

Abstract
Co-administration of the calcineurin inhibitor cyclosporine and the mTOR inhibitors sirolimus or everolimus increases the efficacy of immunosuppression after organ transplantation. However, clinical studies showed enhancement of cyclosporine toxicity. To characterize the biochemical mechanisms involved, we assessed the time-dependent effects of cyclosporine in combination with mTOR inhibitors on energy production (ex vivo 31P-MRS), glucose metabolism (ex vivo 13C-MRS), and reactive oxygen species (ROS) formation (using the fluorescent agent 2′,7′-dichlorofluorescein diacetate) in perfused rat brain slices.
Cyclosporine alone inhibited energy production (ATP: 75±9%), the Krebs cycle (4-13C-glutamate from 1-13C-glucose: 61±27%), and oxidative phosphorylation (NAD+: 62±25%) after 4 h of perfusion. After 10 h, activation of anaerobic glycolysis (3-13C-lactate: 140±17%) compensated for inhibition of mitochondrial energy production and lowered the intracellular pH. ROS formation was increased after 4 h (285±55% of untreated control), but not after 10 h. mTOR inhibitors alone inhibited lactate production. When combined with cyclosporine, sirolimus enhanced cyclosporine-induced inhibition of energy metabolism (ATP: 64±9%) and ROS formation (367±46%). Most importantly, sirolimus inhibited cytosolic glycolysis and therefore compensation for cyclosporine-induced ATP reduction after 10 h. In contrast to sirolimus, everolimus antagonized cyclosporine-induced inhibition of mitochondrial energy metabolism (ATP: 91±7%) and ROS formation (170±49%). The antioxidant tocopherol antagonized all cyclosporine effects on cell metabolism.
Cyclosporine time-dependently inhibited mitochondrial metabolism and increased ROS, followed by compensation involving anaerobic glycolysis. Everolimus antagonized cyclosporine-induced mitochondrial dysfunction, whereas sirolimus inhibited compensatory anaerobic glycolysis, thus enhancing cyclosporine's negative effects. ROS play the key role in mediating the negative effects of cyclosporine on cell energy metabolism.
Keywords: Sirolimus, everolimus, cyclosporine, glucose metabolism, oxidative stress, mitochondrial energy production, glycolysis, metabolomics, MRS
Introduction
The undecapeptide cyclosporine is the basis of many immunosuppressive drug regimens after transplantation, and is used in the treatment of autoimmune diseases (Kahan, 1989; Faulds et al., 1993). Cyclosporine's immunosuppressive mechanism of action involves binding to its cytosolic receptor, cyclophilin (Schreiber & Crabtree, 1992). The cyclosporine/cyclophilin complex inhibits the activity of the Ca2+/calmodulin-regulated protein phosphatase 2B, calcineurin. Inhibition of calcineurin activity inhibits dephosphorylation of the nuclear factor of activated T cells (NFAT) and, subsequently, expression of several cytokines critical for a T-lymphocyte-mediated immune response such as interleukin-2 (Schreiber & Crabtree, 1992).
Cyclosporine has a narrow therapeutic index, and most immunosuppressive protocols include co-administration with other immunosuppressive agents. Recently, the macrolide immunosuppressants sirolimus and its 40-O-(2-hydroxyethyl) derivative everolimus have emerged as promising combination partners for cyclosporine (Neuhaus et al., 2001; Nashan, 2002). Sirolimus and everolimus bind to FK-binding proteins (FKBP) and the sirolimus/FKBP or the everolimus/FKBP complex bind to mTOR, the mammalian target of rapamycin (Gummert et al., 1999). Inhibition of mTOR arrests interleukin-2 driven T-cell proliferation at the G1–S interface. Since sirolimus and everolimus inhibit a later step of the T-cell response than cyclosporine, combination of cyclosporine with mTOR inhibitors results in synergistic immunosuppressive activity (Kahan, 1997; Schuurman et al., 1997; Nashan, 2002).
Another clinical rationale for co-administration of the calcineurin inhibitor cyclosporine and mTOR inhibitors was their different side effect spectra. When used alone, mTOR inhibitors may cause myelosuppression and gastrointestinal dysfunction (Gummert et al., 1999; Nashan, 2002), but lack the typical side effects of calcineurin inhibitors such as nephrotoxicity, hypertension, hepatotoxicity, and a wide range of neurological side effects (Kahan, 1989; Gijtenbeek et al., 1999; Bechstein, 2000; Wijdicks, 2001). The only overlap in their adverse effects is hyperlipidemia. When co-administered in clinical studies, surprisingly, sirolimus enhanced cyclosporine toxicity, most importantly nephrotoxicity (Kahan, 2000; Kahan & Kramer, 2001). In the salt-depleted rat, synergistic renal dysfunction was reported for cyclosporine/sirolimus combinations (Andoh et al., 1996; Bai et al., 2001; Podder et al., 2001). Also, synergistic intestinal dysfunction with sirolimus and cyclosporine at subtherapeutic concentrations was found in the rabbit (Dias et al., 1998). In our previous studies, we found that sirolimus and cyclosporine synergistically inhibited ATP production in vitro in rat brain slices after 3 h of perfusion, as well as in the rat brain in vivo after 6 days of oral administration (Serkova et al., 1999; 2001). Although structurally closely related to sirolimus, everolimus antagonized cyclosporine-induced inhibition of brain energy metabolism in vitro as well as in vivo (Serkova et al., 2000; 2001). However, the mechanism underlying these effects and their time-dependency remain unknown. Cyclosporine is known to inhibit energy production and to increase oxidative stress in the main target organs for cyclosporine toxicity such as kidney and liver (Aupetit et al., 1988; Wolf et al., 1997). To assess the biochemical mechanisms underlying the enhancement of cyclosporine toxicity by sirolimus and to compare those to everolimus, we studied the time dependency of the effects of the study drugs on energy production, glucose metabolism, and formation of reactive oxygen species (ROS) in perfused rat brain slices.
Methods
Chemicals and study drugs
Cyclosporine and sirolimus were purchased from Sigma (St Louis, MO, U.S.A.). Everolimus was kindly provided by Novartis Pharma AG (Basel, Switzerland). In all, 1 g l−1 of cyclosporine and 0.1 g l−1 sirolimus and everolimus stock solutions were prepared in acetonitrile/sulfuric acid (pH=3) 80%/20% v v−1. Stock solutions were stored at −80°C until use. Water-soluble vitamin E (tocopherol) was purchased from Sigma Chemicals (St Louis, MO, U.S.A.). Acetonitrile was from Aldrich Chemicals (Milwaukee, WI, U.S.A.). The fluorescent agent 2′,7′-dichlorofluorescein diacetate (DCFH-DA) used for measurement of radical oxygen species (ROS) was from Molecular Probes (Eugene, OR, U.S.A.).
Preparation of brain slices for magnetic resonance spectroscopy (MRS) experiments
All animal protocols were reviewed and approved by the University of California, San Francisco, and the University of Bremen committees on animal research. Metabolically active brain slices were prepared from 8-day-old Wistar Han rats (from Charles River Inc., Wilmington, MA, U.S.A.). Slices (350 μm thick) were prepared from cerebrocortical regions as described previously (Serkova et al., 1999; 2000). The vitality of the brain slices after preparation was established and monitored by ex vivo 31P-MRS (ATP level) and 13C-MRS (lactate production) for 14 h in a pilot study.
For each 31P-MRS experiment, 24 cerebrocortical slices prepared from 12 rats were pooled. Slices were perfused with low-phosphate Krebs buffered salt solution (BSS) (95% oxygen/5% CO2 at 37°C) in a 20 mm diameter Wilmad NMR tube (Wilmad Glass Co., Buena, NJ, U.S.A.). Brain slices were perfused with the study drugs and their combinations at the following concentrations (all n=4): (I) 500 μg l−1 cyclosporine; (II) 100 μg l−1 sirolimus; (III) 100 μg l−1 everolimus; (IV) 500 μg l−1 cyclosporine+100 μg l−1 sirolimus; (V) 500 μg l−1 cyclosporine+100 μg l−1 everolimus. Study drug concentrations were based on previous studies systematically evaluating the concentration dependency of their effects on cell metabolism in rat brain slices (Serkova et al., 1999; 2000). To evaluate the role of ROS in cyclosporine-induced metabolic effects, brain slices were perfused with the radical scavenger tocopherol: (VI) 500 μmol l−1 tocopherol; (VII) 500 μg l−1 cyclosporine+500 μmol l−1 tocopherol; (VIII) 100 μg l−1 sirolimus+500 μmol l−1 tocopherol; (IX) 500 μg l−1 cyclosporine+100 μg l−1 sirolimus+500 μmol l−1 tocopherol. After an initial perfusion period of 4 h for metabolic recovery and recording of control spectra, brain slices were perfused with the study drugs and their combinations for 10 h.
For ex vivo 13C-MRS experiments with perfused rat brain slices, four cerebrocortical slices from two rats were perfused in an 8-mm diameter tube as described above. The only difference was that the perfusion medium contained 5 mM 1-13C-labeled glucose. After stabilization for 4 h and recording of control spectra, the immunosuppressants or their combinations were added to the perfusion medium for 10 h.
MRS of perfused rat brain slices
31P-MR spectra were recorded using a Nalorac QUEST Model 4.7 T NMR instrument (Martinez, CA, U.S.A.) at a 31P-frequency of 81 MHz in combination with standard composite proton decoupling (CPD, WALTZ-16). The total acquisition time for each 31P MR spectrum was 10 min (128 scans). Each brain slice preparation was used as its own control. 31P-NMR signal intensities of high-energy phosphates were recorded and areas under the peak integrated using the MacFid software (Techmag Inc., Bellair, TX, U.S.A.). In ex vivo experiments, the peak at −10.42 p.p.m. is NAD+ (at physiologically relevant pH). For quantification of NAD+, a subintegration algorithm was used, which allowed for complete separation of the NAD+ from the ATP signal at −10.08 p.p.m. before integration (split integral procedure, MacFid software). Signals were normalized based on the phosphomonoester (PME) signal intensity. This was possible since our previous in vivo as well as ex vivo studies showed no changes in phosphocholine, phosphoethanolamine, and other phospholipid precursors during immunosuppressant exposure (Serkova et al., 2000; 2001). Chemical shifts were referenced to phosphocreatine (PCr) at –2.33 p.p.m. In our experimental setup, the intracellular pH could be calculated from the differences of chemical shifts between intracellular inorganic phosphate (Pi) and PCr, since the chemical shift of Pi, but not of PCr, is dependent on H+ concentrations inside of the cell (Gupta & Wittenberg, 1991). The following standard equation was used to calculate intracellular pH (where δ, in parts per million, is the difference between chemical shifts of Pi versus PCr):
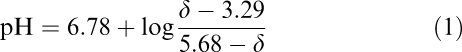 |
Proton-decoupled ex vivo 13C-MRS experiments were carried out using a 600 MHz Bruker high-resolution spectrometer at a 13C frequency of 151 MHz. D2O with trimethylsilyl propionic-2,2,3,3,-d4 acid (TSP, 1 M) in a sealed capillary was placed in the NMR tube to maintain the lock signal. The total acquisition time for each 13C-MR spectrum was 20 min (512 scans). The chemical shifts were referenced to the TSP signal at 0 p.p.m. and the intensities of 13C peaks of metabolites were normalized to the TSP signal.
Measurement of ROS
To evaluate ROS formation during exposure to the study drugs and their combinations, four cerebrocortical slices from two rats (8-day old) were perfused with Krebs BSS in a 20-mm-diameter glass tube as described previously (Serkova et al., 2002). After stabilization, immunosuppressants (final concentration of cyclosporine: 500 μg l−1, sirolimus: 100 μg l−1, everolimus: 100 μg l−1) and tocopherol (500 μmol l−1) were added, alone or in combination, for 4 or 10 h. These perfusion periods were selected based on the results of our MRS studies described above. Brain slices perfused with 1 mM H2O2 for 30 min were used as positive controls for oxidative stress. While being perfused with immunosuppressants for 4 or 10 h, the slices were loaded with the fluorescent agent 2′,7′-dichlorofluorescin diacetate (DCFH-DA) during the last 30 min.
After 4 or 10 h, the brain slices were weighed and homogenized in 5 ml of ice-cold buffer solution containing 1 mg MgCl2·6H2O, 0.7 mg NaH2PO4·H2O, 5 mg glucose, 1 ml HEPES buffer, and 65 μl 100 mM CaCl2. After centrifugation at 1000 × g for 5 min, the pellets were reconstituted in 1 ml buffer. The oxidation of intracellular DCFH to highly fluorescent DCF (dichlorofluorescein) was measured using a Perkin-Elmer LS50B fluorescence spectrophotometer (Perkin-Elmer, Überlingen, Germany) (excitation wavelength: 485 nm; emission wavelength: 525 nm) as described previously (Wang & Joseph, 1999).
Statistical analysis
All results are reported as means±standard deviation (s.d.) for each series of experiments. Results of the controls and brain slices perfused with different study drugs and their combinations (in vitro 1H-MRS and ROS formation experiments) were compared using analysis of variance. Time-dependent differences in the ex vivo 31P and 13C experiments were assessed using a general linear model with repeated-measures analysis. Where appropriate, analysis of variance was used in combination with Scheffé's test as post hoc test (SPSS, version 10.0, SPSS Inc., Chicago, IL, U.S.A.).
Results
Vitality of perfused rat brain slices during perfusion in the MR magnet
31P-MRS studies of brain slices perfused without immunosuppressants showed that high-energy metabolism of brain slices remained unchanged until at least 14 h after preparation. After 4 h of metabolic recovery, areas under the resonance peaks for all phosphorus metabolites were reproducible, with less than 5% variation during the following 10 h of perfusion (n=4). Line widths of all resonances did not change more than 2% over the course of each experiment. After metabolic stabilization during the first 4 h after preparation, the intracellular pH also remained stable over 10 h (7.19±0.05, n=4). 13C-MRS spectra showed an increase in the labeled C3-lactate peak, as well as C4- and C2-glutamate during the initial 4 h. Between 4 and 14 h, intensities of the 13C signals remained stable, indicating steady-state kinetics between synthesis and washout of labeled metabolites from the brain tissue. Based on these results, we allowed brain slices at 4 h of metabolic recovery to reach a steady state before exposing them to the study drugs and their combinations.
Time dependency of the effects of the study drugs and their combinations on energy metabolism of perfused rat brain slices
The following phosphorus metabolites were detectable in the 31P-MR spectra during perfusion of rat brain slices: PMEs, intracellular inorganic phosphate (Pi), PCr, α-, β-, and γ-ATP, and NAD+ (Figure 1).
Figure 1.

Representative ex vivo 31P-MRS of perfused rat brain slices perfused with 500 μg l−1 cyclosporine for 10 h. CsA, cyclosporine; NAD+, nicotinamide adenine dinucleotide; ATP, adenosine triphosphates; PCr, phosphocreatine; Pi, intracellular inorganic phosphate; PME, phosphomonoesthers.
Cyclosporine alone (500 μg l−1) time-dependently reduced concentrations of ATP, PCr, and NAD+. Reduction of ATP (Figure 2a) and PCr concentrations during exposure to cyclosporine reached its maximum after 4 h (Table 1). After 10 h, brain slices were able to compensate for negative cyclosporine-mediated effects on energy metabolism (Figure 2a). The increase in ATP concentrations after 4 h was accompanied by lowering the intracellular pH from 7.21±0.09, before addition of cyclosporine, to pH 6.98±0.10 after 10 h of cyclosporine perfusion (n=4, P<0.05). As shown by our 13C experiments, these compensation processes involved the activation of anaerobic glycolysis (vide infra), resulting in 40% higher lactate production. This explained, at least in part, the significantly lower intracellular pH after 10 h of perfusion with cyclosporine. Cyclosporine reduced NAD+ concentrations with a slight improvement after 10 h (Figure 2b).
Figure 2.

Time dependency of the metabolic effects of cyclosporine alone and in combination with sirolimus, everolimus, and vitamin E on (a) ATP and (b) NAD+ concentrations in perfused rat brain slices calculated from ex vivo 31P-MRS. To facilitate visual comparison, standard deviations are not shown. Data and standard deviations are listed in greater detail in Table 1. CsA, cyclosporine; NAD+, nicotinamide adenine dinucleotide; RAD, everolimus; SRL, sirolimus; VitE, tocopherol.
Table 1.
Real-time effects of immunosuppressants on relative high-energy phosphate concentrations in perfused rat brain slices calculated from ex vivo 31P-MRS: (A) phosphocreatine/PME changes; (B) ATP/PME changes; (C) NAD+/PME changes
| CsA (%) | SRL (%) | RAD (%) | CsA+SRL (%) | CsA+RAD (%) | |
|---|---|---|---|---|---|
| (A) PCr | |||||
| 2 h | 84±5* | 87±6* | 95±10 | 64±12** | 101±8 |
| 4 h | 69±9** | 85±4* | 102±6 | 60±7** | 102±12 |
| 6 h | 87±10 | 82±7* | 97±12 | 62±12** | 97±2 |
| 8 h | 97±12 | 83±9* | 90±7 | 57±10** | 99±7 |
| 10 h | 95±9 | 81±9* | 99±3 | 54±20** | 94±12 |
| (B) ATP | |||||
| 2 h | 84±2* | 90±5 | 104±6 | 69±11** | 97±4 |
| 4 h | 75±9* | 89±7 | 112±9 | 64±9** | 91±7 |
| 6 h | 85±7* | 85±3* | 107±6 | 65±7** | 93±12 |
| 8 h | 88±10 | 80±4** | 115±5* | 60±12** | 109±8 |
| 10 h | 89±4 | 80±7* | 109±7 | 58±8*** | 104±10 |
| (C) NAD+ | |||||
| 2 h | 57±9* | 102±7 | 125±12* | 62±12** | 111±4* |
| 4 h | 48±25* | 98±7 | 125±11* | 71±12* | 135±18* |
| 6 h | 59±14* | 95±12 | 127±24 | 61±17* | 103±8 |
| 8 h | 62±12** | 100±14 | 113±3* | 50±8*** | 89±12 |
| 10 h | 74±14** | 102±8 | 127±9* | 38±7*** | 94±7 |
Results are given as percent from baseline (mean±s.d., n=4). Significance levels: *P<0.05;
P<0.01;
P<0.001.
Rat brain slices were perfused with 500 μg l−1 cyclosporine, 100 μg l−1 sirolimus, 100 μg l−1 everolimus, or their combinations.
CsA, cyclosporine; NAD+, nicotinamide adenine dinucleotide; PCr, phosphocreatine; PME, phosphomonoesters; RAD, everolimus; SRL, sirolimus.
The macrolide sirolimus (100 μg l−1) slightly but significantly decreased the intracellular concentrations of ATP and PCr during 10 h of brain slice perfusion, but had no effect on mitochondrial NAD+ concentrations (Table 1). In contrast to cyclosporine, there was no improvement in the sirolimus-induced reduction of high-energy phosphate concentrations after 10 h. Also, different from cyclosporine, the intracellular pH did not change during sirolimus exposure: 7.26±0.03 after 10 h versus 7.22±0.06 without sirolimus (not statistically significant). When combined with cyclosporine (500 μg l−1 cyclosporine+100 μg l−1 sirolimus), the decrease in PCr, ATP, and NAD+ concentrations was enhanced in comparison to cyclosporine alone (Figure 2). Also, there was no improvement after 10 h (Figure 2). No significant intracellular pH change was detected when cyclosporine and sirolimus were combined, indicating a lack of activation of anaerobic glycolysis (vide infra).
Although everolimus is structurally closely related to sirolimus, addition of 100 μg l−1 everolimus to the perfused rat brain slices did not negatively affect energy metabolism (Table 1). Combination of cyclosporine and everolimus (500 μg l−1 cyclosporine+100 μg l−1 everolimus) improved the cyclosporine-induced reduction of high-energy phosphate metabolism during 4 h of incubation (Figure 2). After 10 h of perfusion with cyclosporine and everolimus, there were no changes in energy metabolism compared to untreated controls. Interestingly, in contrast to the perfusion with cyclosporine alone for 10 h, the intracellular pH remained unchanged, indicating differences between the compensatory mechanisms involved.
Assessment of 1-13C-labeled glucose metabolism in rat brain slices
Since glucose is the major energy source in the brain, we studied the effects of cyclosporine, sirolimus, and everolimus, alone and in combination, on glucose metabolism using 1-13C-labeled glucose and ex vivo 13C-MRS analysis of perfused rat brain slices. In addition to α- and β-[1-13C]glucose at 93.0 and 96.8 p.p.m., 13C-MRS detected the following major labeled intermediates: C3-lactate (at 21 p.p.m.) from glycolysis and C4-glutamate (at 34.3 p.p.m.) from the Krebs cycle through pyruvate dehydrogenase (PDH, Figure 3). Minor signals in the ex vivo 13C-MR spectra were C2-glutamate (at 55.7 p.p.m.) through pyruvate carboxylase, and C3-aspartate (at 52.7 p.p.m.) from the Krebs cycle.
Figure 3.

Representative ex vivo 13C-MRS of perfused rat brain slices after 4 h of perfusion with cyclosporine, cyclosporine+sirolimus, and cyclosporine+everolimus. Asp, aspartate; CsA, cyclosporine; Glu, glutamate; Lac, lactate; RAD, everolimus; SRL, sirolimus.
Addition of 500 μg l−1 cyclosporine to the perfusion medium for 4 h inhibited the production of Krebs cycle intermediates, while lactate production remained unchanged in the first 4 h of perfusion (Figures 3 and 4). Especially, the formation of 13C-labeled C4-glutamate was decreased (61±27%, P<0.05, n=4). After 10 h of cyclosporine perfusion, production of C3-lactate was increased to 140% of the controls (P<0.01, Figure 4). This went parallel with the decrease in intracellular pH and improved energy homeostasis as observed in the 31P-MR spectra (vide supra).
Figure 4.

Effect of immunosuppressants on 1-13C-labeled glucose utilization via glycolysis (C3-lactate) and the Krebs cycle (C4-glutamate) calculated from ex vivo 13C-MRS of perfused rat brain slices. Results are given as normalized intensities of cell metabolite 13C peaks (mean±s.d.; n=4). Significance levels: *P<0.01; **P<0.001. Rat brain slices were perfused with cyclosporine (500 μg l−1), sirolimus (100 μg l−1), everolimus (100 μg l−1), alone or in combination, for up to 10 h. CsA, cyclosporine; Glu, glutamate; Lac, lactate; RAD, everolimus; SRL, sirolimus; VitE, tocopherol.
Perfusion with sirolimus or everolimus (100 μg l−1) significantly reduced glycolytic lactate production (Figure 4).
Perfusion with 500 μg l−1 cyclosporine and 100 μg l−1 sirolimus in combination significantly decreased glucose metabolism, negatively affecting both glycolysis and Krebs cycle (Figures 3 and 4). Inhibition of Krebs cycle and glycolysis reached statistical significance after 4 h of perfusion (C4-glutamate: 48±37% of baseline; C3-lactate: 54±14%; P<0.05 and 0.01, respectively), and was further enhanced after perfusion with the study drugs for 10 h (C4-glutamate: 39±29% of baseline; C3-lactate: 42±26%; both P<0.001). The reduction in glycolysis and Krebs cycle activity paralleled the significant reduction of high-energy phosphate concentrations. In contrast, everolimus antagonized the cyclosporine-induced inhibition of C4-glutamate production after 4 and 10 h (Figures 3 and 4).
ROS formation in rat brain slices after perfusion with immunosuppressants
Perfusion of rat brain slices with hydrogen peroxide (H2O2, 1 mM, positive control) for 30 min increased ROS production to 413±113% of untreated controls (n=5, P<0.0004, Figure 5). Perfusion of rat brain slices with 500 μg l−1 cyclosporine for 4 h resulted in an increased formation of ROS (286±55% of the controls; n=5; P<0.002, Figure 5). After 10 h of perfusion with cyclosporine, however, ROS production decreased to 125±32% of the untreated controls (n=5, not statistically significant). In comparison to the controls, perfusion with 100 μg l−1 sirolimus or everolimus for 4 h and 10 h resulted in significantly higher ROS concentrations (Figure 5).
Figure 5.

ROS formation in rat brain slices after 4 and 10 h of perfusion with immunosuppressants. The values are given as mean±s.d. (n=6). Significance levels: *P<0.05; **P<0.01; ***P<0.001. Rat brain slices were perfused with cyclosporine (500 μg l−1), everolimus (100 μg l−1), sirolimus (100 μg l−1), and/or tocopherol (500 μmol l−1). The fluorescent agent DCF was added for the last 30 min. Slices perfused with 1 mM H2O2 for 30 min served as positive controls. CsA, cyclosporine; RAD, everolimus; SRL, sirolimus, VitE, tocopherol.
Perfusion of the brain slices with cyclosporine in combination with sirolimus yielded the highest ROS concentrations after both 4 and 10 h (Figure 5). In contrast, the combination of cyclosporine and everolimus showed only a tendency towards increased ROS formation without reaching statistical significance.
Role of ROS in the effects of the study drugs and their combinations on cell energy metabolism
To evaluate the potential role of ROS in mediating the negative effects of the study drugs and their combinations on cell energy metabolism, we used the oxygen radical scavenger tocopherol to reduce ROS concentrations. Addition of 500 μmol l−1 tocopherol to the perfusion medium (VE, Figure 5) for 4 or for 10 h did not affect ROS concentrations in controls perfused without the study drugs. In combination with cyclosporine, however, tocopherol completely antagonized cyclosporine-induced ROS formation.
Perfusion of brain slices with 500 μmol l−1 tocopherol alone did not affect high-energy phosphate concentrations, but increased NAD+ (132±12% of the control, n=4, P<0.05 after 4 h of perfusion and 128±12%, n=4, P<0.05 after 10 h of perfusion). Tocopherol antagonized the cyclosporine-induced (500 μg l−1) reduction of brain energy metabolism after 4 h of perfusion (PCr: 96±12% of controls for tocopherol+cyclosporine versus 69±9% for cyclosporine alone; ATP: 98±7% versus 75±9%; NAD+: 94±10% versus 62±25%, all n=4, P<0.01, Figure 2). Tocopherol also protected the intracellular pH (7.20±0.04, n=4) after 10-h perfusion with cyclosporine and tocopherol.
Interestingly, tocopherol did not have any effect on sirolimus-induced decrease of PCr and ATP. On the other hand, tocopherol antagonized cyclosporine+sirolimus-induced reduction of energy metabolism. In comparison to perfusion with medium containing 500 μg l−1 cyclosporine+100 μg l−1 sirolimus for 10 h, addition of 500 μmol l−1 tocopherol to the perfusion medium maintained concentrations of PCr (89±5% with tocopherol versus 54±20% without), ATP (85±9% versus 58±8%), and NAD+ (79±16% versus 38±7%, all n=4, P<0.01) at a higher level. Addition of tocopherol had no effect on high-energy phosphate concentrations in rat brain slices perfused with everolimus or everolimus+cyclosporine.
Tocopherol alone did not affect glucose metabolism in the brain slices. It partially ameliorated cyclosporine-induced decrease in C4-glutamate after 10 h and fully prevented cyclosporine-induced increase of lactate (Figure 4).
Discussion
While the mechanisms of cyclosporine's immunosuppressive action are well understood, understanding the basic biochemical mechanisms resulting in cyclosporine toxicity is incomplete (Gummert et al., 1999). The most likely reason is that conventional biochemical methods that have been used to study the effect of immunosuppressants on cell metabolism were limited to the assessment of selected biochemical pathways and were restricted to single pre-selected time points. In recent years, with the development of MRS-based metabolomics approaches (Nicholson & Wilson, 2003), technology that allows for the simultaneous assessment of all important cellular metabolic pathways including lipid, glucose, and high-energy phosphate metabolism has become available (Leibfritz, 1996). In our study, we took of advantage of one of the major strengths of MRS, its noninvasiveness that allows for monitoring real-time dynamic changes.
Time dependency of cyclosporine's effects on cell metabolism has not been studied before. As shown in our study, this information is critical to understanding the enhancement of the negative effects of cyclosporine on cell energy metabolism by sirolimus.
Cyclosporine time-dependently reduced the concentrations of Krebs cycle intermediates and inhibited mitochondrial oxidative phosphorylation, while lactate production remained unchanged in the first 4 h of perfusion. When perfused for longer than 4 h, lactate concentrations increased, indicating activation of anaerobic glycolysis. This compensatory mechanism resulted in a decrease in cellular pH, reduction in ROS concentrations, and an increase in ATP concentrations to a level not different from baseline.
NAD+ concentrations are controlled by a balance between biosynthesis and hydrolytic breakdown. They are linked to mitochondrial oxidative phosphorylation through mitochondrial redox status, oxygen radical, and ATP concentrations. NAD+ biosynthesis is ATP-dependent (Harvey et al., 1999). NAD+ is an established surrogate marker for mitochondrial oxidative phosphorylation (Erecinska & Wilson, 1982). Our result that NAD+ concentrations were still significantly below baseline (Table 1c) although ATP concentrations increased 10 h after cyclosporine perfusion supports our observation that the compensation for the negative effects of cyclosporine was driven by an extra-mitochondrial mechanism.
Activation of glycolysis as a compensatory mechanism for the inhibition of mitochondrial metabolism by cyclosporine may explain increased basal ganglia glucose metabolism (Meyer, 2002) and lactic acidosis reported in cyclosporine-treated patients (Bechstein, 2000).
Sirolimus did not affect mitochondrial energy metabolism, as indicated by unchanged NAD+ concentrations, but inhibited cytosolic glycolysis. A negative effect of sirolimus on aldolase activity was described previously (Wang et al., 1996). The slight but significant reduction of high-energy phosphates when brain slices were perfused with sirolimus was most likely due to a decrease of the cytosolic glycolysis rates and reduced availability of the Krebs cycle substrate acetyl-CoA. This observation may be specific to the brain since, in contrast to the brain, other organs can generate acetyl-CoA via lipid metabolism. One of the most important results of our study was that, when combined, sirolimus abolished the ability of the cells to compensate for the cyclosporine-induced reduction in mitochondrial energy production by activation of glycolysis after 4 h of perfusion. This was indicated by a reduction in lactate concentrations, unchanged intracellular pH, and a lack of reduction in ROS concentrations (vide infra) when slices were perfused with cyclosporine and sirolimus for 10 h.
The involvement of ROS in cyclosporine toxicodynamics is well established (Uemoto et al., 1989; Wang & Salahudeen, 1994; Wolf et al., 1997; Parra et al., 1998; Cruz & Wolf, 2001). Our study added two important pieces of information: (A) compensatory mechanisms reducing the negative effects of cyclosporine on high-energy phosphate metabolism is associated with a reduction in ROS concentrations and (B) the ROS scavenger tocopherol almost completely antagonizes the negative effects of cyclosporine on cell metabolism. The protective effect of tocopherol against cyclosporine nephrotoxicity has been described (Parra et al., 1998), but the metabolic pathways involved remained unknown. In our study, addition of the antioxidant tocopherol to the cyclosporine-containing perfusion medium completely antagonized the negative effects of cyclosporine on the Krebs cycle and mitochondrial oxidative phosphorylation. This observation confirms our hypothesis that ROS are the key to the negative effects of cyclosporine on mitochondrial metabolism (Serkova et al., 2002). ROS negatively affect PDH, Krebs cycle enzymes, and oxidative phosphorylation. This explains most of the cyclosporine-induced metabolic changes observed in this and other studies (Serkova et al., 1999; 2000; 2001; 2002). These studies generated evidence that ROS maintain and determine the extent of the cyclosporine-mediated inhibition of mitochondrial metabolism. The fact that tocopherol can almost completely antagonize the negative effects of cyclosporine confirms this cause–effect relationship. Interestingly, after 10 h, the ROS concentrations in brain slices perfused with cyclosporine were not different from the controls, while the ROS concentrations in sirolimus or everolimus-perfused slices remained high. We cannot explain the increased ROS concentrations caused by sirolimus or everolimus. The ROS in brain slices during perfusion with sirolimus or everolimus obviously did not have a negative effect on mitochondrial energy metabolism, as indicated by the facts that (A) both drugs had no or less of a negative effect on mitochondrial energy metabolism than cyclosporine, (B) NAD+ concentrations remained unchanged, and (C) tocopherol failed to antagonize the negative effects of sirolimus on energy metabolism. Everolimus even stimulated mitochondrial metabolism. The quantification of ROS formation using a DCF-based assay did not allow for differentiation of ROS concentrations in different cell compartments. A possible explanation is that ROS generated during perfusion with cyclosporine were located in the mitochondria, while those generated during perfusion with sirolimus or everolimus were in a different cell compartment. When combined with cyclosporine, sirolimus prevented the decrease of ROS concentrations after 10 h. Tocopherol antagonized the negative effects on energy metabolism when cyclosporine and sirolimus were combined, again indicating the key role of ROS in the enhancement of the cyclosporine-mediated negative effects on energy metabolism by sirolimus. When everolimus was combined with cyclosporine, no significant change in mitochondrial energy production or ROS formation was found in brain slices after 4 and 10 h. The protective effects of everolimus against the negative effects of cyclosporine on mitochondrial metabolism are different from those of tocopherol since everolimus itself rather increases than reduces ROS concentrations. Like sirolimus, everolimus inhibited anaerobic glycolysis. However, in contrast to sirolimus, in all of our studies, everolimus antagonized the negative effects of cyclosporine on cell energy metabolism (Figure 2) (Serkova et al., 2000; 2001). We found that one of the major differences is that everolimus, but not sirolimus, can cross the mitochondrial membrane at the tested concentrations. Also, everolimus reduced, while sirolimus enhanced, the distribution of cyclosporine into mitochondria (Table 2 ) (Serkova et al., 2001).
Table 2.
Comparison of the effects of cyclosporine, sirolimus, and everolimus on brain metabolism and their presence in brain mitochondria
| Parameter | CsA | SRL | RAD | CsA/SRL | CsA/RAD | Method |
|---|---|---|---|---|---|---|
| Presence in mitochondria | ++ | 0 | + | ++/0 | +/++ | HPLC/MS* |
| Oxid. phosphorylation | ↓ | 0 | ↑ | ↓↓ | 0 | 31P-MRS |
| Krebs cycle (C4-glutamate) | ↓ | 0 | 0 | ↓↓ | 0 | 13C-MRS |
| Glycolysis (C3-lactate) | ↑ | ↓ | ↓ | ↓↓ | 0 | 13C-MRS |
| Intracellular pH | ↓↓ | ↑/0 | 0 | 0 | 0 | 31P-MRS |
| ROS formation | ↑/0 | ↑ | ↑ | ↑↑ | 0 | Fluorescence |
From Serkova et al. (2001). CsA, cyclosporine; HPLC, high-performance liquid chromatography; MRS, magnetic resonance spectroscopy; MS, mass spectrometry; RAD, everolimus; SRL, sirolimus.
Interestingly, several clinical studies showed enhancement of cyclosporine toxicity by everolimus. While everolimus/cyclosporine trough blood concentration ratios targeted in these clinical studies ranged from 1 : 10 to 1 : 25 (Kovarik et al., 2001; 2002; Nashan, 2002), concentration ratios in our studies were only 1 : 3.3 or 1 : 5 (Serkova et al., 2000; 2001). Our results indicate that everolimus, in contrast to sirolimus, has the potential to antagonize cyclosporine toxicity through a yet unidentified mechanism. However, the cyclosporine/everolimus dose and blood concentration ratios seem critical (Serkova & Christians, 2003).
In this study, we chose rat brain slices as a model to study the time-dependent effects of cyclosporine and mTOR inhibitors on cell metabolism to be able to compare our results with our previous studies using the same model (Serkova et al., 1999; 2000; 2002). Based on the results of our previous studies, we also knew that cyclosporine and mTOR inhibitors mainly affect glucose metabolism. Due to the almost exclusive use of glucose as an energy substrate, the brain allowed us to study the effect of the study drugs on cell metabolism without interference by lipid metabolism. However, the clinically most important organ of cyclosporine toxicity in combination with mTOR inhibitors is the kidney. We have meanwhile extended our studies to the kidney and first results showed that the effects of cyclosporine on cell biochemistry described here can be translated to the kidney (Serkova et al., 2003).
In summary, our study showed (A) that the effects of cyclosporine on cell metabolism are time-dependent, with initial inhibition of mitochondrial glucose and high-energy phosphate metabolism and, after several hours, activation of anaerobic glycolysis to compensate for the inhibition of mitochondrial production of high-energy phosphates, (B) that ROS play a key role in the negative effects of cyclosporine on mitochondrial glucose metabolism and oxidative phosphorylation, and that decrease of ROS concentration can antagonize the negative effects of cyclosporine on mitochondrial metabolism, (C) that sirolimus inhibited anaerobic glycolysis and therefore the main mechanism used by the cells to compensate for cyclosporine-induced inhibition of mitochondrial energy metabolism, thus enhancing the negative effects of cyclosporine on cell energy metabolism, and (D) that everolimus has the potential to antagonize the negative effects of cyclosporine on mitochondrial metabolism (Table 2).
Acknowledgments
This study was supported by the German Research Foundation (DFG, Grant SE 985-1/2), the National Institute of Health (RO1 DK065094-01), and by Novartis Pharma AG (Basel, Switzerland). We thank Dr Peter Marbach (Novartis Pharma AG) for his valuable advice.
Abbreviations
- MRS
magnetic resonance spectroscopy
- PCA
perchloric acid
- PCr
phosphocreatine
References
- ANDOH T.F., LINDSLEY J., FRANCESCHINI N., BENNETT W.M. Synergistic effects of cyclosporine and rapamycin in a chronic nephrotoxicity model. Transplantation. 1996;62:311–316. doi: 10.1097/00007890-199608150-00002. [DOI] [PubMed] [Google Scholar]
- AUPETIT B., GHAZI A., BLANCHOUIN N., TOURY R., SHECHTER E., LEGRAND J.-C. Impact on energy metabolism of quantitative and functional cyclosporine-induced damage of kidney mitochondria. Biochim. Biophys. Acta. 1988;936:325–331. doi: 10.1016/0005-2728(88)90008-4. [DOI] [PubMed] [Google Scholar]
- BAI S., BRUNNER L.J., STEPKOWSKI S.M., NAPOLI K.M., KAHAN B.D. Effect of low dose cyclosporine and sirolimus on hepatic drug metabolism in the rat. Transplantation. 2001;71:1585–1592. doi: 10.1097/00007890-200106150-00017. [DOI] [PubMed] [Google Scholar]
- BECHSTEIN W.O. Neurotoxicity of calcineurin inhibitors: impact and clinical management. Transplant. Int. 2000;13:313–326. doi: 10.1007/s001470050708. [DOI] [PubMed] [Google Scholar]
- CRUZ F., WOLF A. Effects of the novel cyclosporine derivative PSC833 on glucose metabolism in rat primary cultures of neuronal and glial cells. Biochem. Pharmacol. 2001;62:129–139. doi: 10.1016/s0006-2952(01)00642-6. [DOI] [PubMed] [Google Scholar]
- DIAS V.C., MADSEN K.L., MULDER K.E., KEELAN M., YATSCOFF R.W., THOMSON A.B. Oral administration of rapamycin and cyclosporine differentially alter intestinal function in rabbits. Dig. Dis. Sci. 1998;43:2227–2236. doi: 10.1023/a:1026610404647. [DOI] [PubMed] [Google Scholar]
- ERECINSKA M., WILSON D.F. Regulation of cellular energy metabolism. J. Membr. Biol. 1982;70:1–14. doi: 10.1007/BF01871584. [DOI] [PubMed] [Google Scholar]
- FAULDS D., GOA K.L., BENFIELD P. Cyclosporin. A review of its pharmacodynamic and pharmacokinetic properties, and therapeutic use in immunoregulatory disorders. Drugs. 1993;45:953–1040. doi: 10.2165/00003495-199345060-00007. [DOI] [PubMed] [Google Scholar]
- GIJTENBEEK J.M., VAN DEN BENT M.J., VECHT C.J. Cyclosporine neurotoxicity. J. Neurol. 1999;246:339–346. doi: 10.1007/s004150050360. [DOI] [PubMed] [Google Scholar]
- GUMMERT J.F., IKONEN T., MORRIS R.E. Newer immunosuppressive drugs: a review. J. Am. Soc. Nephrol. 1999;10:1366–1380. doi: 10.1681/ASN.V1061366. [DOI] [PubMed] [Google Scholar]
- GUPTA R.K., WITTENBERG B.A. 31P-NMR studies of isolated adult heart cells: effect of myoglobin inactivation. Am. J. Physiol. 1991;261:H1155–H1163. doi: 10.1152/ajpheart.1991.261.4.H1155. [DOI] [PubMed] [Google Scholar]
- HARVEY P.A., GREADY J.E., HICKEY H.M., LE COUTEUR D.G., MCLEAN A.J. 31P and 1H NMR spectroscopic studies of liver extracts of carbon tetrachloride-treated rats. NMR Biomed. 1999;12:395–401. doi: 10.1002/(sici)1099-1492(199910)12:6<395::aid-nbm568>3.0.co;2-m. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D. Cyclosporine. N. Engl. J. Med. 1989;321:1725–1738. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D. The synergistic effects of cyclosporine and sirolimus. Transplantation. 1997;63:170. doi: 10.1097/00007890-199701150-00034. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D. Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: a randomized multicentre study. The Rapamune US Study Group. Lancet. 2000;356:194–202. doi: 10.1016/s0140-6736(00)02480-6. [DOI] [PubMed] [Google Scholar]
- KAHAN B.D., KRAMER W.G. Median effect analysis of efficacy versus adverse effects of immunosuppressants. Clin. Pharmacol. Ther. 2001;70:74–81. doi: 10.1067/mcp.2001.116309. [DOI] [PubMed] [Google Scholar]
- KOVARIK J.M., KAHAN B.D., KAPLAN B., LORBER M., WINKLER M., ROUILLY M., GERBEAU C., CAMBON N., BOGER R., RORDORF C. Longitudinal assessment of everolimus in de novo renal transplant recipients over the first post-transplant year: Pharmacokinetics, exposure–response relationships, and influence on cyclosporine. Clin. Pharmacol. Ther. 2001;69:48–56. doi: 10.1067/mcp.2001.112969. [DOI] [PubMed] [Google Scholar]
- KOVARIK J.M., KAPLAN B., TEDESCO SILVA H., KAHAN B.D., DANTAL J., VITKO S., BOGER R., RORDORF C. Exposure–response relationships for everolimus in de novo kidney transplantation: defining a therapeutic range. Transplantation. 2002;72:920–925. doi: 10.1097/00007890-200203270-00016. [DOI] [PubMed] [Google Scholar]
- LEIBFRITZ D. An introduction to the potential of 1H-, 31P- and 13C-NMR spectroscopy. Anticancer Res. 1996;16:1317–1324. [PubMed] [Google Scholar]
- MEYER M.A. Elevated basal ganglia glucose metabolism in cyclosporine neurotoxicity: a positron emission tomography imaging study. J. Neuroimaging. 2002;12:92–93. doi: 10.1111/j.1552-6569.2002.tb00102.x. [DOI] [PubMed] [Google Scholar]
- NASHAN B. Early clinical experience with a novel rapamycin derivative. Ther. Drug Monitor. 2002;24:53–58. doi: 10.1097/00007691-200202000-00010. [DOI] [PubMed] [Google Scholar]
- NEUHAUS P., KLUPP J., LANGREHR J.M. mTOR inhibitors: an overview. Liver Transplant. 2001;7:473–484. doi: 10.1053/jlts.2001.24645. [DOI] [PubMed] [Google Scholar]
- NICHOLSON J.K., WILSON I.D. Understanding ‘global' systems biology: metabonomics and the continuum of metabolism. Nat. Rev. Drug Discov. 2003;8:668–676. doi: 10.1038/nrd1157. [DOI] [PubMed] [Google Scholar]
- PARRA T., DE ARRIBA G., CONEJO J.R., CANTERO M., ARRIBAS I., RODRIGUEZ-PUYOL D., RODRIGUEZ-PUYOL M., CARBALLO F. Cyclosporine increases local glomerular synthesis of reactive oxygen species in rats: effect of vitamin E on cyclosporine nephrotoxicity. Transplantation. 1998;66:1325–1329. doi: 10.1097/00007890-199811270-00011. [DOI] [PubMed] [Google Scholar]
- PODDER H., STEPKOWSKI S.M., NAPOLI K.L., CLARK J., VERANI R.R., CHOU T.C., KAHAN B.D. Pharmacokinetic interactions augment toxicities of sirolimus/cyclosporine combinations. J. Am. Soc. Nephrol. 2001;12:1059–1071. doi: 10.1681/ASN.V1251059. [DOI] [PubMed] [Google Scholar]
- SCHREIBER S.L., CRABTREE G.R. The mechanism of action of cyclosporin A and FK506. Immunol. Today. 1992;13:136–142. doi: 10.1016/0167-5699(92)90111-J. [DOI] [PubMed] [Google Scholar]
- SCHUURMAN H.J., COTTENS S., FUCHS S., JOERGENSEN J., MEERLOO T., SEDRANI R., TANNER M., ZENKE G., SCHULER W. SDZ RAD, a new rapamycin derivative: synergism with cyclosporine. Transplantation. 1997;64:32–35. doi: 10.1097/00007890-199707150-00007. [DOI] [PubMed] [Google Scholar]
- SERKOVA N., CHRISTIANS U. Transplantation: toxicokinetics and mechanisms of toxicity of cyclosporine and macrolides. Curr. Opin. Investig. Drugs. 2003;4:1287–1296. [PubMed] [Google Scholar]
- SERKOVA N., DONOHOE P., GOTTSCHALK S., HAINZ C., NIEMANN C.U., BICKLER P.E., LITT L., BENET L.Z., LEIBFRITZ D., CHRISTIANS U. Comparison of the effects of cyclosporine A on the metabolism of perfused rat brain slices during normoxia and hypoxia. J. Cerebr. Blood Flow Metab. 2002;22:342–352. doi: 10.1097/00004647-200203000-00012. [DOI] [PubMed] [Google Scholar]
- SERKOVA N., KLAWITTER J., NIEMANN C.U. Organ-specific response to inhibition of mitochondrial metabolism by cyclosporine in the rat. Transplant. Int. 2003;16:748–755. doi: 10.1007/s00147-003-0631-1. [DOI] [PubMed] [Google Scholar]
- SERKOVA N., LITT L., JAMES T.L., SADEE W., LEIBFRITZ D., BENET L.Z., CHRISTIANS U. Evaluations of individual and combined neurotoxicity of the immunosuppressants cyclosporine and sirolimus by in vitro multinuclear NMR spectroscopy. J. Pharmacol. Exp. Ther. 1999;289:800–806. [PubMed] [Google Scholar]
- SERKOVA N., LITT L., LEIBFRITZ D., HAUSEN B., MORRIS R.E., JAMES T.L., BENET L.Z., CHRISTIANS U. The novel immunosuppressant SDZ-RAD protects rat brain slices from cyclosporine-induced reduction of high-energy phosphates. Br. J. Pharmacol. 2000;129:485–492. doi: 10.1038/sj.bjp.0703079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SERKOVA N., JACOBSEN W., NIEMANN C.U., BENET L.Z., LEIBFRITZ D., CHRISTIANS U. Sirolimus, but not the structurally related RAD (everolimus), enhances the negative effects of cyclosporine on mitochondrial metabolism in the rat brain. Br. J. Pharmacol. 2001;133:875–885. doi: 10.1038/sj.bjp.0704142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- UEMOTO S., TANAKA K., ASONUMA K., OKAMURA R., KITAKADO Y., MATSUOKA S., OZAKI N., OZAWA K., HASHIDA T., INUI K., HORI R. Effect of cyclosporine on oxidative phosphorylation and adenylate energy charge of regenerating rat liver. Res. Exp. Med. 1989;189:313–320. doi: 10.1007/BF01855036. [DOI] [PubMed] [Google Scholar]
- WANG C., SALAHUDEEN A.K. Cyclosporine nephrotoxicity: attenuation by an antioxidant-inhibitor of lipid peroxidation in vitro and in vivo. Transplantation. 1994;58:940–946. doi: 10.1097/00007890-199410270-00014. [DOI] [PubMed] [Google Scholar]
- WANG H., JOSEPH J.A. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic. Biol. Med. 1999;27:612–616. doi: 10.1016/s0891-5849(99)00107-0. [DOI] [PubMed] [Google Scholar]
- WANG X., LUO H., PERKS A., WU J. Rapamycin inhibits aldolase A expression during human leukocytes activation. J. Cell. Biochem. 1996;63:239–251. doi: 10.1002/(sici)1097-4644(19961101)63:2<239::aid-jcb11>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- WIJDICKS E.F. Neurotoxicity of immunosuppressive drugs. Liver Transplant. 2001;7:937–942. doi: 10.1053/jlts.2001.27475. [DOI] [PubMed] [Google Scholar]
- WOLF A., TRENDELENBURG C.F., DIEZ-FERNANDEZ C., PRIETO P., HOUY S., TROMMER W.E., CORDIER A. Cyclosporine A-induced oxidative stress in rat hepatocytes. J. Pharmacol. Exp. Ther. 1997;280:1328–1334. [PubMed] [Google Scholar]