

Abstract
Titanium tetrakis(amido) complexes catalyze the intramolecular hydroamination of alkynes and allenes more efficiently than Cp-based species. We report here that electron-withdrawing and sterically demanding bis(sulfonamido) ligands lead to enhanced catalytic activity. Zirconium analogues have also been prepared, and the tosyl-substituted complex 20 has been structurally characterized. As in the titanium series, bis(sulfonamido) zirconium catalysts are more efficient in the intramolecular hydroamination of allenes than bis(cyclopentadienyl) complex Cp2ZrMe2 (23). Furthermore, these compounds transform 1,3-disubstituted aminoallenes with high stereoselectivity to the Z-allylamines and allow the hydroamination of a trisubstituted allene. Titanium bis(sulfonamido) imido complex 27 was synthesized. It converts aminoallene 10 to cylic imine 11 with a rate comparable to that of tetrakis(amide) 15, supporting the hypothesis of a catalytically active titanium imido intermediate.
Introduction
The direct addition of a N–H bond across a carbon–carbon multiple bond, the hydroamination reaction, constitutes an atom-economical method for the synthesis of substituted amines.1,2 While a general procedure for the hydroamination of unactivated alkenes remains elusive,3,4 appreciable progress has been made in developing analogous transformations of alkynes.5,6 The first catalytic intermolecular hydroamination of alkynes used highly toxic mercury and thallium compounds.7,8 More recently, reactions relying on alkali metal bases,9 organolanthanides10,11 and -actinides12 as well as late transition metals, such as palladium,13 rhodium,14 and ruthenium,15,16 have been developed. However, the extreme air- and water-sensitivity of the former and the high costs of the latter compounds are significant drawbacks to these procedures.
In contrast to many other potential catalysts, titanium and zirconium complexes are readily available and inexpensive. In the early 1990s, we reported the catalytic activity of zirconocene amido complexes in the hydroamination of alkynes.17,18 Subsequently, Doye disclosed the intermolecular hydroamination of alkynes using Cp2TiMe219 (1) as the precatalyst.20–24 Detailed mechanistic investigations of this reaction in our group revealed that the catalytically active species is generated via an unexpected Cp/amido ligand exchange.25,26 The isolated monocyclopentadienyl titanium amido complex Cp(ArNH)(py)Ti=NAr (Ar = 2,6-(Me)2C6H3) (2)27 exhibits enhanced catalytic activity in the hydroamination of alkynes and allenes.25 Therefore, we wondered if the efficiency could be further increased by the replacement of the remaining Cp ligand in 2 by another amide.28 A report from Odom’s group29 revealed that hydroamination reactions can be mediated by commercially available Ti(NMe2)4 (3) and, hence, confirmed our hypothesis. On the basis of these results, Odom and co-workers developed efficient titanium dipyrrolylmethane catalysts for the hydroamination of alkynes.30–33 We, however, were intrigued by the potential of using titanium bis(sulfonamido) complexes34 to accomplish regioselective hydroamination reactions. Herein, we (a) disclose a comprehensive investigation of titanium- and zirconium-catalyzed intramolecular hydroamination reactions of alkynes and allenes, (b) show that the reactivity and regioselectivity of these cyclization reactions strongly depend on the transition metal as well as the ligand set, and (c) provide evidence for a titanium imido intermediate in the catalytic cycle.
Results and Discussion
Hydroamination of Alkynes.
At the outset of our studies, we evaluated the catalytic activity of various titanium complexes in the intramolecular hydroamination of alkyne 4 to cyclic imine 5 (eq 1).
(1).
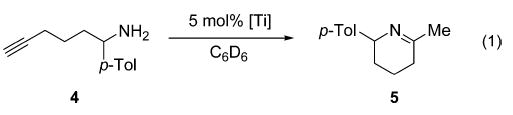
Titanium tetrakis(alkoxide) complexes, such as TADDOL-compound 635 or salen-complex 736 (Figure 1), afforded no product even at elevated temperature (Table 1, entries 1 and 2). Replacing two alkoxides with dimethylamides, as in BINOL-or TADDOL-complexes 8 and 9, gave rise to catalytic activity, but the conversion was rather slow even at 75 °C (entries 3 and 4).37,38 Commercially available Ti(NMe2)4 (3) efficiently converted aminoalkyne 4 to imine 5 even at room temperature (entry 5).
Figure 1.
Titanium alkoxo complexes.
Table 1.
Hydroamination of Aminoalkyne 4
| entry | cat. | T/°C | t/h | yield (NMR, %) |
|---|---|---|---|---|
| 1 | 6 | 105 | 16 | – |
| 2 | 7 | 105 | 2 | – |
| 3 | 8 | 75 | 3 | 45 |
| 4 | 9a | 75 | 16 | 35 |
| 5 | 3 | 25 | 30 | 85 |
Prepared in situ from 3 and the corresponding diol.
Hydroamination of Allenes.
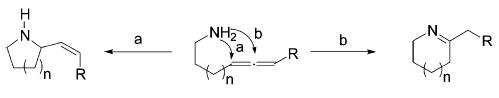
Since the hydroamination of alkynes is easily effected by titanium tetrakis(amide) 3,28,37 these substrates are of limited value for probing the reactivity of potentially more powerful catalysts. The hydroamination of allenes, however, is more challenging25 and hence constitutes a more stringent assay of catalytic activity. Additionally, the hydroamination of aminoallenes can result in two different regioisomers (Scheme 1). Whereas Ag-, Hg-, and Pd-based precatalysts exclusively provide allylamines via pathway a,39–43 lanthanide complexes convert monosubstituted aminoallenes into mixtures of the two regioisomers (pathways a and b). However, it is noteworthy that lanthanide catalysts convert 1,3-disubstituted aminoallenes exclusively to the corresponding allylamine.44–46
Scheme 1.
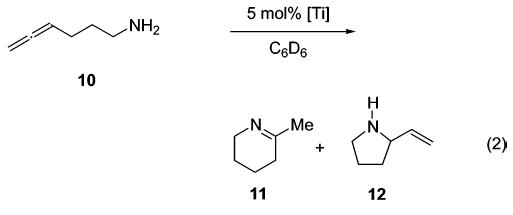
We studied the conversion of aminoallene 10 using 5 mol % of various titanium catalyst precursors (eq 2). The Cp-based complexes Cp2TiMe2 (1) and CpTi(NHAr)(NAr)py (Ar: 2,6-dimethylphenyl) (2) have been applied successfully in numerous intermolecular hydroamination reactions of allenes.25 Unexpectedly, the intramolecular transformation proceeded relatively slowly at 75 °C, and harsher reaction conditions were necessary to guarantee a quantitative conversion of substrate 10 (Table 2, entries 1 and 2). On the other hand, tetrakis(amide) 3 accomplishes the formation of imine 11 selectively at room temperature, although a temperature of 75 °C is necessary to achieve a practical reaction rate (entry 3). Since titanium bis-(sulfonamides) such as 13 are readily available following Walsh’s procedure,34 we also explored the use of tosyl-substituted complex 13 in the hydroamination of aminoallene 10. The chelating bis(sulfonamido) ligand resulted in a significantly increased reactivity, leading to selective and quantitative product formation even at room temperature (entry 4). The chelating N,N′-dimethyl-substituted complex 14, however, exhibits a reactivity profile similar to that of Ti(NMe2)4 (3) (entry 5). Therefore, the bidentate nature of the ligand is not the sole source of enhanced reactivity. The improved performance of 13 might instead be a consequence of a weak coordination of the sulfur-bound oxygen atoms to the metal center34 or of the different electronic properties of the bis-(sulfonamido) ligand (vide infra). Furthermore, the six-membered ring product is formed exclusively using titanium tetrakis(amide) complexes.
Table 2.
Hydroamination of Aminoallene 10
| yield (NMR, %) | |||||
|---|---|---|---|---|---|
| entry | cat. | T/°C | t/h | 11 | 12 |
| 1 | Cp2TiMe2 | 75 | 12 | 9 | --- |
| 1 | 135 | 3 | 74 | 10 | |
| 2 |
|
75 | 4 | 11 | 4 |
| 2a | 135 | 2 | 72 | 10 | |
| 3 | Ti(NMe2)4 | 25 | 26 | 41 | --- |
| 3 | 75 | 3 | quant. | --- | |
| 4 |
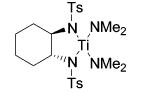
|
||||
| 13 | 25 | 5 | quant. | --- | |
| 5 |
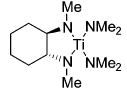
|
25 | 24 | 18 | --- |
| 14b | 75 | 3 | 72 | --- | |
Ar = 2,6-(CH3)2–C6H3
Prepared in situ from 3 and the corresponding bis(amine).
(2).
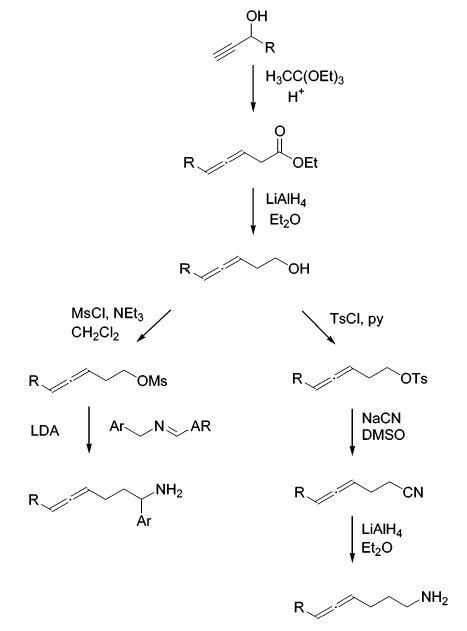
Having discovered the increased reactivity as well as the improved regioselectivity of bis(sulfonamide)-based catalyst 13, we studied the substrate scope provided by 13 in intramolecular hydroamination reactions of allenes. Differently substituted aminoallenes were synthesized following the general sequence depicted in Scheme 2.
Scheme 2.
When the α-position of a monosubstituted aminoallene bore an aromatic group, the regioselectivity of the cyclization depended upon the nature of the amido ligands (Table 3). While 3 generated a mixture of regioisomers (5–10% allylamine), the chelated titanium complex 13 formed the cyclic imines as the sole products, thereby allowing the isolation of these compounds by simple filtration through K2CO3. The examples listed in Table 3 demonstrate the superior activity of 13, which led to completion at significantly reduced reaction times. This system tolerates a range of substituents on the aromatic ring and provides the respective products in good yields, thus offering the possibility for further transformations (entries 5–8). More important, the scope of this procedure is not limited to the generation of relatively strain-free six-membered rings, but can also be extended to the formation of seven-membered ring systems (entry 9).47
Table 3.
Hydroamination of Aryl-Substituted Aminoallenes (5 Mol % of Catalyst in C6D6, 75°C)
| isolated yielda |
||||||
|---|---|---|---|---|---|---|
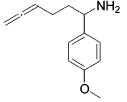
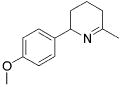
| entry | substrate | major product | cat. | t / h | imine | allyamine |
| 1 |
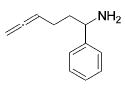
|
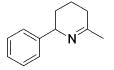
|
3 | 22 | (95) | (5) |
| 2 | 13 | 1 | 84 | --- | ||
| 3 |
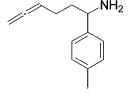
|
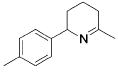
|
3 | 9 | (90) | (10) |
| 4 | 13 | 5 | 79 | --- | ||
| 5 |
|
|
3 | 4 | (92) | (8) |
| 6 | 13 | 1.5 | 95 | --- | ||
| 7 |
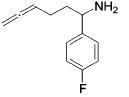
|
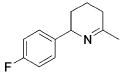
|
13 | 10 | 93 | --- |
| 8 |
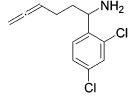
|
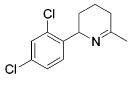
|
13 | 2 | 88 | --- |
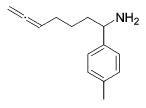
| 9 |
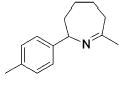
|
|
13b | 3 | (93) | (7) |
| 10 |
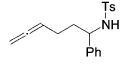
|
--- | 13b | 24 | --- | --- |
Yields in parentheses refer to ratio of imine/allylamine by NMR after complete and selective conversion of the starting material (versus internal standard)
10 mol %.
Steric Influence of the Bis(sulfonamido) Ligand.
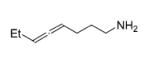
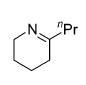
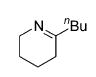
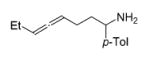
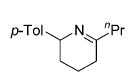
The influence of steric congestion around the metal center of the bis(sulfonamido) catalyst was studied by comparing the reactivity of p-tolyl-substituted precursor 13 with that of mesityl-substituted complex 1534 (Figure 2). Both precatalysts efficiently convert aminoalkynes and monosubstituted aminoallenes regioselectively to six-membered cyclic imines (Table 4, entries 1–4). They are also capable of affecting the more demanding hydroamination of 1,3-disubstituted allenes,48 yielding exclusively the corresponding cyclic imines (entries 5–8). The data obtained thus far suggest that the efficacy of the sterically more hindered catalyst 15 exceeds that of tosyl-substituted complex 13 (entry 1 versus 2 and 5 versus 6).
Figure 2.
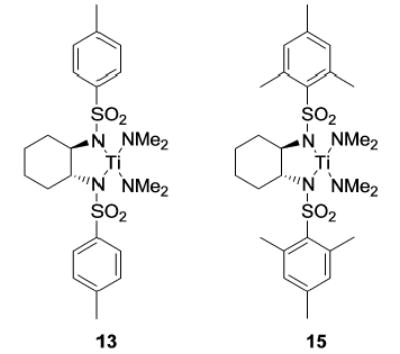
Titanium bis(sulfonamido) complexes.
Table 4.
Hydroamination of Substituted Aminoallenes with 13 and 15 (5 Mol % of Catalyst in C6D6)
| entry | cat. | substrate | product | T / °C | t / h | yield (NMR, %) |
|---|---|---|---|---|---|---|
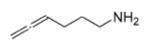
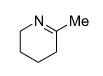
| 1 | 13 |
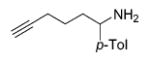
|
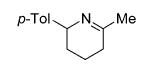
|
25 | 11 | 50 |
| 2 | 15 | 25 | 4 | 78 | ||
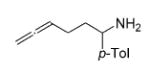
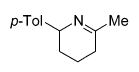
| 3 | 15 |
|
|
25 | 3 | quant. |
| 4 | 15 |
|
|
75 | 0.5 | 8 |
| 5 | 13 |
|
|
75 | 2 | 70 |
| 6 | 15 | 75 | 2 | 96 | ||
| 7 | 15 |
|
|
75 | 7 | quant. |
| 8 | 15 |
|
|
105 | 2 | quant. |
93% isolated yield.
Electronic Influence of the Bis(sulfonamido) Ligand.
To study the influence of the electronic nature of the bis-(sulfonamido) ligand on the catalytic activity, we synthesized the methyl- and trifluoromethyl-substituted analogues 16 and 17 from Ti(NMe2)4 (3) and the corresponding bis(amide) (eq 3).
(3).
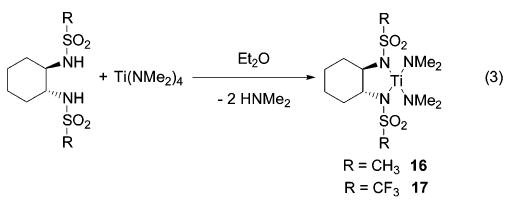
While bis(triflamide) 17 converted aminoalkyne 4 to imine 5 selectively and efficiently even at room temperature (47% conversion after 15 min), no transformation of 4 was achieved by the methyl-substituted complex 16 at this temperature (90 min, 1H NMR). These experiments illustrate the importance of the electron-withdrawing nature of the sulfonamido ligands on the catalytic activity. Furthermore, bis(triflamido) complex 17 converts aminoallenes selectively to the corresponding cyclic imines (Table 5). The transformation of 1,3-disubstituted allene 18 demonstrates the increased efficacy of bis(triflamide) 17, yielding 87% of imine 19 at 75 °C after 20 h. Note that complex 13 affords only 12% of 19 under the same reaction conditions.
Table 5.
Hydroamination Reactions of Allenes Using Catalyst 17 (5 Mol % of Catalyst in C6D6)
| entry | substrate | product | T / °C | t / h | yield (NMR, %) |
|---|---|---|---|---|---|
| 1 |
|
|
25 | 6.5 | 92 |
| 10 | 11 | ||||
| 2 |
|
|
75 | 20 | 87 |
| 18 | 19 |
Zirconium Analogues.
Previously we have carried out nitrogen–carbon bond-forming reactions using zirconium imido complexes.17,18,49 Therefore, we were interested in comparing the synthesis and catalytic activity of titanium complexes 13 and 15 with their zirconium analogues. The corresponding tosyl-and mesitylsulfonyl-substituted complexes 20 and 21 (Figure 3) were obtained starting from Zr(NMe2)4 (22) as outlined for the respective titanium complexes.
Figure 3.
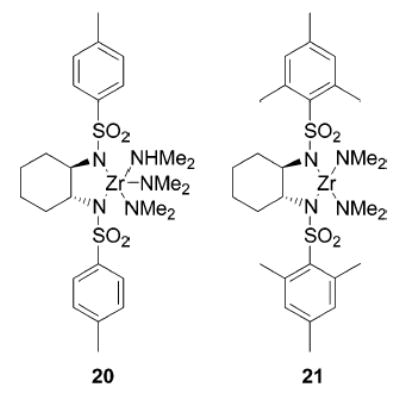
Zirconium bis(sulfonamido) complexes.
Whereas mesitylsulfonyl complex 21 showed the expected NMR features, tosyl-substituted compound 20 exhibited resonances for two separate sets of p-tolyl groups. In addition, 1 equiv of HNMe2 was detected. A crystal structure analysis was performed and unambiguously confirmed the structure of 20 shown in Figure 4. This confirms that coordination of the sulfur-bound oxygen atoms to the metal center has occurred.
Figure 4.
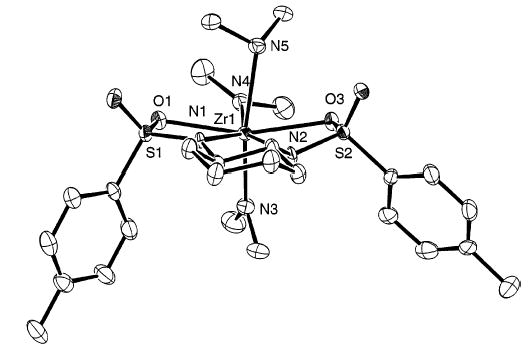
ORTEP diagram of complex 20.
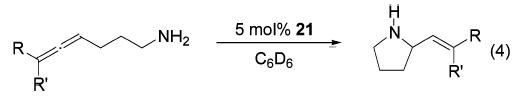
Initially, we explored the catalytic reactivity of a variety of different zirconium species in the hydroamination of parent aminoallene 10 (eq 2). The cyclopentadienyl-based complex 23 converted the aminoallene to products 11 and 12 slowly even at a temperature of 135 °C (Table 6, entry 1). Abstraction of a methyl group with B(C6F5)3 prior to the addition of the substrate led to an acceleration of the reaction at 135 °C. The cationic zirconium complex gave an inverted regioselectivity, yielding the allylamine as the major product (entry 2). In analogy to the titanium series, a more pronounced effect was observed upon switching from a Cp to an amido ligand set, illustrated by a lower reaction temperature of 75 °C (entry 3). The reaction was even faster using bis(sulfonamido) complexes (entries 4 and 5). While the tosyl-substituted sulfonamide complex 20 exhibited a catalytic activity comparable to that of Zr(NMe2)4 (22) (entry 4), mesitylsulfonyl-substituted compound 21 was a precursor to a significantly more efficient catalyst (entry 5). This improved performance can be rationalized by the increased steric hindrance around the metal center, as observed in the titanium series. In contrast to the analogous titanium species, the use of zirconium complexes yields significant amounts of allylamine 12. The introduction of substituents at the terminal position of the allene moiety inverts the regioselectivity of the cyclization reaction (eq 4). Several examples demonstrate that the regioselectivity changes from 1:16 (Table 6, entry 5) for the parent substrate to 4:1 and 7:1 (Table 7, entries 1 and 2) in the case of 1,3-disubstituted aminoallenes. Furthermore, the diastereoselectivity is excellent for the conversion of 1,3-disubstituted substrates, yielding the corresponding Z-isomers 24 and 25 exclusively. The scope of this transformation is further highlighted by the hydroamination of a trisubstituted allene (Table 7, entry 3). In this case, a good regioselectivity of 11:1 favoring allylamine 26 is obtained.
Table 6.
Hydroamination of Allene 10 Using Zirconium Complexes (5 Mol % of Catalyst in C6D6)
| yield (NMR, %)
|
|||||
|---|---|---|---|---|---|
| entry | cat. | T/°C | t/h | 11 | 12 |
| 1 | Cp2ZrMe2 (23) | 135 | 18 | 16 | 3 |
| 2 | Cp2ZrMe2 (23)/B(C6F5)3 | 135 | 18 | 34 | 66 |
| 3 | Zr(NMe2)4 (22) | 75 | 24 | 35 | 2 |
| 20 | 75 | 3 | 10 | – | |
| 4 | 135 | 3 | 84 | 12 | |
| 5 | 21 | 75 | 4 | 94 | 6 |
Table 7.
Synthesis of Allylamines Using Zirconium Catalysts 21
| entry | product | T / °C | yield (%)a | Z/E | regioselectivity |
|---|---|---|---|---|---|
| 1 |
|
75 | 62b | > 20 / 1 | 4 / 1 |
| 24 | |||||
| 2 |
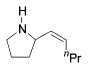
|
75 | 68 | > 20 / 1 | 7 / 1 |
| 25 | |||||
| 3 |
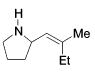
|
135c | 88 | 1.8 / 1 | 11 / 1 |
| 26 |
Isolated as the trifluoroacetamide derivative.
By 1H NMR.
10 mol % 21.
(4).
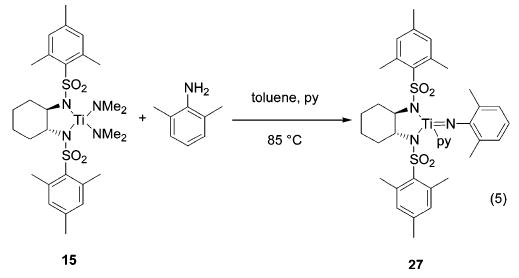
Mechanistic Considerations.
The mechanism of the Cp2-TiMe2-catalyzed hydroamination reaction has been investigated experimentally25,50 and theoretically.26 These studies suggest that the catalytically active species is a titanium imido complex. To gain insight into the mechanism of the reaction mediated by tetrakis(amido) precursors, we performed the stoichiometric reaction between titanium catalyst 15 and a primary amine (eq 5). To avoid a potential dimerization of the imido species,25,26 we chose the sterically hindered 2,6-dimethylaniline. Monitoring the reaction by 1H NMR revealed the formation of imido complex 27 accompanied by liberation of HNMe2. Pyridine was added to stabilize the imido compound, and the resulting adduct was isolated in 61% yield, contaminated with free amine. All attempts to remove the remaining impurities met with no success, which prevented satisfactory elemental analysis. Difficulties associated with removing amine contaminants from early transition metal imido species have been reported previously.25,51,52 Imido complex 27 (5 mol %) converts aminoallene 10 quantitatively to imine 11 after 3 h at room temperature and, hence, leads to a reaction with a rate comparable to that observed using precatalyst 15.
(5).
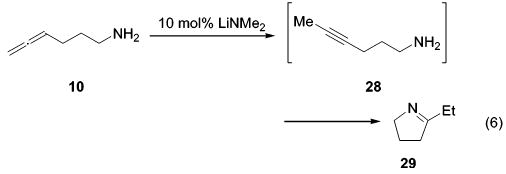
Knochel and co-workers have shown that intramolecular hydroamination reactions of alkynes can be mediated by bases.9,53,54 However, catalytic amounts of LiNMe2 (10 mol %) isomerized aminoallene 10 quantitatively in 1 h at ambient temperature to internal alkyne 28 (observed by 1H and 13C NMR). Alkyne 28 was subsequently converted to the five-membered cyclization product 29 (eq 6). Imine 29 was observed neither in the titanium- nor in the zirconium-catalyzed reactions. Therefore, it seems unlikely that the catalytic activity originates from traces of free amide.
(6).
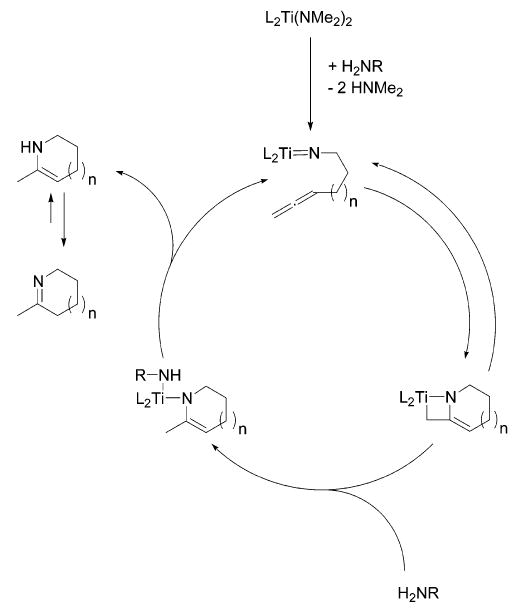
On the basis of our results, we propose the catalytic cycle depicted in Scheme 3 for the hydroamination reactions discussed in this work (illustrated for titanium and an allene). In the crucial steps of this mechanism, a formal [2 + 2] cycloaddition of the imido species with the allene is followed by a stepwise protonation of the azametallacyclobutane to regenerate the actual catalysts as well as to release the hydroamination product.
Scheme 3.
Conclusion
Titanium tetrakis(amido) complexes catalyze the intramolecular hydroamination of alkynes and allenes more efficiently than do Cp-based species. The catalytic activity can be increased through the use of electron-withdrawing or sterically demanding bis(sulfonamido) ligands. Analogous zirconium complexes have been synthesized, and one of them has been structurally characterized. These complexes catalyze the intramolecular hydroamination of allenes more efficiently than Cp2ZrMe2 (23). More importantly, these compounds convert 1,3-disubstituted aminoallenes with good stereoselectivity to the Z-allylamines and allow the hydroamination of a trisubstituted allene moiety. Finally, titanium bis(sulfonamido) imido complex 27 cyclizes aminoallene 10 at a rate comparable to that observed using tetrakis(amide) 15, supporting the hypothesis that in the catalytic cycle a titanium imido species is an important intermediate.
Experimental Section
General.
All syntheses were carried out under N2 using predried glassware. All hydroamination reactions and complex synthesis were performed in a N2-filled Vacuum Atmospheres inert atmosphere box. Et2O, pyridine, THF, and d6-benzene were dried by distillation over Na and transferred under N2. CH2Cl2, benzene, n-pentane, and toluene were dried by passing through a single column of activated alumina.55,56 Flash chromatography: Merck silica gel 60 (230–400 mesh). NMR:
Spectra were recorded on Bruker AMX300 or AMX400 spectrometers in the solvents indicated; chemical shifts (δ) are given in ppm relative to TMS, coupling constants (J) in Hz. 1,3,5-Trimethoxybenzene (Aldrich) was employed as an internal standard in NMR tube reactions. IR: Mattson Galaxy FTIR 3000, wavenumbers in cm−1; only selected data are reported. Mass spectra were performed by the University of California, Berkeley Micro-Mass Facility.
Herein, selected representative procedures are reported. A fully detailed account is provided in the Supporting Information.
General Procedure for the Synthesis of Titanium and Zirconium Complexes.57
Under an N2 atmosphere, the appropriate ligand (1 mmol) was stirred in Et2O (10 mL) at room temperature. To this mixture was added Ti(NMe2)4 (1 mmol) as a solution in Et2O (1 mL) in one portion. On addition of the titanium complex, the mixture turned red-brown, and the ligand dissolved to give a clear solution. If necessary, the solution was filtered after the ligand had dissolved. When no undissolved ligand remained, stirring was discontinued and the solution was cooled to −35 °C. The solution was decanted, washed with cold Et2O (1 mL, −35 °C), and the product was dried in vacuo.
Following the general procedure, 20 was obtained as white crystals in 46% yield.
1H NMR (C6D6, 400 MHz): δ 8.03 (2H, d, J = 8.2 Hz), 7.96 (2H, d, J = 8.2 Hz), 6.87 (2H, d, J = 7.9 Hz), 6.81 (2H, d, J = 8.1 Hz), 3.45–3.35 (2H, m), 3.41 (6H, s), 3.23 (6H, s), 2.40 (6H, d, J = 6.1 Hz), 2.20 (1H, m), 1.99 (1H, m), 1.88 (3H, s), 1.87 (3H, s), 1.70 (1H, m), 1.34 (1H, m), 1.26 (1H, m), 1.20–0.85 (3H, m). 13C NMR (C6D6, 100 MHz): δ 142.7, 142.4, 129.8, 129.5, 128.1, 127.9, 126.8, 126.1, 65.2, 63.7, 47.6, 44.6, 42.8, 40.3, 32.8, 24.7, 21.1, 21.1. IR (CH2Cl2): 3363, 3284, 3065, 3054, 2938, 2859, 1599, 1451, 1428, 1337, 1262, 1162, 1093, 904 cm−1. C26H43N5O4S2Zr (645.01) Anal. Calcd: C, 48.42; H, 6.72; N, 10.86. Found: C, 48.36; H, 6.66; N, 10.51.
Typical Procedure for the Hydroamination/Cyclization Reaction.
A solution of 1-(4-fluoro-phenyl)-hexa-4,5-dienyl-1-amine (121 mg, 0.63 mmol) and 13 (18 mg, 0.03 mmol) in benzene (3 mL) was heated for 10 h to 75 °C. The solution was cooled and treated with 20 drops of methanolic NaOH (10%). The mixture was stirred for 0.5 h at room temperature and concentrated in vacuo. The remaining residue was extracted with n-hexane (30 mL) and filtered through K2CO3 to afford 2-(4-fluoro-phenyl)-6-methyl-2,3,4,5-tetrahydro-pyridine (112 mg, 93%) as a pale yellow oil with a purity of >95% by NMR.
2-(4-Fluoro-phenyl)-6-methyl-2,3,4,5-tetrahydro-pyridine.
1H NMR (CD2Cl2, 300 MHz): δ 7.23 (m, 2H), 7.01 (tm, 2H, J = 8.9 Hz), 4.43 (m, 1H), 2.30–2.10 (m, 2H), 1.98 (d, 3H, J = 2.0 Hz), 1.95–1.60 (m, 3H), 1.40–1.20 (m, 1H). 13C NMR (CD2Cl2, 100 MHz): δ 168.6, 128.4, 128.3, 114.8, 114.5, 60.8, 30.6, 29.9, 27.3, 19.0. MS (EI) m/z (relative intensity): 191 (81) [M+], 163 (14), 162 (14), 148 (11), 121 (100), 109 (10). HRMS (EI) m/z: calcd for C12H14FN, 191.1110; found, 191.1109.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (Grant No. GM-25459 to R.G.B.). L.A. thanks the Deutscher Akademischer Austauschdienst (DAAD) for a postdoctoral fellowship. The structural study of 20 was carried out by Drs. A. G. Oliver and F. J. Hollander of the U. C. Berkeley College of Chemistry X-ray Diffraction Facility (CHEXRAY). The Center for New Directions in Organic Synthesis is supported by Bristol-Myers Squibb as a sponsoring member and Novartis as a supporting member.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for new compounds (PDF). This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Müller TE, Beller M. Chem Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]
- 2.Nobis M, Driessen-Hölscher B. Angew Chem, Int Ed. 2001;40:3983. doi: 10.1002/1521-3773(20011105)40:21<3983::aid-anie3983>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 3.As early as 1993, the catalytic anti-Markovnikov addition of amines to olefins was mentioned as one of the top 10 challenges for catalysis: Haggin J.Chem Eng News 1993716 [Google Scholar]
- 4.For leading references for the hydroamination of activated alkenes, see: (a) Casalnuovo AL, Calabrese JC, Milstein D.J Am Chem Soc 19881106738 [Google Scholar]; b Dorta R, Egli P, Zürchner F, Togni A. J Am Chem Soc. 1997;119:10857. [Google Scholar]; c Beller M, Eichberger M, Trauthwein H. Angew Chem, Int Ed Engl. 1997;36:2225. [Google Scholar]; d Kawatsura M, Hartwig JF. J Am Chem Soc. 2000;122:9546. [Google Scholar]; e Kawatsura M, Hartwig JF. Organometallics. 2001;20:1960. [Google Scholar]; f Fadini L, Togni A. Chem Commun. 2003:30. doi: 10.1039/b210680a. [DOI] [PubMed] [Google Scholar]
- 5.Shimada T, Yamamoto Y. J Am Chem Soc. 2002;124:12670. doi: 10.1021/ja027683y. [DOI] [PubMed] [Google Scholar]
- 6.For a review, see: Pohlki F, Doye S.Chem Soc Rev 200332104. [DOI] [PubMed] [Google Scholar]
- 7.Barluenga J, Aznar F. Synthesis. 1977:195. [Google Scholar]
- 8.Barluenga J, Aznar F, Liz R, Rodes R. J Chem Soc, Perkin Trans 1. 1980:2732. [Google Scholar]
- 9.Seayad J, Tillack A, Hartung CG, Beller M. Adv Synth Catal. 2002;344:795. [Google Scholar]
- 10.Li Y, Marks TJ. Organometallics. 1996;15:3770. [Google Scholar]
- 11.Li Y, Marks TJ. J Am Chem Soc. 1998;120:1757. [Google Scholar]
- 12.Haskel A, Straub T, Eisen MS. Organometallics. 1996;15:3773. [Google Scholar]
- 13.Yamamoto Y, Radhakrishnan U. Chem Soc Rev. 1999;28:199. [Google Scholar]
- 14.Hartung CG, Tillack A, Trautwein H, Beller M. J Org Chem. 2001;66:6339. doi: 10.1021/jo010444t. [DOI] [PubMed] [Google Scholar]
- 15.Uchimaru Y. Chem Commun. 1999:1133. [Google Scholar]
- 16.Tokunaga M, Eckert M, Wakatsuki Y. Angew Chem, Int Ed. 1999;38:3222. doi: 10.1002/(sici)1521-3773(19991102)38:21<3222::aid-anie3222>3.0.co;2-7. [DOI] [PubMed] [Google Scholar]
- 17.Baranger AM, Walsh PJ, Bergman RG. J Am Chem Soc. 1993;115:2753. [Google Scholar]
- 18.Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708. [Google Scholar]
- 19.Siebenreicher H, Doye S. J Prakt Chem. 2000;342:102. [Google Scholar]
- 20.Haak E, Bytschkov I, Doye S. Angew Chem, Int Ed. 1999;38:3389. [PubMed] [Google Scholar]
- 21.Bytschkov I, Doye S. Eur J Org Chem. 2001:4411. [Google Scholar]
- 22.Pohlki F, Heutling A, Bytschkov I, Hotopp T, Doye S. Synlett. 2002:799. [Google Scholar]
- 23.Siebenreicher H, Doye S. Eur J Org Chem. 2002:1213. [Google Scholar]
- 24.For titanium-based hydroamination catalysts, see also: (a) Tillack A, Castro IG, Hartung CG, Beller M.Angew Chem, Int Ed 2002412541. [DOI] [PubMed] [Google Scholar]; b Ong TG, Yap GPA, Richeson DS. Organometallics. 2002;21:2839. [Google Scholar]
- 25.Johnson JS, Bergman RG. J Am Chem Soc. 2001;123:2923. doi: 10.1021/ja005685h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Straub BF, Bergman RG. Angew Chem, Int Ed. 2001;40:4632. doi: 10.1002/1521-3773(20011217)40:24<4632::aid-anie4632>3.0.co;2-v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McGrane PL, Livinghouse T. J Org Chem. 1992;57:1323. [Google Scholar]
- 28.Ackermann L, Bergman RG. Org Lett. 2002;4:1475. doi: 10.1021/ol0256724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shi Y, Ciszewski JT, Odom AL. Organometallics. 2001;20:3967. [Google Scholar]
- 30.Shi Y, Hall C, Ciszewski JT, Cao C, Odom AL. Chem Commun. 2003:586. doi: 10.1039/b212423h. [DOI] [PubMed] [Google Scholar]
- 31.Harris SA, Ciszewski JT, Odom AL. Inorg Chem. 2001;40:1987. doi: 10.1021/ic001238r. [DOI] [PubMed] [Google Scholar]
- 32.Li Y, Turnas A, Ciszewski JT, Odom AL. Inorg Chem. 2002;41:6298. doi: 10.1021/ic0259484. [DOI] [PubMed] [Google Scholar]
- 33.Cao C, Ciszewski JT, Odom AL. Organometallics. 2001;20:5011. [Google Scholar]
- 34.Pritchett S, Gantzel P, Walsh PJ. Organometallics. 1999;18:823. [Google Scholar]
- 35.Seebach D, Plattner DA, Beck AK, Wang YM, Hunziker D, Petter W. Helv Chim Acta. 1992;75:2171. [Google Scholar]
- 36.Flores-Lopéz LZ, Parra-Hake M, Somanathan R, Walsh PJ. Organometallics. 2000;19:2153. [Google Scholar]
- 37.Duncan D, Livinghouse T. Organometallics. 1999;18:4421. [Google Scholar]
- 38.For the catalytic activity of isolated alkoxo(imido) titanium complexes, see: Hill JE, Profilet RD, Fanwick PE, Rothwell IP.Angew Chem Int Ed Engl 199029664 [Google Scholar]
- 39.Arseniyadis S, Gore J. Tetrahedron Lett. 1983;24:3997. [Google Scholar]
- 40.Kinsman R, Lathbury D, Vernon P, Gallagher T. J Chem Soc, Chem Commun. 1987:243. [Google Scholar]
- 41.Fox DNA, Gallagher T. Tetrahedron. 1990;46:4697. [Google Scholar]
- 42.Al-Masum M, Meguro M, Yamamoto Y. Tetrahedron Lett. 1997;38:6071. [Google Scholar]
- 43.Meguro M, Yamamoto Y. Tetrahedron Lett. 1998;39:5421. [Google Scholar]
- 44.Arredondo VM, McDonald FE, Marks TJ. J Am Chem Soc. 1998;120:4871. [Google Scholar]
- 45.Arredondo VM, Tian S, McDonald FE, Marks TJ. J Am Chem Soc. 1999;121:3633. [Google Scholar]
- 46.Arredondo VM, McDonald FE, Marks TJ. Organometallics. 1999;18:1949. [Google Scholar]
- 47.Note that the synthesis of seven-membered rings was not achieved using Cp2TiMe2: Bytschkov I, Doye S.Tetrahedron Lett 2002433715 [Google Scholar]
- 48.All attempts to convert acyclic disubstituted allenes using cyclopentadienyl-based titanium catalysts have met with no success.25
- 49.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pohlki F, Doye S. Angew Chem, Int Ed. 2001;40:2305. [PubMed] [Google Scholar]
- 51.Arney DJ, Bruck MA, Huber SR, Wigley DE. Inorg Chem. 1992;31:3749. [Google Scholar]
- 52.Blake AJ, Collier PE, Dunn SC, Li WS, Mountford P, Shishkin OV. J Chem Soc, Dalton Trans. 1997:1549. [Google Scholar]
- 53.Koradin C, Dohle W, Rodriguez AL, Schmid B, Knochel P. Tetrahedron. 2003;59:1571. [Google Scholar]
- 54.Rodriguez AL, Koradin C, Dohle W, Knochel P. Angew Chem, Int Ed. 2000;39:2488. doi: 10.1002/1521-3773(20000717)39:14<2488::aid-anie2488>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 55.Alaimo PJ, Peters DW, Arnold J, Bergman RG. J Chem Educ. 2001;78:64. [Google Scholar]
- 56.Pangborn AB, Giardello MA, Grubbs RH, Rosen RK, Timmers FJ. Organometallics. 1996;15:1518. [Google Scholar]
- 57.A general procedure for the synthesis of such titanium complexes has been reported.34