

Abstract
Endoplasmic reticulum (ER) stress has been found to be associated with neurodegenerative diseases and diabetes mellitus. Whether ER stress is involved in the development of heart disease is not known. Cardiac-specific expression of monocyte chemoattractant protein-1 (MCP-1) in mice causes the development of ischemic heart disease. Here we report that microarray analysis of gene expression changes in the heart of these transgenic mice revealed that a cluster of ER stress-related genes was transcriptionally activated in the heart during the development of ischemic heart disease. The gene array results were verified by quantitative real-time PCR that showed highly elevated transcript levels of genes involved in unfolded protein response such as ER and cytoplasmic chaperones, oxidoreductases, protein disulfide isomerase (PDI) family, and ER-associated degradation system such as ubiquitin. Immunoblot analysis confirmed the expression of chaperones, PDI, and ubiquitin. Immunohistochemical analyses showed that ER stress proteins were associated mainly with the degenerating cardiomyocytes. A novel ubiquitin fold modifier (Ufm1) that has not been previously associated with ER stress and not found to be induced under any condition was also found to be upregulated in the hearts of MCP mice (transgenic mice that express MCP-1 specifically in the heart). The present results strongly suggest that activation of ER stress response is involved in the development of ischemic heart disease in this murine model.
Keywords: monocyte chemoattractant protein-1, transgenic, myocardial inflammation
A variety of human disorders, including neurodegenerative diseases and diabetes mellitus, are known to involve endoplasmic reticulum (ER) stress. ER stress leads to accumulation of unfolded proteins in ER (17, 31), which in turn evokes the unfolded protein response (UPR) (33). This response attempts to reduce the amount of misfolded proteins by inducing the production of the ER chaperones, such as BiP/Grp78 and Grp94, that promote protein folding, and by reducing general protein synthesis (12) and enhancing the degradation of misfolded proteins via a ubiquitin-proteasome system termed ER-associated degradation (ERAD) (3, 39). The persistent accumulation of misfolded proteins beyond the capacity of ER quality control causes cellular dysfunction and cell death involved in several human disorders. Involvement of this process in heart disease has not been extensively studied (41), and whether it is involved in ischemic heart disease is unknown.
Ischemic heart disease, a leading cause of death, is widely recognized to be an inflammatory disease, and infiltration of monocytes/macrophages is thought to make a significant contribution to the pathophysiology of this disease (16). The major chemotactic factor for monocytes/macrophages, monocyte chemoattractant protein-1 (MCP-1), a member of the small inducible chemokine gene family, plays a role in the recruitment of monocytes to the sites of injury and infection. Recent studies revealed that MCP-1 plays an important role in the development of ischemic heart disease (6, 7, 14). However, the molecular changes induced by MCP-1 during the development of the disease are not known. The progression of such molecular changes can be best studied in an animal model. To investigate the molecular mechanisms involved in the induction of ischemic heart disease by the chronic inflammatory processes, we developed transgenic mice that express MCP-1 specifically in the heart (MCP mice; 18). The MCP mice manifest myocardial inflammation and develop thrombolic vasculopathy, cardiac dilatation, interstitial fibrosis, myocyte vacuolization, and mortality with congestive heart failure by ~6 mo of age (18, 23). The clinical, histological, and molecular features that occur in the hearts of these animals closely resemble the changes observed in human ischemic cardiomyopathy (1, 18, 23). This model is therefore suitable for elucidating how chronic inflammation contributes to the development of ischemic heart disease and to investigate whether ER stress response is involved in this process.
The aim of this study was to investigate whether the gene expression changes associated with ER stress are involved in the development of ischemic heart disease by using the MCP mouse model. Here we report that DNA microarray analysis of gene expression profile changes revealed transcriptional activation of a set of ER stress-related genes during the development of heart disease in this model. A recently identified ubiquitin fold modifier 1 (Ufm1) gene that had not been previously reported to be induced by any condition was also found to be upregulated. Quantitative real-time PCR validated the conclusions reached from the gene array analyses. The expression of ER stress-related proteins was also demonstrated by immunoblotting and immunohistochemical studies. These findings, for the first time, strongly suggest that ER stress response activation is involved in the development of ischemic heart disease induced by chronic inflammation.
MATERIALS AND METHODS
Animals used
The animal protocols used in this study were approved by the Animal Care and Use Committee of University of Central Florida and the Ohio State University. Hearts from five male animals for both wild-type and MCP mice from each age of 2, 4, 8, 16, and 24 wk were used. The hearts were removed, rinsed free of blood, and immediately either snap frozen in liquid nitrogen or fixed with buffered 10% (vol/vol) formalin for histopathological analyses. Frozen samples then underwent RNA and protein isolation for further microarray, real-time PCR, and Western blot analysis.
DNA microarray analysis
Total RNA was extracted from left ventricular specimens with RNA Kit (Invitrogen, Carslbad, CA). Generation of cDNA for Affymetrix arrays and hybridization conditions were set up according to the manufacture’s protocol (Affymetrix, Santa Clara, CA). Briefly, 10 μg of total RNA were used to synthesize double-stranded cDNA with a SuperScript synthesis kit (Bio-Rad, Hercules, CA), incorporating a T7 promoter sequence by using oligo(dT)-T7 primer. Biotin labeling of cDNA and hybridization with Murine Genome-U74A GeneChip [which has 6,000 functionally characterized sequences and 6,000 expressed sequence tags from the UniGene database (Affymetrix)] was done as recommended by the manufacturer. Data analysis was performed by using Affymetrix GeneChip 3.1 software under default parameter setting at the Heart and Lung Institute at Ohio State University.
Real-time quantitative PCR analysis
Total RNA samples used in microarray analysis were also used for quantitative real-time PCR. Primers used for real-time RT-PCR are listed in Table 1. The relative expression levels of each targeted gene were normalized by subtracting the corresponding mouse β-actin threshold cycle (CT) values by using the ΔΔCT comparative method (26).
Table 1.
Primer sequences used in real-time PCR
| Sequence No. | Gene Name | Primer Sequences (Mouse) | Accession No. | Product Size, bp |
|---|---|---|---|---|
| 1 | Asparagine synthetase | Forward: GAACACAGACAGCGTGGTG | NM_012055 | 152 |
| Reverse: CGGAGAACATCAAACAGGT | ||||
| 2 | PDI | Forward: AACGGGAGAAGCCATTGTA | AW045202 | 156 |
| Reverse: AGGTGTCATCCGTCAGCTCT | ||||
| 3 | Ribophorin | Forward: GCCAGGAAGTGGTGTTTGTT | D31717 | 158 |
| Reverse: CCAGAGGATTGGGTTCTTCA | ||||
| 4 | Cai | Forward: CTCTCCGGGAATTTGTCACA | NM009787 | 280 |
| Reverse: ATGTCGTTGGCAGTAGCATC | ||||
| 5 | ERO1 | Forward: AGCAAACAGCACCAAAGAA | NM026184 | 129 |
| Reverse: TGGTCCTGCGAATCATCATA | ||||
| 6 | CHOP | Forward: TATCTCATCCCCAGGAAACG | NM007837 | 258 |
| Reverse: CTGCTCCTTCTCCTTCATGC | ||||
| 7 | ATF6 | Forward: TGGGAGTGAGCTGCAAGTGT | XM129579 | 276 |
| Reverse: ATAAGGGGGAACCGAGGAG | ||||
| 8 | TRB3 | Forward: CGCTTTGTCTTCAGCAACTG | AF358868 | 354 |
| Reverse: CAAGTCGCTCTGAAGGTTCC | ||||
| 9 | BiP/Grp78 | Forward: ACCTGGGTGGGGAAGACTTT | NM022310 | 232 |
| Reverse: TCTTCAAATTTGGCCCGAGT | ||||
| 10 | Grp94 | Forward: TGGGTCAAGCAGAAAGGAG | NM011631 | 212 |
| Reverse: TCTCTGTTGCTTCCCGACTT | ||||
| 11 | HSP25 | Forward: CCTCTTCGATCAAGCTTTCG | NM013560 | 276 |
| Reverse: GCCTTCCTTGGTCTTCACTG | ||||
| 12 | HSP40 | Forward: CGGTTCCGAATCAAAGTTGT | NM018808 | 148 |
| Reverse: GGGAAATATGGACCTTGTG | ||||
| 13 | HSP70 | Forward: TGCAGCAGGACATCAAGTTC | AJ002387 | 157 |
| Reverse: TACGCCTCAGCAGTCTCCTT | ||||
| 14 | Ufm1 | Forward: CCGTTCACAGCAGTGCTAAA | NM026435 | 130 |
| Reverse: CCGTGCTTCAGGAAAACA | ||||
| 15 | β-Actin | Forward: GAAACAGCTTCAGCCCAAA | NM_007393 | 216 |
| Reverse: ACGTCAGGTGACTGCTGAA |
PDI, protein disulfide isomerase; ERO1, endoplasmic reticulum oxidoreductase; CHOP, C/EBP homologous protein; ATF6, activating transporter factor; TRB3, tribbles-related protein 3; HSP25, HSP40, HSP70, 25-kDa, 40-kDa, and 70-kDa heat shock protein; Ufm1, ubiquitin fold modifier 1.
Protein extraction and immunoblot analysis
The heart samples were homogenized in a buffer containing 20 mM Tris · HCl, pH 6.8, 1 mM EDTA, 1% SDS, 1 mM PMSF, and 1× protease inhibitor cocktail (Roche, Mannheim, Germany). Equal amounts of protein from each sample were separated by 10% (Grp78 and Grp94) or 12.5% [protein disulfide isomerase (PDI), 25-kDa heat shock protein (HSP25), 40-kDa heat shock protein (HSP40)] SDS-PAGE. The amount of proteins loaded was HSP40, 100 μg; Grp78, Grp94, ubiquitin, and PDI, 50 μg; HSP25, 10 μg. Percentage gel used and amount of protein loaded were dependent on the molecular weight of the protein and the sensitivity of the antibody, respectively. The separated proteins were transferred to polyvinylidene difluoride membranes (Immobilon-P, Millipore, MA), and the membranes were incubated with polyclonal antibody to HSP25, 40-kDa heat shock protein (HSP40), Bip/Grp78, Grp94, PDI (Stressgene Biotechnologies, Canada), or ubiquitin (Dako, Carpinteria, CA) followed by horseradish peroxidase-conjugated secondary antibodies. Immunoreactive bands were visualized with the SuperSignal West Pico enhanced chemiluminescence kit according to the manufacturer’s specifications (Pierce, Rockford, IL). Equal protein loading was confirmed by staining the gel with Coomassie blue and probing with GAPDH antibody (Novus Biological, Littleton, CO) because this protein level did not change. Band intensities were analyzed using a densitometer equipped with a multianalyst software program (Alpha Innotech, San Leandro, CA).
Immunohistochemistry
Hearts from MCP mice and wild-type mice were cut into three slices from the base to the apex and embedded in paraffin by using standard protocols. The transverse sections (5 μm) were incubated with antibodies against HSP25, HSP40, HSP70, Grp78, Grp94, PDI (Stressgene Biotechnologies), or ubiquitin (Dako). The sections were then incubated with horseradish peroxidase-conjugated secondary antibodies followed by diaminobenzidine and counterstained with hematoxylin.
Statistical analysis
The experimental data were analyzed by using SPSS 10.0 software under Windows XP. At least five animals were analyzed for each data point, and each experiment was repeated at least three times. All values are presented as means ± SE. The statistical significance of differences between wild-type control and MCP-1 transgenic group was calculated by using Student’s t-test. A value of P < 0.05 was considered significant.
RESULTS
Gene expression profile changes reveal induction of ER stress-associated genes during the development of ischemic heart disease
To identify the gene expression changes that occur during the development of ischemic heart disease caused by chronic inflammation, gene expression profile changes of the hearts of the MCP mice were examined during progression of the disease. RNA from hearts of MCP mice of 2, 4, 8, 16, and 24 wk of age with age and sex-matched wild-type controls were used for this analysis using Murine Genome-U74A chips. Among the genes whose expression was upregulated was a set of genes that are thought to be associated with ER stress (Table 2). The largest group of upregulated genes was represented by genes encoding ER resident chaperones and disulfide isomerases such as Cai, endoplasmic reticulum oxidoreductase (ERO1), Bip/Grp78, and PDI. The second major group of upregulated genes includes genes involved in protein transport. This group includes members of the Sec61 complex (Sec51 and Sec63) and translocation-associated protein. Asparagine synthetase was also significantly induced. Genes involved in the regulation of ER Ca2+ homeostasis such as calumenin and ribophorin were also induced. The transcription factors included two established UPR genes: C/EBP homologous protein (CHOP) and X box-binding protein 1 were persistently activated in the hearts of MCP mice. Upregulated genes also included genes involved in ER-associated degradation such as ubiquitin-conjugating enzyme and proteosome 26S subunit. The expression of the chaperone HSP40 was also upregulated in the hearts of MCP mice. These data show that ER stress response pathway members were upregulated at the transcript level in the hearts of MCP mice as the myocardial pathology and dysfunction developed, indicating that ER stress was activated in this mouse model of ischemic heart disease.
Table 2.
ER stress-related genes with fold changes in the hearts of MCP mice
|
*Fold Change with Age
|
||||||
|---|---|---|---|---|---|---|
| Gene Name | Accession No. | 2 wk | 1 mo | 2 mo | 4 mo | 6 mo |
| CHOP | X67803 | 1.8 | 6.7 | 6.5 | 3.9 | 3.2 |
| BiP/GRP78 | AJ002387 | 4.2 | 112 | 3.3 | 2.4 | 2.1 |
| PDI | AW045202 | 6.3 | 24 | 6 | 4.3 | 3.4 |
| PDI-related protein P5 | A1842377 | 3.2 | 9.5 | 4.3 | 4.2 | 3.6 |
| DnaJ (HSP40) homologous protein | AW120711 | 5.3 | 17 | 8.1 | 3.9 | 2.5 |
| Similar to ER-associated DnaJ | AW122551 | 2.5 | 7.6 | 4.9 | 3.3 | 2.4 |
| Calumenin | U81829 | 1.1 | 31 | 1.7 | 1.7 | 1.5 |
| Asparagine synthetase | U38940 | 1.4 | 23 | 7.6 | 3.7 | 2.1 |
| ATF6 | AB012276 | 1.7 | 11 | 10 | 5.4 | 2.7 |
| X-box binding protein | AW123880 | 1.8 | 13 | 2.1 | 1.1 | 1.8 |
| Defender against apoptotic death | U81052 | 1.3 | 47 | 1.7 | 1.4 | 1.6 |
| Stress associated ER protein 1 | A1843466 | 4.5 | 12.2 | 4.2 | 3.9 | 7.7 |
| Ribophorin | D31717 | 1.8 | 14 | 2.6 | 1.8 | 1.5 |
| Ribophorin 1 | A1846708 | 3.7 | 6.8 | 3.1 | 2.4 | 2.4 |
| Oligosccharyl transferase | D89063 | 4.1 | 8.6 | 3.1 | 2.5 | 2.3 |
| Translocation-associated protein | AW227650 | 1.8 | 22 | 2.2 | 1.8 | 2.1 |
| ER-integral membrane protein P34 | AW125178 | 1.1 | 17 | 2.2 | 1.9 | 1.9 |
| Secretary complex (Sec61) | U11027 | 1.5 | 31 | 2.5 | 1.9 | 1.5 |
| Secretary complex (Sec53) | AF007267 | 1.3 | 3 | 5.2 | 7.1 | 5 |
| Secretary complex (sec23b) | A1848343 | 2.8 | 5.1 | 6.7 | 7.3 | 3.7 |
| Ubiquitin-conjugating enzyme | X92665 | 2.1 | 3.3 | 2 | 1.3 | 1 |
| Proteosome 26S subunit, non-ATPase | M64298 | −1 | 15 | 1.8 | 2.2 | 2.5 |
| Lysozyme M | M21050 | 5 | 15 | 1.8 | 2.2 | 2.5 |
| Lysozyme P | X51547 | 10 | 26 | 3.7 | 4.5 | 7.5 |
| Cai/ERp72 | NM_009787 | 4.86 | 1.18 | 2.18 | ||
| ERO1 | NM-026184 | 3.83 | 3.92 | 1.75 | 1.92 | |
| Ufm1 | NM_026435 | 4.08 | 1.51 | 1.15 | ||
ER, endoplasmic reticulum; MCP mice, transgenic mice that express monocyte chemoattractant protein-1 specifically in the heart.
Fold change expression in the MCP mice/expression in the wild-type mice.
Real-time PCR verification of upregulation of the ER stress-associated genes in the hearts of MCP mice
To test for the validity of the microarray findings, the mRNA levels of some of the upregulated ER stress-associated genes were measured by quantitative real-time PCR. Consistent with the results obtained by the microarray analysis, mRNA levels of BiP/ Grp78, Grp94, PDI, Cai/ERp72, asparagine synthetase, ribophorin, and ERO1, CHOP, activating transporter factor (ATF6), and tribbles-related protein 3 (TRB3) were found to be significantly upregulated in the hearts of the MCP mice compared with age- and sex-matched wild-type controls (Fig. 1). The mRNA for BiP/Grp78, Grp94, PDI, ERO1, asparagine synthetase, and ribophorin were at high levels at 2 mo of age with subsequent decreases at 4 and 6 mo of age. At all time points their expression levels were significantly higher than that in the age- and sex-matched wild-type controls. Consistent with the gene array data, mRNA levels of cytosolic chaperones HSP25, HSP40, and HSP70 were also found to be upregulated in the hearts of MCP mice (Fig. 2). These results demonstrate activation of ER stress response in the hearts of MCP mice during the development of myocardial deterioration and dysfunction.
Fig. 1.
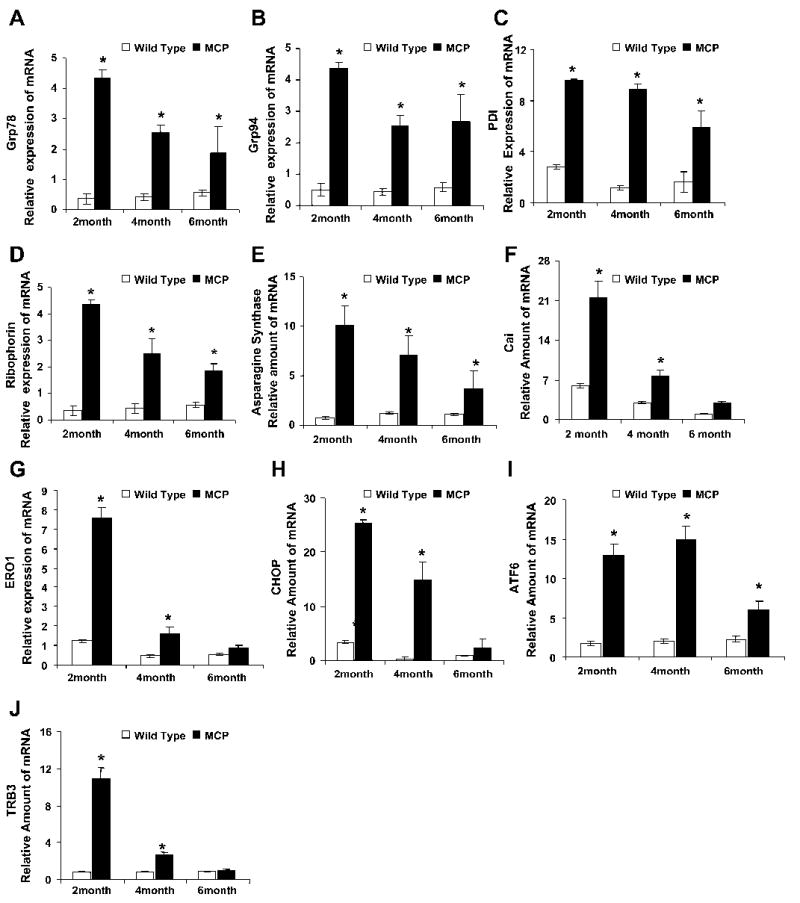
Elevated levels of transcripts of endoplasmic reticulum (ER) stress-associated genes in the hearts of transgenic mice that express monocyte chemoattractant protein-1 (MCP-1) specifically in the heart (MCP mice). Relative mRNA levels of ER stress-associated genes Bip/Grp78 (A), Grp94 (B), protein disulfide isomerase (PDI; C), ribophorin (D), asparagine synthetase (E), Cai (F), endoplasmic reticulum oxidoreductase (ERO1; G), C/EBP homologous protein (CHOP; H), ATF6 (I) and tribbles-related protein 3 (TRB3; J) were measured by quantitative real-time PCR. A total of 5 samples for each time point was used, and each sample was run in triplicate for real-time PCR. Expression level of all the samples was normalized by β-actin. *Significantly different from wild-type controls (P < 0.05).
Fig. 2.
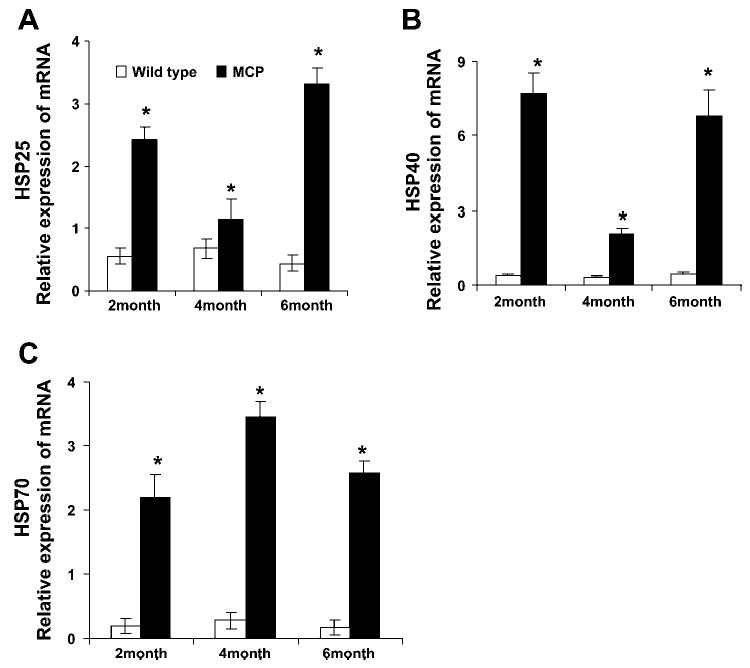
Elevated levels of transcripts of cytosolic chaperones in the hearts of MCP mice. Relative mRNA levels of cytosolic chaperones, 25-kDa heat shock protein (HSP25; A), 40-kDa heat shock protein (HSP40; B), and 70-kDa heat shock protein (HSP70; C), were measured by quantitative real-time PCR. A total of 5 samples for each time point was used, and each sample was run in triplicate for real-time PCR. The expression level of all the samples was normalized by β-actin. *Significantly different from wild-type controls (P < 0.05).
Immunoblot analysis of ER stress-associated proteins in the hearts of MCP mice
To test whether the transcript level changes observed in the hearts of MCP mice were reflected in ER stress-associated protein levels, the hearts of MCP mice and age- and sex-matched control mice were examined by immunoblot analysis. The levels of Bip/Grp78, Grp94, and PDI were elevated in the hearts of MCP mice compared with wild-type controls (Fig. 3, A–D), indicating that ER stress response was activated in the hearts of MCP mice. The expression of cytosolic chaperones HSP25 and HSP40 were also induced (Fig. 3, A, E, and F). Because these chaperones play crucial roles in protein folding and are induced on stress, it is clear that inflammatory changes in the heart of MCP mice elicited a stress response not only in the ER but also in the cytosol.
Fig. 3.
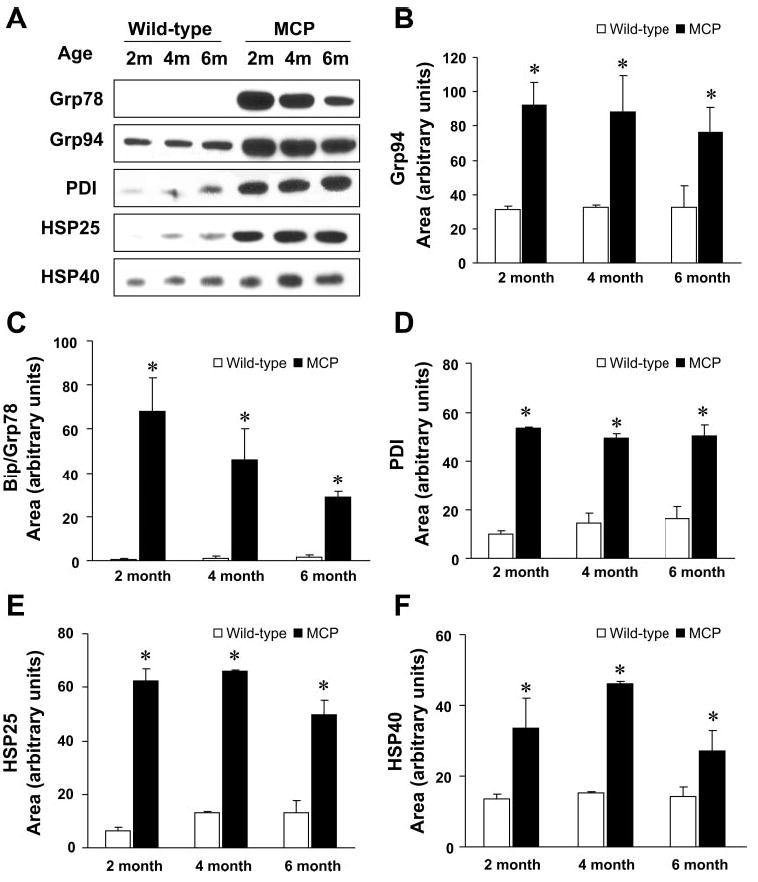
Immunoblot analysis of ER resident chaperones and cytosolic chaperones in the hearts expressing MCP-1. A: Western blots showing levels of BiP/Grp78, Grp94, PDI, HSP25, and HSP40 proteins in the hearts of 2-, 4-, and 6-mo-old wild-type controls and MCP mice. The expression level of all the samples was normalized to GADPH (B–F). Quantitative analyses of levels of Bip/Grp78, Grp94, PDI, HSP25, and HSP40 proteins by densitometry. *Significantly different from wild-type controls (P < 0.05).
Immunohistochemical evidence for increased expression of ER chaperones in the cardiomyocytes in the hearts of MCP mice
To determine the localization of ER stress-associated proteins in the hearts of MCP mice, immunohistochemical staining was performed. Strong staining for Grp78 was observed in the hearts of MCP mice, with intense staining found in cardiomyocytes undergoing degenerative changes as indicated by the presence of vacuoles. The cardiomyocytes with less visible indications of degeneration showed weaker immunoreactivity, whereas the hearts of wild-type mice showed a much weaker staining for Bip/Grp78 in both myocardium and interstitial infiltrating inflammatory cells (Fig. 4, A and D). A similar pattern of immunoreactivity for Grp94 was observed in the hearts of MCP mice (Fig. 4, B and E). A strong staining for PDI was found in many cardiomyocytes of MCP mice but not in wild-type controls (Fig. 4, C and F). The strongest staining for PDI was found in cardiomyocytes undergoing degenerative changes such as vacuolation with weaker staining in cardiomyocytes with less obvious degenerative changes (Fig. 4F). The expression of cytosolic chaperones HSP25, HSP40, and HSP70 were also markedly elevated in the cardiomyocytes of MCP mice compared with age- and sex-matched wild-type controls (Fig. 5, A–F). The degenerating vacuolated cardiomyocytes showed the strongest staining.
Fig. 4.
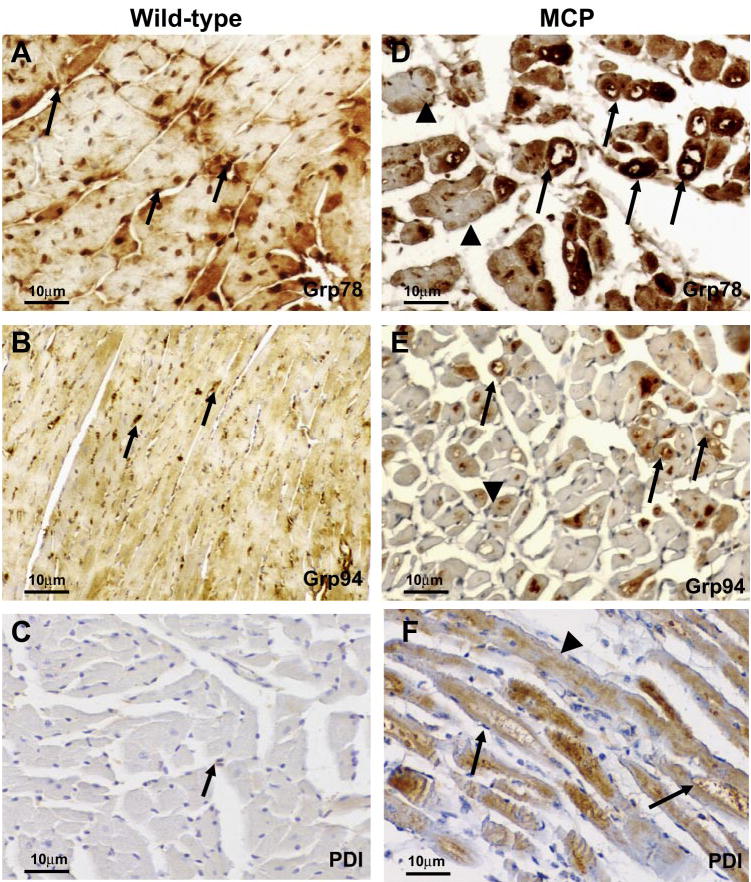
Immunohistochemical staining of ER-stress associated proteins Bip/Grp78, Grp94, and PDI in the hearts of wild-type controls and MCP mice of 6 mo of age. A–C: immunoreactivity (brown) in myocardium from wild-type controls. Note the absence or very weak cytoplasmic staining in cardiomyocytes and positive staining of Grp78 or Grp94 in interstitial infiltrating cells (arrow). D–F: immunoreactivity (brown) in myocardium from MCP mice. Note the strong staining in cardiomyocytes undergoing degenerative changes such as vacuolation (arrow) and the weaker immunoreactivity in cardiomyocytes with normal appearance (arrowheads). Original magnification, × 400.
Fig. 5.
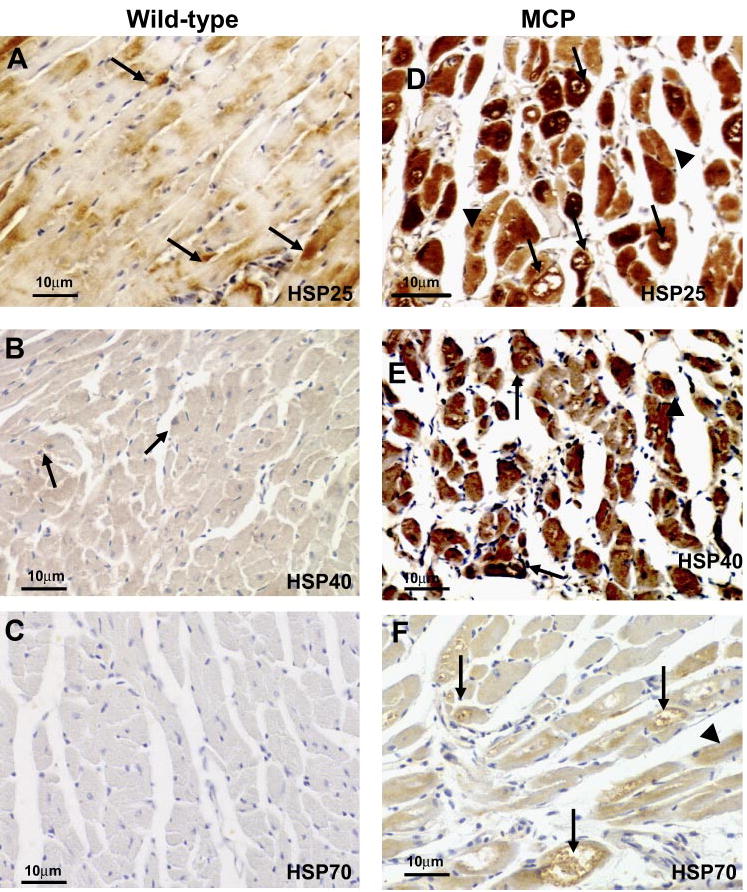
Immunohistochemical staining of heat-shock proteins HSP25, HSP40, and HSP70 in the hearts of wild-type controls and MCP mice of 6 mo of age. A–C: immunore-activity (brown) in myocardium from wild-type controls. Note the absence or very weak cytoplasmic staining in cardiomyocytes. D–F: immunoreactivity (brown) in myocardium from MCP mice. Note the strong staining in cardiomyocytes undergoing degenerative changes such as vacuolation (arrow) and the weaker immunoreactivity in cardiomyocytes with less degeneration (arrowheads). Original magnification, ×400.
Induction of ER-associated protein degradation in the hearts of MCP mice
A severe form of ER dysfunction would result in accumulation of misfolded proteins, which tend to form toxic aggregates. Besides UPR, the ERAD is known to be induced to activate degradation of unfolded or misfolded proteins via the ubiquitinproteasome system (17, 24). To test for such a possibility, we examined the accumulation of ubiquitinylated proteins in the hearts of MCP mice by immunohistochemistry and Western blot analysis. The immunoreactivity for ubiquitin was evident in the cardiomyocytes of MCP mice but not in the wild-type controls, demonstrating induction of ER stress in the cardiomyocytes (Fig. 6, A and B). Western blotting revealed marked accumulation of ubiquitinylated proteins in the hearts of the MCP mice compared with the wild-type controls (Fig. 6C). This implies that the accumulation of unfolded proteins induced by ER dysfunction exceeds the capacity of the proteasome to degrade them, strongly suggesting that ER functioning is disturbed in the hearts of MCP mice that are undergoing the development of ischemic heart disease.
Fig. 6.
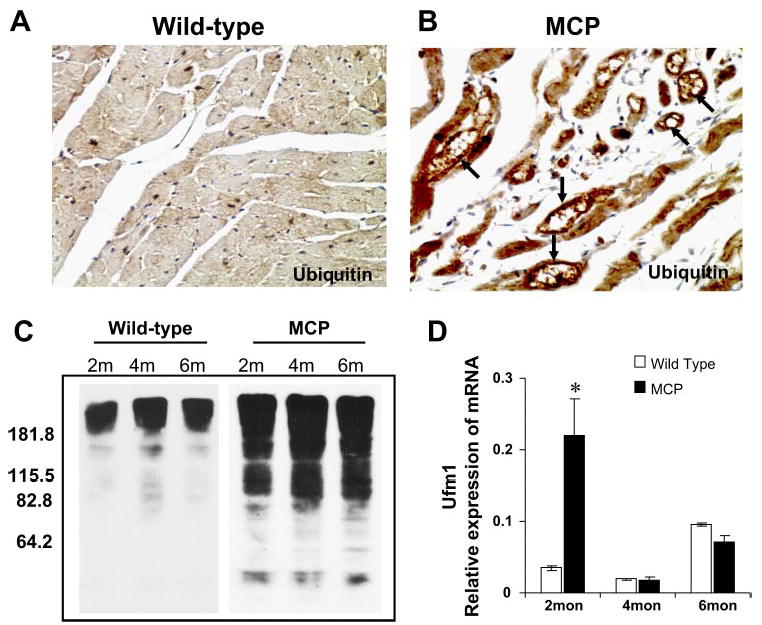
Accumulation of ubiquitinylated proteins in the hearts of wild-type and MCP mice. Immunohistochemical staining of ubiquitin in the hearts of wild-type controls (A) and MCP (B) mice of 6 mo of age. Note the absence or weaker cytoplasmic staining (brown) in cardiomyocytes of wild-type controls and the strong staining in cardiomyocytes undergoing degenerative changes such as vacuolation (arrow). Original magnification, ×400. C: Western blotting of heart homogenates from wild-type controls and MCP mice of 2, 4, and 6 mo of age showing marked accumulation of ubiquitinylated proteins in the hearts of MCP mice. D: elevated expression of ubiquitin fold modifier 1 (Ufm1) in the hearts of MCP mice determined by quantitative real-time PCR. *Significantly different from wild-type controls (P < 0.05).
One of the transcriptionally upregulated genes found in the MCP mice would encode a small protein that had not been previously found in any organism but shows homology to a recently identified human ubiquitin fold modifier designated Ufm1 that was not induced by any condition (19). Quantitative real-time PCR analysis confirmed that this murine Ufm1 was transcriptionally upregulated just before the onset of the clinical symptoms of left ventricular dysfunction (Fig. 6D).
DISCUSSION
In this study we found that two major components of the ER stress response pathway, UPR and ERAD, were highly activated in the hearts of MCP mice during the development of ischemic heart disease. Two main groups of genes that belong to UPR were upregulated in the MCP mice. One group includes peptide-binding molecular chaperones Bip/Grp78 and Grp94 (17, 24). Bip/Grp78 and Grp94, the most well characterized among them, interact transiently with protein-folding intermediates to prevent aggregation of a protein by keeping it in a folding-competent state (17, 21, 24). Interaction between the chaperones and proteins ensures that only proteins that are properly assembled and folded leave the ER compartment and thus attempt to alleviate the threat of cell death (17). The second group that was upregulated during the development of ischemic heart disease is composed of the disulfide isomerase family, including PDI, Cai/ERp72, ERp59, and ERp29 (17). These are involved in protein folding by functioning as oxidoreductases in the formation/isomerization of disulfide bonds and thereby increase the rate at which proteins attain their final folded conformation (9, 21). In addition, PDI, Cai/ERp72, ERp59, and ERp29 also have peptide-binding activity (9, 21). Cai is an ER luminal protein that is both a stress protein and a member of the protein disulfide isomerase (PDI) family of proteins (35). ERO1, which was found to be significantly upregulated in the present study at 2 mo in transgenic mice, is probably involved in the delivery of oxidizing equivalents required for the ER-located PDI (8). In humans, Ero1α and Ero1β are upregulated by hypoxia, suggesting that the ERO1 protein undergoes functional specialization in response to the different demands of oxygen tension and high-throughput protein folding. A variety of endoplasmic proteins including ERO1 and chaperones are induced by energy depletion caused by severe cellular hypoxia (anoxia) or by glucose deprivation (2). Induction of hypoxia-associated genes observed in the present study is consistent with the finding that MCP mice develop vasculopathy and ischemic degeneration of cardiomyocytes (23). It has been observed that CHOP sensitizes cells to ER stress-mediated death by directly regulating target genes in the nucleus. By activating ERO1α, CHOP promotes oxidizing conditions in the ER, which contribute to death (22). ATF6, which is a resident transmembrane protein of ER, has been found to be upregulated in the MCP transgenic mice. It has been proposed that ER stress signaling, especially that mediated by ATF6, plays a decisive role in the induction of apoptosis in muscle tissues through activation of caspase-12 (27). TRB3 (tribbles-related protein 3) was found to be up-regulated in the present study; it has been reported that TRB3 plays a important role in cell death in conjunction with CHOP where CHOP is also induced by other stress signals such as oxidative stress, amino acid deprivation, and hypoxia. Under prolonged ER stress, the excess TRB production probably would ultimately lead to cell death, as previously suggested (29). The present results are consistent with the conclusion that MCP mice develop ischemic heart disease.
ER-associated degradation (ERAD) constitutes further evidence for the ER stress response. Accumulation of unfolded or misfolded proteins impairs the uniquitin-proteasome protein degradation system, which sensitizes cells to ER stress (5, 37). Hyperubiquitination of proteins have been observed in human dilated cardiomyopathy (5) and was also found in mutant KDEL receptor transgenic mice, in which the ER quality control system was impaired and the mice developed dilated cardiomyopathy (11). We observed increased ubiquitination of proteins in the hearts of the MCP mice with disease progression, and the ubiquitinylated proteins were located primarily in those cardiomyocytes showing marked vacuolization. This finding suggests that increased levels of unfolded or misfolded proteins probably saturated the uniquitin-proteasome system and caused prolonged ER stress in cardiomyocytes that were no longer able to escape degeneration and death.
One of the genes transcriptionally activated during the development of inflammation-induced heart failure encodes a novel protein, Ufm1, that was recently identified in HEK293 cells and mouse tissues (19). This protein was suggested to function with a unique set of alternate ubiquitin conjugating enzyme complexes, although the biological role of this protein is not known (19). Agents known to induce ER stress, and other stress inducers such as high temperature and heavy metals, were reported to be unable to induce this protein. The present inflammation-induced heart disease represents the first instance of induction of this newly discovered member of the ubiquitin family of proteins.
The magnitude of the ER reorganization upon massive accumulation of unfolded proteins invokes not only ER-specific but also general cellular stress mechanisms (4, 40). HSPs are molecular chaperones in the cytosol and are involved in various cellular metabolic processes, including protein synthesis, folding, assembly, and degradation, thereby preventing intracellular accumulation of unfolded or misfolded proteins (25, 32). In the present study, we observed that expression of HSPs, such as HSP70, HSP40, and HSP25, was also markedly increased at both mRNA and protein levels in the hearts of MCP mice compared with aged-matched wild-type controls. Even though the elevated levels of these proteins may assist the folding of proteins into appropriate conformations, re-fold misfolded proteins, and rescue previously aggregated proteins (10, 15), in the face of prolonged stress caused by chronic myocardial inflammation, these efforts fail to protect the cardiac cells.
Although the precise molecular mechanisms that lead to the development of ischemic heart disease in the MCP mouse model are not well understood, all of the data so far obtained indicated the following sequence of events. Cardiac-specific MCP-1 expression induces the infiltration of monocytes/macrophages within myocardium where they undergo apoptosis with the release of Fas ligand (FasL) that cause death of vascular endothelial and smooth muscle cells, leading to thrombotic occlusive arteriolar vasculopathy that results in ischemia, which triggers a series of events (28). The cardiomyocytes become exposed to proinflammatory cytokines, altered redox state, and oxidative stress, which in turn triggers the stress response in the ER as well as in the cytosol. ER stress proteins and HSPs are generated to protect the cells from the adverse effect of stress. However, with the chronic inflammation, these efforts fail, despite the production of the high levels of such proteins, and the cells succumb to the death-inducing processes. Thus, in the early stages of disease, the MCP mice hearts show high levels of expression of ER stress proteins throughout the myocardium. As the protection fails, some of cardiomyocytes are unable to withstand the prolonged stress and undergo degenerative process that results in the formation of highly vacuolated cardiomyocytes. As the degenerative process is in its advanced stages, the ER stress proteins synthesis no longer stays at high levels. Thus ER stress is involved in the initiation of the disease.
Neurodegenerative diseases are known to involve cell death initiated by ER stress and are thus regarded as ER stress-associated diseases or conformational diseases (13, 20, 34, 36). However, the involvement of ER stress in heart disease has not been studied extensively (41). Studies in transverse aortic constriction (TAC) mice have demonstrated that pressure overload by TAC induces prolonged ER stress, which contributes to cardiac myocyte apoptosis during progression of cardiac hypertrophy to failure (30). The present report that activation of ER stress is associated with the development of ischemic heart disease, and further studies on the association of stress proteins with ischemic heart disease in other models and humans, may reveal early biomarkers for the ischemic heart disease.
Acknowledgments
We thank Isagani Santos and Charlene Ross for the care of the transgenic animals used in this study.
Footnotes
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grant HL-69458. Part of this work was done in the Ohio State University.
References
- 1.Beltrami CA, Finato N, Rocco M, Feruglio GA, Puricelli C, Cigola E, Quaini F, Sonnenblick EH, Olivetti G, Anversa P. Structural basis of end-stage failure in ischemic cardiomyopathy in humans. Circulation. 1994;89:151–163. doi: 10.1161/01.cir.89.1.151. [DOI] [PubMed] [Google Scholar]
- 2.Bernhard G, Karl-Heinz H, Roland HW, Christiane L, Helmut E, Meyerand AK. The cellular oxygen tension regulates expression of the endoplasmic oxidoreductase ERO1-Lα. Eur J Biochem. 2003;270:2228–2235. doi: 10.1046/j.1432-1033.2003.03590.x. [DOI] [PubMed] [Google Scholar]
- 3.Brodsky JL, McCracken AA. ER protein quality control and proteasome-mediated protein degradation. Semin Cell Dev Biol. 1999;10:507–513. doi: 10.1006/scdb.1999.0321. [DOI] [PubMed] [Google Scholar]
- 4.Bush KT, Goldberg AL, Nigam SK. Proteasome inhibition leads to a heat-shock response, induction of endoplasmic reticulum chaperones, and thermotolerance. J Biol Chem. 1997;272:9086–9092. doi: 10.1074/jbc.272.14.9086. [DOI] [PubMed] [Google Scholar]
- 5.Chung KKK, Dawson VL, Dawson TM. The role of the ubiquitin-proteasomal pathway in Parkinson’s disease and other neurodegenerative disorders. Trends Neurosci. 2001;24(Suppl 1):S7–S14. doi: 10.1016/s0166-2236(00)01998-6. [DOI] [PubMed] [Google Scholar]
- 6.De Lemos JA, Morrow DA, Sabatine MS, Murphy SA, Gibson GM, Antman EM, McCabe CH, Cannon CP, Braunwald E. Association between plasma levels of monocyte chemoattractant protein-1 and long-term clinical outcomes in patients with acute coronary syndrome. Circulation. 2003;107:690–695. doi: 10.1161/01.cir.0000049742.68848.99. [DOI] [PubMed] [Google Scholar]
- 7.Deo R, Khera A, McGuire DK, Murphy SA, Meo Neto Jde P, Morrow DA, de Lemos JA. Association among plasma levels of monocyte chemoattractant protein-1, transitional cardiovascular risk factor, and subclinical atherosclerosis. J Am Coll Cardiol. 2004;44:1812–1818. doi: 10.1016/j.jacc.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 8.Dias-Gunasekara S, Gubbens J, van Lith MC, Dunne Williams JA, Kataky R, Scoones D, Lapthorn A, Bulleid NJ, Benham AM. Tissue-specific expression and dimerization of the endoplasmic reticulum oxidoreductase Ero1 (beta) J Biol Chem. 2005;280:33066–33075. doi: 10.1074/jbc.M505023200. [DOI] [PubMed] [Google Scholar]
- 9.Ferrari DM, Soling HD. The protein disulphideisomerase family: unraveling a string of folds. Biochem J. 1999;339:1–10. [PMC free article] [PubMed] [Google Scholar]
- 10.Glover JR, Lindquist S. HSP104, HSP70, and HSP40: a novel chaperone system that rescue previously aggregated proteins. Cell. 1998;94:73–82. doi: 10.1016/s0092-8674(00)81223-4. [DOI] [PubMed] [Google Scholar]
- 11.Hamada H, Suzuki M, Yuasa S, Mimura N, Shinozuka N, Takada Y, Suzuki M, Nishino T, Nakaya H, Koseki H, Aoe T. Dilated cardiomyopathy caused by aberrant endoplasmic reticulum quality control in mutant KDEL receptor transgenic mice. Mol Cell Biol. 2004;24:8007–8017. doi: 10.1128/MCB.24.18.8007-8017.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Harding HP, Zhang Y, Ron D. Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 1999;397:271–274. doi: 10.1038/16729. [DOI] [PubMed] [Google Scholar]
- 13.Harding HP, Ron D. Endoplasmic reticulum stress and the development of diabetes: a review. Diabetes. 2002;51:S455–S461. doi: 10.2337/diabetes.51.2007.s455. [DOI] [PubMed] [Google Scholar]
- 14.Hayashidani S, Tsutsui H, Shiomi T, Ikeuchi M, Matsusaka H, Suematsu N, Wen J, Egashira K, Takeshita A. Anti-monocyte chemoattractant protein-1 gene therapy attenuates left ventricular remodeling and failure after experimental myocardial infarction. Circulation. 2003;108:1–7. doi: 10.1161/01.CIR.0000092890.29552.22. [DOI] [PubMed] [Google Scholar]
- 15.Hendricks JP, Hartl FU. Molecular chaperone function of heat-shock proteins. Annu Rev Biochem. 1993;62:349–384. doi: 10.1146/annurev.bi.62.070193.002025. [DOI] [PubMed] [Google Scholar]
- 16.Ikeda U. Inflammation and coronary artery disease. Curr Vasc Pharmacol. 2003;1:65–70. doi: 10.2174/1570161033386727. [DOI] [PubMed] [Google Scholar]
- 17.Kaufman RJ. Stress signaling from the lumen of endoplasmic reticulum: coordination of gene transcriptional and translational controls. Genes Dev. 1999;13:1211–1233. doi: 10.1101/gad.13.10.1211. [DOI] [PubMed] [Google Scholar]
- 18.Kolattukudy PE, Quach T, Bergese S, Breckenridge S, Hensley J, Altschuld R, Gordillo G, Klenotic S, Orosz C, Parker-Thornburg J. Myocarditis induced by targeted expression of the MCP-1 gene in murine cardiac muscle. Am J Pathol. 1998;152:101–111. [PMC free article] [PubMed] [Google Scholar]
- 19.Komatsu M, Chiba T, Tatsumi K, Iemura S, Tanida I, Okazaki N, Ueno T, Kominami E, Natsume T, Tanaka K. A novel protein-conjugating system for Ufm1, a ubiquitin-fold modifier. EMBO J. 2004;23:1977–1986. doi: 10.1038/sj.emboj.7600205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kopito RR, Ron D. Conformational disease. Nat Cell Biol. 2000;2:E207–E209. doi: 10.1038/35041139. [DOI] [PubMed] [Google Scholar]
- 21.Lee AS. The glucose-regulated proteins: stress induction and clinical applications. Trends Biochem Sci. 2001;26:504–510. doi: 10.1016/s0968-0004(01)01908-9. [DOI] [PubMed] [Google Scholar]
- 22.Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, Nagata K, Harding HP, Ron D. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moldovan NI, Goldschmidt-Clermont PJ, Parker-Thornburg J, Shapiro SD, Kolattukudy PE. Contribution of monocytes/macrophages to compensatory neovascularization: the drilling of metalloelastase-positive tunnels in ischemic myocardium. Circ Res. 2000;87:378–384. doi: 10.1161/01.res.87.5.378. [DOI] [PubMed] [Google Scholar]
- 24.Mori K. Tripartite management of unfolded proteins in the endoplasmic reticulum. Cell. 2000;101:451–454. doi: 10.1016/s0092-8674(00)80855-7. [DOI] [PubMed] [Google Scholar]
- 25.Morimoto RI. Cells in stress: transcriptional activation of heat shock genes. Science. 1993;259:1409–1410. doi: 10.1126/science.8451637. [DOI] [PubMed] [Google Scholar]
- 26.Muller PY, Janovjak H, Miserez AR, Dobbie Z. Processing of gene expression data generated by quantitative real-time RT-PCR. Biotechniques. 2002;32:1372–1379. [PubMed] [Google Scholar]
- 27.Nakanishi K, Sudo T, Morishima N. Endoplasmic reticulum stress signaling transmitted by ATF6 mediates apoptosis during muscle development. J Cell Biol. 2005;169:555–560. doi: 10.1083/jcb.200412024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Niu J, Azfer A, Deucher MF, Goldschmidt-Clermont PJ, Kolattukudy PE. Targeted cardiac expression of soluble Fas prevents the development of heart failure in mice with cardiac-specific expression of MCP-1. J Mol Cell Cardiol. 2006;40:810–820. doi: 10.1016/j.yjmcc.2006.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ohoka N, Yoshii S, Hattori T, Onozaki K, Hayashi H. TRB3, a novel ER stress-inducible gene, is induced via ATF4-CHOP pathway and is involved in cell death. EMBO J. 2005;24:1243–1255. doi: 10.1038/sj.emboj.7600596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okada KI, Minamino T, Tsukamoto Y, Liao Y, Tsukamoto O, Takashima S, Hirata A, Fujita M, Nagamachi Y, Nakatani T, Yutani C, Ozawa K, Ogawa S, Tomoike H, Hori M, Kitakaze M. Prolonged endoplasmic reticulum stress in hypertrophic and failing heart after aortic constriction: possible contribution of endoplasmic reticulum stress to cardiac myocyte apoptosis. Circulation. 2004;110:705–712. doi: 10.1161/01.CIR.0000137836.95625.D4. [DOI] [PubMed] [Google Scholar]
- 31.Oyadomari S, Takeda K, Takiguchi M, Gotoh T, Matsumoto M, Wada T, Akira S, Araki E, Mori M. Nitro oxide-induced apoptosis in pancreatic beta cells is mediated by the endoplasmic reticulum stress pathway. Proc Natl Acad Sci USA. 2001;98:10845–10850. doi: 10.1073/pnas.191207498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Parsel DA, Lindquist S. The function of heat shock proteins in stress tolerance: degradation and reactivation of damaged proteins. Annu Rev Cell Biol. 1993;9:437–496. doi: 10.1146/annurev.ge.27.120193.002253. [DOI] [PubMed] [Google Scholar]
- 33.Patil C, Walter P. Intracellular signaling from the endoplasmic reticulum to the nucleus: the unfolded protein response in yeast and mammals. Curr Opin Cell Biol. 2001;13:349–355. doi: 10.1016/s0955-0674(00)00219-2. [DOI] [PubMed] [Google Scholar]
- 34.Paschen W. Endoplasmic reticulum: a primary target in various acute disorders and degenerative diseases of the brain. Cell Calcium. 2003;34:365–383. doi: 10.1016/s0143-4160(03)00139-8. [DOI] [PubMed] [Google Scholar]
- 35.Sharma M, Benharouga M, Hu W, Lukacs GL. Conformational and temperature-sensitive stability defects of the delta F508 cystic fibrosis transmembrane conductance regulator in post-endoplasmic reticulum compartments. J Biol Chem. 2001;276:8942–8950. doi: 10.1074/jbc.M009172200. [DOI] [PubMed] [Google Scholar]
- 36.Sherman MY, Goldberg AL. Cellular defenses against unfolded proteins: a cell biologist thinks about neurodegenerative diseases. Neuron. 2001;29:15–32. doi: 10.1016/s0896-6273(01)00177-5. [DOI] [PubMed] [Google Scholar]
- 37.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P. Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell. 2000;101:249–258. doi: 10.1016/s0092-8674(00)80835-1. [DOI] [PubMed] [Google Scholar]
- 38.Weeks J, Morrison A, Mullen R, Wait R, Barton P, Dunn MJ. Hyperubiquitination of proteins in dilated cardiomyopathy. Proteomics. 2003;3:208–216. doi: 10.1002/pmic.200390029. [DOI] [PubMed] [Google Scholar]
- 39.Wiertz EJ, Tortorella D, Bogyo M, Yu J, Mothes W, Jones TR, Rapoport TA, Ploegh HL. Sec61-mediated transfer of a membrane protein from the endoplasmic reticulum to the proteasome for destruction. Nature. 1996;384:432–438. doi: 10.1038/384432a0. [DOI] [PubMed] [Google Scholar]
- 40.Wyttenbach A, Carmichael J, Swartz J, Furlong RA, Narain Y, Rankin J, Rubinsztein DC. Effects of heat shock, heat shock protein 40 (HDL-2), and proteasome inhibition on protein aggregation in cellular models of Huntington’s disease. Proc Natl Acad Sci USA. 2000;97:2898–2903. doi: 10.1073/pnas.97.6.2898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Xu C, Bailly-Maitre B, Reed JC. Endoplasmic reticulum stress: Cell life and death decisions. J Clin Invest. 2005;115:2656–2664. doi: 10.1172/JCI26373. [DOI] [PMC free article] [PubMed] [Google Scholar]