam: North–Holland; 1971. [Google Scholar]
- 7.Šrajer V, Teng T Y, Ursby T, Pradervand C, Ren Z, Adachi S, Schildkamp W, Bourgeois D, Wulff M, Moffat K. Science. 1996;274:1726–1729. doi: 10.1126/science.274.5293.1726. [DOI] [PubMed] [Google Scholar]
- 8.Schlichting I, Berendzen J, Phillips G N, Jr, Sweet R M. Nature (London) 1994;371:808–812. doi: 10.1038/371808a0. [DOI] [PubMed] [Google Scholar]
- 9.Teng T-Y, Šrajer V, Moffat K. Nat Struct Biol. 1994;1:701–705. doi: 10.1038/nsb1094-701. [DOI] [PubMed] [Google Scholar]
- 10.Hartman H, Zinser S, Komninos P, Schneider R T, Nienhaus G U, Parak F. Proc Natl Acad Sci USA. 1996;93:7013–7016. doi: 10.1073/pnas.93.14.7013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Case D A, Karplus M. J Mol Biol. 1979;132:343–368. doi: 10.1016/0022-2836(79)90265-1. [DOI] [PubMed] [Google Scholar]
- 12.Henry E R, Levitt M, Eaton W A. Proc Natl Acad Sci. USA. 1985;82:2034–2038. doi: 10.1073/pnas.82.7.2034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gibson Q H, Regan R, Elber R, Olson J S, Carver T E. J Biol Chem. 1992;267:22022–22034. [PubMed] [Google Scholar]
- 14.Lim M, Jackson T A, Anfinrud P A. Nat Struct Biol. 1997;4:209–214. doi: 10.1038/nsb0397-209. [DOI] [PubMed] [Google Scholar]
- 15.Brunori M, Cutruzzolà F, Savino C, Travaglini-Allocatelli C, Vallone B, Gibson H. Biophys J. 1999;76:1259–1269. doi: 10.1016/S0006-3495(99)77289-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Travaglini-Allocatelli C, Cutruzzolà F, Brancaccio A, Vallone B, Brunori M. FEBS Lett. 1994;352:63–66. doi: 10.1016/0014-5793(94)00918-x. [DOI] [PubMed] [Google Scholar]
- 17.Phillips G N, Jr, Arduini R M, Springer B A, Sligar S G. Proteins. 1990;7:358–365. doi: 10.1002/prot.340070407. [DOI] [PubMed] [Google Scholar]
- 18.Kabsch W. J Appl Crystallogr. 1993;26:795–800. [Google Scholar]
- 19.Murshudov G N, Vagin A A, Dodson E J. Acta Crystallogr D. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 20.Murshudov G N, Lebedev A, Vagin A A, Wilson K S, Dodson E J. Acta Crystallogr D. 1999;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 21.Jones T A, Zou J Y, Cowan S W, Kjelgaard M. Acta Crystallogr A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- 22.Perrakis A, Sixma T K, Wilson K S, Lamzin V S. Acta Crystallogr D. 1997;53:448–455. doi: 10.1107/S0907444997005696. [DOI] [PubMed] [Google Scholar]
- 23.Quillin M L, Arduini R M, Olson J S, Phillips G N., Jr J Mol Biol. 1993;234:140–153. doi: 10.1006/jmbi.1993.1569. [DOI] [PubMed] [Google Scholar]
- 24.Vojtechovsky J, Chu K, Berendzen J, Sweet R M, Schlichting I. Biophys J. 1999;77:2153–2174. doi: 10.1016/S0006-3495(99)77056-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kachalova G S, Popov A N, Bartunik H D. Science. 1999;284:473–476. doi: 10.1126/science.284.5413.473. [DOI] [PubMed] [Google Scholar]
- 26.Tilton R F, Jr, Kuntz I D, Petsko G A. Biochemistry. 1984;23:2849–2857. doi: 10.1021/bi00308a002. [DOI] [PubMed] [Google Scholar]
- 27.Elber R, Roitberg A, Simmerling C, Goldstein R F, Verhiver G, Li H, Ulitsky A. In: Statistical Mechanics, Protein Structure, and Protein Substrate Interactions. Doniach S, editor. New York: Plenum; 1994. pp. 112–124. [Google Scholar]
- 28.Scott E E, Gibson Q H. Biochemistry. 1997;36:11909–11917. doi: 10.1021/bi970719s. [DOI] [PubMed] [Google Scholar]
- 29.Davenport H E. Proc R Soc London Ser B. 1949;136:255–270. doi: 10.1098/rspb.1949.0024. [DOI] [PubMed] [Google Scholar]
- 30.Jongeward K A, Magde D, Taube J, Masters J C, Traylor T G, Sharma V S. J Am Chem Soc. 1988;110:380–387. [Google Scholar]
- 31.Ansari A, Jones C M, Henry E R, Hofrichter J, Eaton W A. Biochemistry. 1994;33:5128–5145. doi: 10.1021/bi00183a017. [DOI] [PubMed] [Google Scholar]
- 32.Frauenfelder H, Petsko G A, Tsernoglou D. Nature (London) 1979;280:558–563. doi: 10.1038/280558a0. [DOI] [PubMed] [Google Scholar]
- 33.Huang X, Boxer S G. Nat Struct Biol. 1994;1:226–229. doi: 10.1038/nsb0494-226. [DOI] [PubMed] [Google Scholar]
- 34.Brunori M, Cutruzzolà F, Savino C, Travaglini-Allocatelli C, Vallone B, Gibson Q H. Trends Biochem Sci. 1999;24:253–255. doi: 10.1016/s0968-0004(99)01421-8. [DOI] [PubMed] [Google Scholar]
- 35.Austin R H, Beeson K W, Eisenstein L, Frauenfelder H, Gunsalus I C. Biochemistry. 1975;14:5355–5373. doi: 10.1021/bi00695a021. [DOI] [PubMed] [Google Scholar]