

Abstract
We describe a procedure for a space-time description of protein structures. The method is capable of determining populations of conformational substates, and amplitudes and directions of internal protein motions. This is achieved by fitting static and dynamic NMR data. The approach is based on the jumping-among-minima concept. First, a wide conformational space compatible with structural NMR data is sampled to find a large set of substates. Subsequently, intrasubstate motions are sampled by using molecular dynamics calculations with force field energy terms. Next, the populations of substates are fitted to NMR relaxation data. By diagonalizing a second moment matrix, directions and amplitudes of motions are identified. The method was applied to the adhesion domain of human CD2. We found that very few substates can account for most of the experimental data. Furthermore, only two types of collective motions have high amplitudes. They represent transitions between a concave (closed) and flat (open) binding face and resemble the change upon counter-receptor (CD58) binding.
Many experimental observations indicate that proteins continuously fluctuate, and many proteins change their conformation when performing their function, most significantly when binding ligands (1). NMR data contain implicitly information about the details of internal motions. However, dynamics studies with this technique primarily have been used to obtain information about the time scales of internal motions and order parameters S2 as rough estimates of the extents of motions (2, 3). Few attempts have been made to move beyond this. For example, Brüschweiler and Case (4) have developed a collective NMR relaxation model. They used a set of low-frequency normal mode coordinates to fit order parameter S2 as defined by the model free approach of Lipari and Szabo (5). Ulyanov et al. (6) considered multiple substates in the PARSE (probability assessment via relaxation rates of a structural ensemble) procedure. They attempted to reproduce two-dimensional nuclear Overhauser data by assuming multiple conformers, and probabilities of conformers were determined. Nilges and coworkers (7, 8) have studied protein motions with principle component analysis and observed that relatively few concerted motions can explain the relaxation properties the proteins studied.
Here we describe our attempts to make a more complete use of dynamic data and to develop a strategy for obtaining a space-time description of protein structures by fitting both structural nuclear Overhauser constraints and dynamic 15N relaxation data. The goal was to obtain the coordinates of the most populated conformational states and the amplitudes and directions of the dominant internal motions. We assume that dynamic protein structures can be described by a distribution of conformational substates, and motions within and between the substates. Intrasubstate motions are fast and can be simulated reasonably well with molecular dynamics (MD) approaches. Intersubstate motions are much slower and not readily accessible to MD calculations. However, they represent the dominant motions with the largest amplitudes. Therefore, we based our analysis on the jumping-among-minima (JAM) concept (9). Intrasubstate motions are simulated with MD calculations using common force fields, and intersubstate motions are simulated with a procedure that averages the contributions of all the substates. The weights (populations) of the substates that reproduce best the experimental data are fitted to the experimental dynamics data. With this achieved, a second-moment matrix for the deviations of the coordinates from the average positions is defined. The matrix is diagonalized, and eigenvalues and eigenvectors define the amplitudes and directions of internal collective motions (JAM modes).
We apply this approach to the adhesion domain of human CD2 (hCD2), a transmembrane glycoprotein found on T lymphocytes and natural killer cells (10). CD2 mediates cell–cell recognition by binding to the counter-receptor CD58 that is found on the surface of a variety of target cells, such as antigen-presenting cells. CD2 contains two Ig superfamily domains. Solution and crystal structures have been determined for the adhesion domains or both extracellular domains of rat CD2 (11, 12) and hCD2 (13–15). Recently, the structures of the adhesion domain of the counter-receptor hCD58 (16) and the complex of the adhesion domains of hCD2 and hCD58 also have been solved (17). In the crystal structures of rat and human CD2, a crystallographic dimer is observed with a conformation similar to the CD2/CD58 heterodimer (12, 14, 17) and more different from free CD2 in solution. Thus, the conformational changes of either protein upon counter-receptor binding have become known. The adhesion domain of hCD2 forms a nine-stranded Ig superfamily V-set fold. The dynamics of hCD2 have been characterized with 15N NMR relaxation experiments (18). Here, we have present a space-time description of the adhesion domain of CD2 based on the JAM approach. Surprisingly, a very small number of JAM modes dominate the dynamics of the adhesion domain of hCD2, and these appear to be directly related to the main function of the protein, interaction with the counter-receptor, hCD58.
Methods
Principle of Approach.
In the JAM approach (9), the ensemble average of an arbitrary dynamical variable A, 〈A〉, is given by 〈A〉 = ∑k=1M fk〈A〉k, where k is the index for the conformational substate. 〈A〉k represents the ensemble average of variable A within the substate k, and fk is the statistical weight of the substate. The average 〈A〉k is obtained by molecular simulation sampling motions within the substate k. The statistical weights fk are determined to reproduce the experimentally determined value of 〈A〉. This equation can be easily extended to matching several dynamic parameters simultaneously. In this paper, the statistical weights are fitted to reproduce NMR order parameters, S2, as an initial application (5). Order parameters were calculated by using the averaging scheme of our JAM model, which is based on the method by Henry and Szabo (19) and is shortly described below. The order parameter, S, is given by,
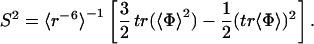 |
1 |
The elements of the tensor Φ(t) are defined as,
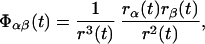 |
2 |
where r(t) = |r(t)|. A vector r(t) is a bond vector between two nuclei, such as proton and nitrogen. rα(t) and rβ(t) denote the x, y, or z components of r(t). In the JAM model, Eq. 1 can be rewritten as,
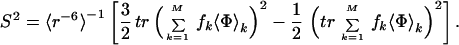 |
3 |
The average of tensor Φ within the kth substate, 〈Φ〉k, is determined from a molecular simulation. Once a set of fk is determined, we can calculate various quantities, such as directions and amplitudes of atomic fluctuations. This is achieved by defining a second moment matrix C with the elements Cij. It describes the fluctuations of all the xl and is defined as,
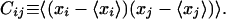 |
4 |
The ith diagonal element of C is a mean-square fluctuation along the ith coordinate from the average conformation. Therefore, the matrix C gives the complete information on the magnitudes of the atomic fluctuations. In the JAM model (9), C is expressed as:
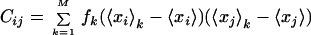 |
5 |
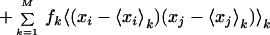 |
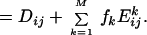 |
The matrix Ek = (Eijk), the second moment matrix within the kth substate, is obtained directly from molecular simulation in each conformational substate, and the matrix D = (Dij), which represents the distribution of conformational substates, is determined from fk and the average positions of the substates. To extract large-amplitude motions, collective coordinates are determined by solving the standard eigenvalue problem of either C or D. The former is called principal component analysis (20), the latter JAM analysis (9).
Sequence of Refinement Steps.
The space-time refinement consists of three stages. In stage 1, an ensemble of structures is generated that satisfy the spatial restraints. This is achieved with five independent MD runs that include restraint energy terms, covalent energy terms, and quadratic repulsive energy terms between nonbonded atom pairs. To sample a large conformational space, relatively weak constraints were used. We checked the efficiency of the sampling in these MD simulations by using principal component analysis. We found that the conformational space highly overlapped between the different simulations. This indicates that these simulations sample rather completely the conformational space compatible with the spatial NMR constraints (data not shown). In stage 2, intrasubstate fluctuations are simulated by using MD calculations with full force field. In the first cycle, 50 conformations were sampled from the former five MD trajectories at every 50 psec, and the restraint energy was minimized. Starting from these minimum-energy conformations, MD simulations are carried out in vacuo by using the full force fields without restraint energy terms. It should be noted that intrasubstate fluctuations in solution occurring on a short time scale (mostly consisting of vibrational or damped oscillation modes) are compatible with those determined by the simulation in vacuo (9). However, the longer time-scale behavior is rather different. In each MD simulation, after 10-psec equilibration, 15-psec trajectory was stored and used to calculate time correlation functions for the intrasubstate motions. In stage 3, statistical weights fk are determined to minimize the difference between experimental and calculated order parameters. Stages 2 and 3 are repeated until the fitting of the order parameters converges. In work described here, this cycle was repeated twice. For the second cycle of stages 2 and 3, the 30 substates with the highest statistical weights fk were selected. In addition, 35 substates, which are relatively close in conformational space to the substates with relatively high values of fk (larger than 0.01), were added to the ensemble. We examined how the refinement results depend on the initial values of fc and the number of the order parameters used in the refinement. We found that the final set of fk values does not depend on the initial set. Furthermore, a limited variation of the number of order parameters used had only small effect on the final fk values.
Results and Discussion
Data Used for Space-Time Refinement of hCD2 Adhesion Domain.
The space-time refinement of hCD2 was based on the following experimental data. We started out randomly choosing one of an ensemble of 18 structures previously determined with static NMR constraints. The spatial constraints used for the space-time refinement described here were adapted from Wyss et al. (15). They consisted of 50 distance restraints for hydrogen bonding pairs, 945 nuclear Overhauser restraints (860 intrapolypeptide, 46 intraglycan, and 39 polypeptideglycan), and 192 dihedral restraints (92 φ, 19 χ1, 13 β-methylene, and 68 glycan). The dynamic constrains were adapted from previously performed NMR relaxation parameters of NH groups (18). Using relaxation rates, RN(Nz), RN(Nx,y), and RN(Hz → Nz), spectral densities, Jeff(0), J(ωN), and Jave(ωH) were determined by using spectral density mapping methods (21, 22). First, the three spectral densities are fitted to the original model free approach of Lipari and Szabo (5) assuming isotropic tumbling with a single overall correlation time and a single exponential decay for internal motion. If the fitting results are not satisfactory, an extended model with a chemical exchange term λRex was applied. Of the 102 order parameters obtained from the experimental data Wyss et al. (18), 66 main-chain order parameters with the lowest experimental errors were used for the refinement.
Two Substates Are Dominant.
The application of this procedure to the adhesion domain of hCD2 showed the rather surprising results that very few substates can account for the mobility data in hCD2. In the final stage of this space-time refinement, 65 substates were used. Among them, only two substates have dominantly large statistical weights. The most populated substate 1 has the statistical weight f1 = 0.42 and substate 2 has a weight f2 = 0.24. Thus, the probability of the state point staying in either substate 1 or 2 is 0.66. The structures of the six most populated substates are shown in Fig. 1. To illustrate the populations, the thickness of the bonds is proportional to the values of fk.
Figure 1.

Stereo diagram of the dominant substates of the adhesion domain of hCD2 in solution. The six most populated substates are shown. The relative population of each substate is indicated with the thickness of the lines. Substates 1 and 2 are colored white and yellow, respectively, the others are colored light blue. The instantaneous structures closest to the average structures of each substate are drawn. The β-strands are labeled A–G. The figure was prepared with the program molmol (27).
CD58-Binding Face Is Very Flexible.
With the fk values determined, we calculated the second-moment matrix C that contains the mean square atomic fluctuations as the diagonal elements. These can be compared with experimentally measured values from NMR and x-ray data. In Fig. 2, the rms fluctuation (RMSF) for each residue averaged over the main-chain atoms is compared with values derived from crystallographic B factor. The curves are in good agreement although the values calculated from the NMR data are generally smaller. Six loops, the B–C, C–C′, C′–C", C"–D, and D–E loops are more flexible, whereas the A–B and E–F loops are rigid. This may be functionally important because the loops near the binding site are flexible whereas two loops relatively far from it are not. This suggests that the regions near the binding site are designed to be flexible even in the free CD2 prepared for the conformational change upon binding, whereas other regions are designed to be more rigid. It also should be noted that the crystal structure of hCD2 contains the N-terminal V-set adhesion domain and a second C-type domain. Therefore, the RMSF near the C terminus in the crystal is relatively low. Except for this region, the RMSF in crystal is much larger than that in present refinement. It is reasonable because the RMSF in the refinement originated only from internal motion, whereas B factor consists of internal contribution, external contribution, and crystal defects (23).
Figure 2.

Comparison of rms fluctuations of backbone atoms, obtained from the diagonal elements of the second moment matrix C (solid line) and B factor of the x-ray structure of hCD2 (dotted line) (14). The latter was obtained by using the relation that mean square fluctuations of the lth atom are given by 〈Δxl2〉 = 〈(xi − 〈xi〉)2 + (xi+1 − 〈xi+1〉)2 + (xi+2 − 〈xi+2〉)2〉 = (3/8π2)Bl (dashed line).
The Motions in Free hCD2 Resemble the Conformational Change upon CD58 Binding.
The eigenvectors and eigenvalues of the D component in the second moment matrix (Eq. 5) yield collective coordinates and amplitudes of motions along these coordinates. The collective coordinates are linear combinations of the Cartesian atomic coordinates and form an orthogonal set (9). Pictorially, the axis of the first JAM mode is defined as the direction along which conformational substates are most broadly distributed. That of the second JAM mode is orthogonal to the first one, and along this axis, substates are distributed second most. Among 65 JAM modes, the first and second one dominantly contributed to the total intersubstate mean square fluctuation (MSF) by 77.1%. In other words, conformational substates are mostly distributed in the two-dimensional space spanned by the first and second JAM modes. The first JAM mode, which contributes to the total MSF by 63.5%, is directly involved with the conformational transition between substate 1 and 2. The second JAM mode contributes to total MSF by 13.6%. The motions along these two axes are typical dynamic domain motions. We further analyzed the first two modes with the program dyndom (24), which identifies rigid substructures and linker regions. The results are shown in Fig. 3. The moving domain in the first JAM mode consists of a part of the B–C loop, a part of the F strand, and the F–G loop. The moving domain of the second JAM mode is on the C′–C" loop. The CD58 binding site consists of the C–C′, C′–C", and F–G loops and the regions between (25, 26), which includes most of the moving domains of both JAM modes. In Fig. 4, projections of the positions of the significantly populated conformational substates onto the two-dimensional space spanned by the first and second JAM modes are shown. The collective coordinates of the two crystal structures of the homodimer and the heterodimer (14, 25, 26) also are shown. If CD2 moves along the positive direction in the first JAM mode, or along the negative direction in the second JAM mode (Fig. 4), the binding site is more opened. Therefore, substate 2 is more opened than substate 1 with respect to mode 1, and both crystal structures have a more open binding site than all substates of the free protein in solution. This is shown in close detail in Fig. 5. Thus, it seems that the directions of the collective motions as described above are closely related to the conformational changes occurring upon binding the counter-receptor CD58. It seems that the dominant change is along the first JAM mode. There are significant changes along the second and several other JAM modes as well, however. The motions in the free protein resemble the conformational change upon CD58 binding although the conformational fluctuations in the free state have smaller amplitudes than the changes occurring upon CD58 binding. In the first cycle of the refinement, the conformations relatively close to the CD58-binding form were included in stage 3; however, their fk values were negligibly small. The binding process may use conformations that are populated in free CD2 and sufficient to make an energetically favorable initial complex, followed by additional small changes to form the final complex structure.
Figure 3.

Dynamic domain motions in (a) the first and (b) second JAM mode. The residues shown in blue, red, and green represent fixed-domain, moving-domain, and bending residues, respectively, as determined by dyndom (24). The arrows represent the axes of domain motions. The moving-domains rotate around these axes. The residues in the fixed domain are (a) Gly-11–Gln-25, Lys-34–Tyr-81, Lys-91–Asn-92, Leu-94–Lys-101, and (b) Gly-11–Phe-47, Phe-54–Lys-101. Those in moving domain are (a) Met-26–Ile-33, Lys-82–Gly-90, Val-93 and (b) Arg-48–Thr-53. Bending residues are (a) Gln-25–Met-26, Asp-32–Lys-34, Tyr-81–Lys-82, Gly-90–Leu-94 and (b) Gln-46–Arg-48, Thr-53–Phe-54. The residues that act as a mechanical hinge are (a) Asp-32, Ile-33, Lys-34, Lys-82 and (b) Gln-46, Phe-47, Arg-48, Thr-53, and Phe-54. The figure was created by using the programs dyndom (24) and rasmol (28).
Figure 4.

Statistical weights fk of each substate projected onto the two-dimensional space spanned by the first and second JAM coordinates. The bars shown by white and yellow represent fk of substates 1 and 2, respectively. The magenta and blue marks represent the positions of the crystal structure of the hCD2 dimer (14) and the complex with hCD58 (17), in this JAM mode subspace. The axes of the first and second JAM coordinates are scaled by the SDs of 0.90 Å and 0.42 Å, respectively.
Figure 5.

The CD2 surface flattens upon CD58 binding (in stereo). Substates 1 and 2 of free CD2 are shown in white and yellow, respectively. The crystal structure of the hCD2, which is a crystallographic homodimer (14) and the CD2/CD58 heterodimer (17) are shown in dark and light blue, respectively. The figure was prepared with insight ii (Molecular Simulations, Waltham, MA).
Conclusion
We have developed a method that can determine the average structure, the distribution of conformational substates, as well as directions and amplitudes of internal motions in a protein. We have applied this method to the adhesion domain of hCD2 and have shown that the fluctuations in the free protein resemble the onset of the counter-receptor-binding event. Here, we have fitted the dynamic structures to the order parameters, S2, derived from 15N relaxation experiments. Extension of this approach to fitting in addition nuclear Overhauser intensities to a dynamic model is straight forward, although computationally challenging. The time scale of the motions analyzed here is in the ps to ns range, which is faster than the overall motions of the protein. Experimentally, we have observed slower motions in the μs to ms range as well, in particular for the CD58-binding site (18). To rationalize motions at this slow time scale is still beyond the approach presented here.
Acknowledgments
From April 1998 to March 1999, A.K. was a Monbusho-Sponsored Japanese Overseas Research Fellow at Harvard Medical School. This work was supported by National Science Foundation Grant MCB 931 6938 and National Institutes of Health Grant GM47467.
Abbreviations
- MD
molecular dynamics
- JAM
jumping among minima
- hCD
human CD
Footnotes
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.030540397.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.030540397
References
- 1.Gerstein M, Lesk A M, Chothia C. Biochemistry. 1994;33:6739–6749. doi: 10.1021/bi00188a001. [DOI] [PubMed] [Google Scholar]
- 2.Wagner G. Curr Opin Struct Biol. 1994;3:748–754. doi: 10.1016/0959-440x(94)90265-8. [DOI] [PubMed] [Google Scholar]
- 3.Kay L E. Biochem Cell Biol. 1998;76:145–152. doi: 10.1139/bcb-76-2-3-145. [DOI] [PubMed] [Google Scholar]
- 4.Brüschweiler R, Case D A. Phys Rev Lett. 1994;72:940–943. doi: 10.1103/PhysRevLett.72.940. [DOI] [PubMed] [Google Scholar]
- 5.Lipari G, Szabo A. J Am Chem Soc. 1982;104:4546–4559. [Google Scholar]
- 6.Ulyanov N B, Schmitz U, Kumar A, James T L. Biophys J. 1995;68:13–24. doi: 10.1016/S0006-3495(95)80181-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abseher R, Horstink L, Hilbers C W, Nilges M. Proteins. 1998;31:370–382. [PubMed] [Google Scholar]
- 8.Horstink L, Abseher R, Nilges M, Hilbers C W. J Mol Biol. 1999;287:569–577. doi: 10.1006/jmbi.1999.2629. [DOI] [PubMed] [Google Scholar]
- 9.Kitao A, Hayward S, Go N. Proteins. 1998;33:496–517. doi: 10.1002/(sici)1097-0134(19981201)33:4<496::aid-prot4>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 10.Davis S J, Ikemizu S, Wild M K, van der Merwe P A. Immunol Rev. 1998;163:217–236. doi: 10.1111/j.1600-065x.1998.tb01199.x. [DOI] [PubMed] [Google Scholar]
- 11.Driscoll P C, Cyster J G, Campbell I D, Williams A F. Nature (London) 1991;353:762–765. doi: 10.1038/353762a0. [DOI] [PubMed] [Google Scholar]
- 12.Jones E Y, Davis S J, Williams A F, Harlos K, Stuart D I. Nature (London) 1992;360:232–239. doi: 10.1038/360232a0. [DOI] [PubMed] [Google Scholar]
- 13.Wyss D F, Withka J M, Knoppers M H, Sterne K A, Recny M A, Wagner G. Biochemistry. 1993;32:10995–11006. doi: 10.1021/bi00092a008. [DOI] [PubMed] [Google Scholar]
- 14.Bodian D L, Jones E Y, Harlos K, Stuard D I, Davis S J. Structure (London) 1994;2:755–766. doi: 10.1016/s0969-2126(94)00076-x. [DOI] [PubMed] [Google Scholar]
- 15.Wyss D F, Choi J S, Li J, Knoppers M H, Willis K J, Arulanandam A R N, Smolyar A, Reinherz E L, Wagner G. Science. 1995;269:1273–1278. doi: 10.1126/science.7544493. [DOI] [PubMed] [Google Scholar]
- 16.Sun Z, Dotsch V, Kim M, Li J, Reinherz E, Wagner G. EMBO J. 1999;18:2941–2949. doi: 10.1093/emboj/18.11.2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wang J, Smolyar A, Tan K, Liu J, Kim M, Sun Z, Wagner G, Reinherz E. Cell. 1999;97:791–803. doi: 10.1016/s0092-8674(00)80790-4. [DOI] [PubMed] [Google Scholar]
- 18.Wyss D F, Dayie K T, Wagner G. Protein Sci. 1997;6:534–542. doi: 10.1002/pro.5560060303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Henry E R, Szabo A. J Chem Phys. 1985;82:4753–4761. [Google Scholar]
- 20.Kitao A, Hirata F, Go N. Chem Phys. 1991;158:447–472. [Google Scholar]
- 21.Peng J W, Wagner G. Biochemistry. 1992;31:8571–8586. doi: 10.1021/bi00151a027. [DOI] [PubMed] [Google Scholar]
- 22.Lefevre J-F, Dayie K T, Peng J W, Wagner W. Biochemistry. 1996;35:2674–2686. doi: 10.1021/bi9526802. [DOI] [PubMed] [Google Scholar]
- 23.Kidera A, Inaka K, Matsushima M, Go N. J Mol Biol. 1992;225:477–486. doi: 10.1016/0022-2836(92)90933-b. [DOI] [PubMed] [Google Scholar]
- 24.Hayward S, Berendsen H J C. Proteins. 1998;30:144–154. [PubMed] [Google Scholar]
- 25.Arulanandam A R N, Withka J M, Wyss D F, Wagner G, Kister A, Pallai P, Recny M A, Reinherz E L. Proc Natl Acad Sci USA. 1993;90:11613–11617. doi: 10.1073/pnas.90.24.11613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Arulanandam A R N, Kister A, McGregor M J, Wyss D F, Wagner G, Reinherz E L. J Exp Med. 1994;180:1861–1871. doi: 10.1084/jem.180.5.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Koradi R, Billeter M, Wuthrich K. J Mol Graphics. 1996;14:51–55. doi: 10.1016/0263-7855(96)00009-4. [DOI] [PubMed] [Google Scholar]
- 28.Sayle R, Milner-White E J. Trends Biochem Sci. 1995;20:374–376. doi: 10.1016/s0968-0004(00)89080-5. [DOI] [PubMed] [Google Scholar]