

Abstract
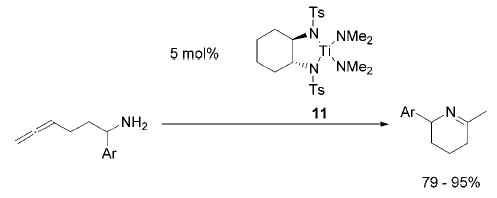
Tetrakisamido titanium complexes are significantly more active than Cp2TiMe2 (1) in the intramolecular hydroamination of aminoalkynes and aminoallenes. In the latter case, the regioselectivity of the transformation depends on the nature of the precatalyst, yielding the most selective and reactive catalysis with the bis(sulfonamido) complex 11.
The direct addition of an N–H bond across a carbon–carbon multiple bond, the hydroamination reaction, is the most atom economical way to synthesize substituted amines.1 Although appreciable progress has been made,2 a general procedure for this transformation remains elusive.
In the early 1990s, we reported the catalytic activity of zirconocene amido complexes in the hydroamination of alkynes.3 Doye subsequently disclosed the intermolecular hydroamination of alkynes using Cp2TiMe24 (1) as the precatalyst.5 Detailed mechanistic investigations of this reaction in our group revealed that the catalytically active species is generated via a Cp/amide ligand exchange. This conversion of the titanocene species (Cp2TiL2) into a monocyclopentadienyl titanium amido complex (CpTi(N-RH)Ln)6 led to the development of a titanium complex with enhanced catalytic activity in the hydroamination of alkynes and allenes.7 Therefore, we became interested in studying the catalytic reactivity of noncyclopentadienyl-supported titanium precursors. The recent report by Odom and co-workers8 that Ti(NMe2)4 (2) catalyzes the hydroamination of alkynes prompted us to disclose our preliminary results concerning the development of a highly active precatalyst for intramolecular hydroaminations of alkynes and allenes.
To compare the reactivity of the bis(cyclopentadienyl)-based precursor Cp2TiMe2 (1) with the tetrakisamide pre-catalyst Ti(NMe2)4 (2), we initially investigated the intramolecular hydroamination of alkynes (Scheme 1).6,9 The reactions were performed in d6-benzene and monitored by 1H NMR spectroscopy and GC/MS. In the presence of 5 mol % of 1 the formation of the expected cyclization product 4 was not observed after 12 h at 75 °C. To achieve a conversion of the terminal alkyne 3, a higher temperature (135 °C) was necessary. The conversion of the internal alkyne 5 was easier and a selective formation of 6 was achieved at 75 °C, while no catalytic activity was observed at room temperature. The commercially available tetrakisamide-based precursor 2 (5 mol %) was much more effective, providing both products quantitatively at room temperature (41 h and 30 min for 4 and 6, respectively).
Scheme 1.
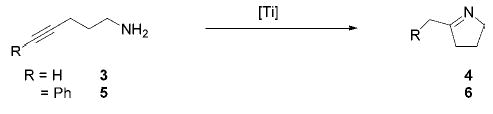
Hydroamination of Aminoalkynes
Having illustrated the increased reactivity of tetrakisamido titanium precatalyst 2, we wished to optimize the efficiency of this precursor. Because the hydroamination of alkynes is easily effected by tetrakisamido titanium complexes, these substrates are of limited value for probing the reactivity of potentially more powerful catalysts. The hydroamination of aminoallenes, however, is more challenging and hence provides an appropriate assay of catalytic activity. Another factor that has to be considered in the hydroamination of aminoallenes is the regioselectivity (Scheme 2). Whereas Ag-, Hg-, or Pd-based precatalysts provide exclusively allylamines via pathway b,10 lanthanide complexes convert monosubstituted aminoallenes into mixtures of the two regioisomers (pathways a and b).11
Scheme 2.
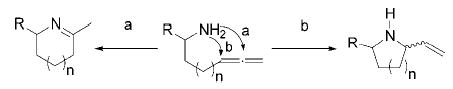
Regioselectivity in the Hydroamination of Aminoallenes
We investigated the conversion of aminoallene 7 using 5 mol % of different titanium and zirconium catalyst precursors (Table 1). Once again, the bis(cyclopentadienyl) complex 1 forms the hydroamination product 8 slowly at 75 °C (entry 1) and harsher reaction conditions are necessary to guarantee a quantitative conversion of substrate 7 (entry 2). By switching to the tetrakisamide precatalyst 2, the formation of imine 8 is selectively12 accomplished at room temperature (entry 3), although a temperature of 75 °C is appropriate to achieve a practical reaction rate (entry 4).
Table 1.
Hydroamination of Aminoallene 7 (5 mol % of Catalyst)

| yield (%)a |
|||||
|---|---|---|---|---|---|
| Entry | cat. | T/°C | t/h | imine | 5-exo |
| 1 | 1 | 75 | 12 | 9 | ---b |
| 2 | 1 | 135 | 3 | 74 | 10 |
| 3 | 2 | 25 | 26 | 41 | ---b |
| 4 | 2 | 75 | 3 | quant.c | ---b |
| 5 | 9 | 75 | 24 | 35 | 2 |
| 6 | 11 | 25 | 5 | quant.c | ---b |
NMR conversion versus 1,3,5-C6H3(OMe)3.
Not observed.
Complete and selective conversion of 7.
Previously we have carried out reactions with allenes using zirconium imido complexes.3,13 Therefore we investigated the reactivity of the zirconium analogue of 2, Zr(NMe2)4 (9). Although 9 exhibits catalytic activity, the reaction is much slower at 75 °C and less selective than the one using the titanium precursor 2 (entry 5).
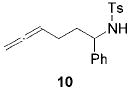
Complex 2 is significantly more reactive than 1 in intramolecular hydroamination reactions. However, a direct insertion of an alkyne into a titanium–nitrogen bond of a LnTiNMe2 species and subsequent protonation can lead to the formation of undesirable dimethylamine addition side products.8 Electron-poor secondary tosylamido complexes show no tendency to react in a similar manner. This is illustrated by the fact that the amidoallene 10 was deprotonated by 2 to form the corresponding titaniumamido complex and HNMe2. No cyclization was observed (Scheme 3).
Scheme 3.
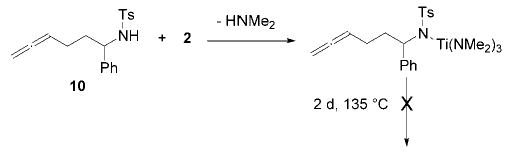
Attempted Hydroamination of Amidoallene 10
As the asymmetric catalytic hydroamination remains our ultimate goal, we studied the reactivity of the titanium bis-(sulfonamide) 11 (Figure 1), which was prepared in one step from 2 following Walsh’s procedure.14 Gratifyingly, the chelating bissulfonamide ligand results in a significantly increased reactivity, leading to selective product formation at room temperature (entry 6).15
Figure 1.
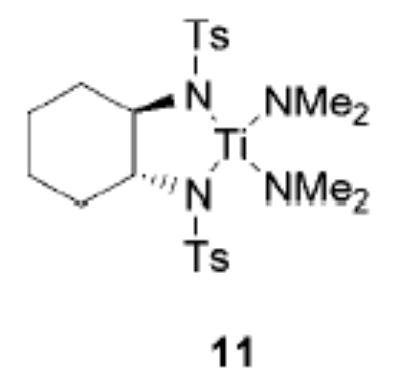
Precatalyst 11.
Furthermore, it is noteworthy that the six-membered ring product 8 is formed exclusively using titanium tetrakisamido precatalysts 2 and 11.
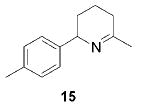
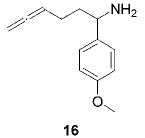
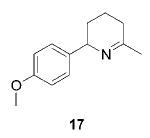
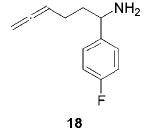
When the α position of the amine is substituted with an aromatic group, the regioselectivity of the cyclization depends on the nature of the amide ligands (Table 2). While 2 generates a mixture of regioisomers (5–10% 5-exo product), the chelated titanium complex 11 forms the cyclic imines as the sole products, thereby allowing the isolation of these compounds by simple filtration through K2CO3.16 Furthermore, the examples compiled in Table 2 demonstrate the increased efficacy of 11; the reaction times are significantly reduced.17
Table 2.
Hydroamination of Substituted Aminoallenes (5 mol % of Catalyst, 75 °C)
| yielda |
||||
|---|---|---|---|---|
| substrate | major product | cat. (t/b) | Imine | 5-exo |
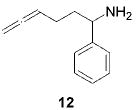
|
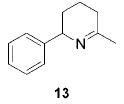
|
2 (22) | (95) | (5) |
| 11 (1) | 84 | ---b | ||
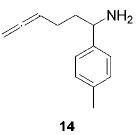
|
|
2 (9) | (90) | (10) |
| 11 (5) | 79 | ---b | ||
|
|
2 (4) | (92) | (8) |
| 11 (1.5) | 95 | ---b | ||
|
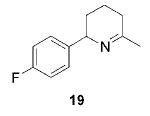
|
11 (10) | 93 | ---b |
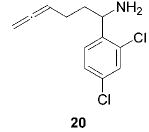
|
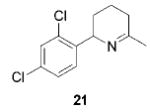
|
11 (2) | 88 | ---b |
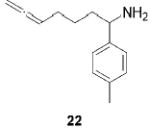
|
|
11c (36) | (60) | (4) |
|
--- | 11c (24) | ---b | |
Isolated yield, NMR conversion in parentheses.
Not observed by 1H NMR.
10 mol % of 11, 135 °C.
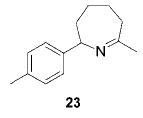
Having established the increased reactivity as well as the improved regioselectivity of the bis(sulfonamide)-based catalyst 11, we studied the scope of the intramolecular hydroamination reaction of aminoallenes. As depicted in Table 2, this system tolerates methoxy – and halogen–arene bonds, providing the respective products 17, 19, and 21 in good yields. More importantly, the scope of this procedure is not limited to the synthesis of favorably formed six-membered rings but can be instead extended to the formation of larger ring systems such as 23.
The enhanced regioselectivity of 11 may be due to the sterically demanding bis(sulfonamide) ligand, which disfavors cycloaddition of the titanium imido species with the internal double bound of the allene (Scheme 4).
Scheme 4.
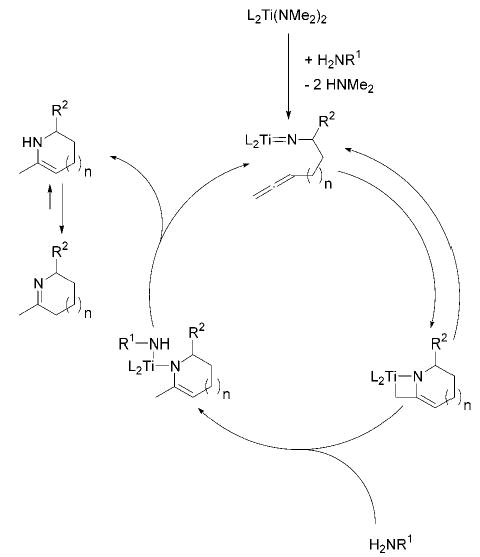
Postulated Mechanism for the Intramolecular Hydroamination of Aminoallenes (L = Cp or Amide Ligand; R1 = Allenyl; R2 = H or Aryl)
In summary, we have shown that titanium and zirconium tetrakisamide-based transition metal complexes can be used for the efficient intramolecular hydroamination of aminoalkynes and aminoallenes. The regioselectivity of this transformation depends on the nature of the precursor, yielding the most selective and reactive catalysis with the bis(sulfonamido) complex 11. Studies concerning the scope of this procedure, especially using 1,3-disubstituted amino-allenes, are currently ongoing.
Acknowledgments
This work was supported by the U.S. National Institutes of Health (Grant GM-25459 to R.G.B.). L.A. thanks the Deutscher Akademischer Austauschdienst (DAAD) for a postdoctoral fellowship.
Footnotes
Supporting Information Available: Experimental procedures and characterization data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.a Müller TE, Beller M. Chem Rev. 1998;98:675. doi: 10.1021/cr960433d. [DOI] [PubMed] [Google Scholar]; b Nobis M, Driessen-Hölscher B. Angew Chem, Int Ed. 2001;40:3983. doi: 10.1002/1521-3773(20011105)40:21<3983::aid-anie3983>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- 2.a Kawatsura M, Hartwig JF. J Am Chem Soc. 2000;122:9546. [Google Scholar]; b Kawatsura M, Hartwig JF. Organometallics. 2001;20:1960. [Google Scholar]; c Löber O, Kawatsura M, Hartwig JF. J Am Chem Soc. 2001;123:4366. doi: 10.1021/ja005881o. [DOI] [PubMed] [Google Scholar]; d Ryu JS, Marks TJ, McDonald FE. Org Lett. 2001;3:3091. doi: 10.1021/ol010129t. [DOI] [PubMed] [Google Scholar]; e Straub T, Haskel A, Neyroud TG, Kapon M, Botoshansky M, Eisen MS. Organometallics. 2001;20:5017. [Google Scholar]; f Yamamoto Y, Radhakrishnan U. Chem Soc Rev. 1999;28:199. [Google Scholar]; g Senn HM, Blöchel PE, Togni A. J Am Chem Soc. 2000;112:4098. [Google Scholar]; h Müller TE, Berger M, Grosche M, Herdtweck E, Schmidtchen FP. Organometallics. 2001;20:4384. [Google Scholar]; i Minami T, Okamoto H, Ikeda S, Tanaka R, Ozawa F, Yoshifuji M. Angew Chem, Int Ed. 2001;40:4501. doi: 10.1002/1521-3773(20011203)40:23<4501::aid-anie4501>3.0.co;2-k. [DOI] [PubMed] [Google Scholar]; j Hartung CG, Tillack A, Trautwein H, Beller M. J Org Chem. 2001;66:6339. doi: 10.1021/jo010444t. [DOI] [PubMed] [Google Scholar]; k Hartung CG, Breindl C, Tillack A, Beller M. Tetrahedron. 2000;56:5157. [Google Scholar]
- 3.a Walsh PJ, Baranger AM, Bergman RG. J Am Chem Soc. 1992;114:1708. [Google Scholar]; b Baranger AM, Walsh PJ, Bergman RG. J Am Chem Soc. 1993;115:2753. [Google Scholar]
- 4.Siebenreicher H, Doye S. J Prakt Chem. 2000;342:102. [Google Scholar]
- 5.a Haak E, Bytschkov I, Doye S. Angew Chem, Int Ed. 1999;38:3389. [PubMed] [Google Scholar]; b Bytschkov I, Doye S. Eur J Org Chem. 2001:4411. [Google Scholar]
- 6.McGrane PL, Jensen M, Livinghouse T. J Am Chem Soc. 1992;114:5459. [Google Scholar]
- 7.Johnson JS, Bergman RG. J Am Chem Soc. 2001;123:2923. doi: 10.1021/ja005685h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Shi Y, Ciszewski JT, Odom AL. Organometallics. 2001;20:3967. [Google Scholar]
- 9.a McGrane PL, Livinghouse T. J Org Chem. 1992;57:1323. [Google Scholar]; b McGrane PL, Livinghouse T. J Am Chem Soc. 1993;115:11485. [Google Scholar]
- 10.a Arseniyadis S, Gore J. Tetrahedron Lett. 1983;24:3997. [Google Scholar]; b Kinsman R, Lathbury D, Vernon P, Gallagher T. J Chem Soc, Chem Commun. 1987:243. [Google Scholar]; c Fox DNA, Gallagher T. Tetrahedron. 1990;46:4697. [Google Scholar]; d Al-Masum M, Meguro M, Yamamoto Y. Tetrahedron Lett. 1997;38:6071. [Google Scholar]; e Meguro M, Yamamoto Y. Tetrahedron Lett. 1998;39:5421. [Google Scholar]
- 11.a Arredondo VM, McDonald FE, Marks TJ. J Am Chem Soc. 1998;120:4871. [Google Scholar]; b Arredondo VM, McDonald FE, Marks TJ. Organometallics. 1999;18:1949. [Google Scholar]; c Arredondo VM, Tian S, McDonald FE, Marks TJ. J Am Chem Soc. 1999;121:3633. 1,3-Disubstituted aminoallenes, however, are converted exclusively to the 5-exo product using lanthanide precatalysts. [Google Scholar]
- 12.Note that the intermolecular hydroamination of alkynes using 2 yields preferentially the Markovnikov product.8
- 13.Sweeney ZK, Salsman JL, Andersen RA, Bergman RG. Angew Chem, Int Ed. 2000;39:2339. doi: 10.1002/1521-3773(20000703)39:13<2339::aid-anie2339>3.0.co;2-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pritchett S, Gantzel P, Walsh PJ. Organometallics. 1999;18:823. [Google Scholar]
- 15.The improved reactivity of 11 may be a consequence of the weak coordination of the sulfur-bound oxygen atoms to the metal center.14 However, the different electronic properties as well as the bidentate nature of the bis(sulfonamide) ligand can also play an important role.
- 16.Representative procedure: A solution of 18 (121 mg, 0.63 mmol) and 11 (18 mg, 0.03 mmol) in benzene (3 mL) was heated for 10 h to 75 °C. The solution was cooled and treated with 20 drops of methanolic NaOH (10%). The mixture was stirred for 0.5 h at room temperature and concentrated in vacuo. The remaining residue was extracted with n-hexane (30 mL) and filtered through K2CO3 to afford 19 (112 mg, 93%) as a pale yellow oil. 1H NMR (CD2Cl2, 300 MHz): δ 7.23 (m, 2H), 7.01 (tm, 2H, J = 8.9 Hz), 4.43 (m, 1H), 2.30–2.10 (m, 2H), 1.98 (d, 3H, J = 2.0 Hz), 1.95–1.60 (m, 3H), 1.40–1.20 (m, 1H). 13C NMR (CD2Cl2, 100 MHz): δ168.6, 128.4, 128.3, 114.8, 114.5, 60.8, 30.6, 29.9, 27.3, 19.0. MS (EI) m/z (relative intensity) 191 (81) [M+], 163 (14), 162 (14), 148 (11), 121 (100), 109 (10). HR-MS (EI) m/z calcd for C12H14FN 191.1110, found 191.1109.
- 17.As the sterogenic centers of the imine products are not generated during the catalytic hydroamination reaction, a potential kinetic resolution has yet not been intensively investigated. Instead, the conversion of 1,3-disubstituted aminoallenes11 should constitute an appropriate tool to explore asymmetric catalysis.