

Abstract
The pharmacology of monoacylglycerol lipase (MAGL) is not well understood. In consequence, the abilities of a series of analogues of 2-arachidonoylglycerol (2-AG) to inhibit cytosolic 2-oleoylglycerol and membrane-bound anandamide hydolysis by MAGL and fatty acid amide hydrolase (FAAH), respectively, have been investigated.
2-AG and its 1-regioisomer (1-AG) interacted with MAGL with similar affinities (IC50 values 13 and 17 μM, respectively). Shorter homologues of 2-AG (2-linoleoylglycerol and 2-oleoylglycerol) had affinities for MAGL similar to 2-AG. This pattern was also seen when the arachidonoyl side chain of arachidonoyl trifluoromethylketone was replaced by an oleoyl side chain.
Arachidonoyl serinol (IC50 value 73 μM) was a weaker inhibitor of MAGL than 2-AG. The IC50 values of noladin ether towards MAGL and FAAH were 36 and 3 μM, respectively. Arachidonoyl glycine interacted with FAAH (IC50 value 4.9 μM) but only weakly interacted with MAGL (IC50 value >100 μM).
α-Methyl-1-AG had similar potencies towards MAGL and FAAH (IC50 values of 11 and 33 μM, respectively). O-2203 (1-(20-cyano-16,16-dimethyl-eicosa-5,8,11,14-tetraenoyl) glycerol) and O-2204 (2-(20-hydroxy-16,16-dimethyl-eicosa-5,8,11,14-tetraenoyl) glycerol) were slightly less potent, but again affected both enzymes equally. α-Methyl-1-AG, O-2203 and O-2204 interacted only weakly with cannabinoid CB1 receptors expressed in CHO cells (Ki values 1.8, 3.7 and 3.2 μM, respectively, compared with 0.24 μM for 1-AG) and showed no evidence of central cannabinoid receptor activation in vivo at doses up to 30 mg kg−1 i.v.
It is concluded that compounds like α-Methyl-1-AG, O-2203 and O-2204 may be useful as leads for the discovery of selective MAGL inhibitors that lack direct effects upon cannabinoid receptors.
Keywords: 2-Arachidonoyl glycerol, anandamide, monoacylglycerol lipase, fatty acid amide hydrolase, endocannabinoid
Introduction
Cannabinoid receptors, the main target for cannabinoid drugs such as Δ9-tetrahydrocannabinol, derived from Cannabis sativa, were first identified in the early 1990s. Since then, enormous progress has been made towards understanding the biology, chemistry, and pharmacology of the endogenous ligands (endocannabinoids) for these receptors (for a recent review, see De Petrocellis et al., 2004). In particular, recent data implicating endocannabinoid involvement in pain processing, neuroprotection and cell proliferation (see e.g. Pertwee, 2001; Bifulco & Di Marzo, 2002; Fowler, 2003, for reviews) has underlined the possible importance of the endocannabinoid system as a target for drug development.
The most investigated endocannabinoid ligands have been anandamide (AEA) and 2-arachidonoylglycerol (2-AG). Both are synthesised by neurons on demand, through stimulus-dependent cleavage of membrane phospholipid precursors (see De Petrocellis et al., 2004). In the brain, endocannabinoids have been shown to act as retrograde transmitters controlling the release of GABA and glutamate (see Gerdeman & Lovinger, 2003). It is not clear, however, which endocannabinoid is responsible for this effect, and indeed, whether the same endocannabinoid is involved in different brain regions and animal species. One possible way of investigating this would be the use of agents selectively preventing the breakdown of AEA or 2-AG (see Kim & Alger, 2004, where this approach has been utilised for retrograde signalling in rat hippocampal slices). Both AEA and 2-AG are very rapidly metabolised (see Járai et al., 2000; Wiley et al., 2000), owing to the presence of efficient metabolising enzymes. For AEA, the principal metabolic pathway is hydrolysis to arachidonic acid by a ∼63 kDa enzyme termed fatty acid amide hydrolase (FAAH, Deutsch & Chin, 1993; Cravatt et al., 1996). This enzyme, which has a wide substrate specificity, has been well characterised pharmacologically (for reviews, see Cravatt & Lichtman, 2002; Fowler, 2004), and selective inhibitors such as URB597 (3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate) have been described in the literature (Kathuria et al., 2003). Mice lacking FAAH show increased brain levels of AEA, together with both reduced pain sensitivity (Cravatt et al., 2001) and seizure thresholds (Clement et al., 2003).
2-AG can be metabolised by FAAH, indeed at a faster rate than AEA (Di Marzo et al., 1998; Goparaju et al., 1998), and in some cell lines inhibition of FAAH leads to increased levels of 2-AG (see e.g. Ligresti et al., 2003). However, in the brain, the enzyme monoacylglycerol lipase (MAGL) is a more important metabolic enzyme for this endocannabinoid (Goparaju et al., 1999; Dinh et al., 2002; Saario et al., 2004; see also Lichtman et al., 2002). MAGL, a 33 kDa protein with close sequence similarities between rat, mouse and man (Karlsson et al., 1997; 2001; Dinh et al., 2002; Ho et al., 2002), was initially investigated owing to its participation in the metabolism of triacylglycerols, and shown to be able to metabolise a number of monoacylglycerols with, in the case of the arachidonoyl and oleoyl compounds, similar rates of metabolism for the 2-glycerols and the 1(3)-regioisomers (Tornqvist & Belfrage, 1976; Sakurada & Noma, 1981; Saario et al., 2004), a property shared with FAAH (Di Marzo et al., 1998, see also Ikeda et al., 1977). In contrast to FAAH, which is a membrane-bound enzyme with a pH optimum in the region of 9–10 (see e.g. Schmid et al., 1985; Di Marzo et al., 1998; Goparaju et al., 1998), MAGL activity has been described for both cytosolic and membrane-bound locations, with a pH optimum generally reported in the area of pH 7–8 (Tornqvist & Belfrage, 1976; Sakurada & Noma, 1981; Somma-Delpéro et al., 1995; Di Marzo et al., 1999), and with a more discrete distribution in the brain than FAAH (Dinh et al., 2002).
MAGL has been shown to be inhibited by nonspecific agents such as p-chloromercuribenzoic acid, N-ethylmaleimide and diisopropyl fluorophosphate (Tornqvist & Belfrage, 1976; Sakurada & Noma, 1981), and to have a much lower sensitivity to inhibition by ‘standard' FAAH inhibitors such as phenylmethylsulphonyl fluoride, arachidonoyl trifluoromethyl ketone (ATMK), URB597 and arachidonoyl-serotonin than FAAH (Bisogno et al., 1997; Di Marzo et al., 1999; Goparaju et al., 1999; Dinh et al., 2002; Saario et al., 2004). However, a detailed pharmacological study on the comparative effects of arachidonoyl-based compounds upon FAAH and MAGL has not yet been reported. This has been addressed in the present study.
Methods
Compounds
Radiolabelled arachidonoylethanolamide [ethanolamine 1-3H] ([3H]AEA, 60 Ci mmol−1) and 2-Mono-oleoylglycerol [glycerol-1,2,3-3H] ([3H]2-OG, 20 Ci mmol−1) were obtained from American Radiolabelled Chemicals Inc. (St Louis, MO, U.S.A.). 2-Arachidonoylglycerol (2-AG), 1-arachidonoylglycerol (1-AG), α-Me-1-AG (α-methyl-1-arachidonoylglycerol, compound O-1428), compounds O-1502, O-2203 and O-2204 were synthesised by co-authors Razdan and Mahadevan using previously published procedures (Han & Razdan, 1999; Ng et al., 1999; Dasse et al., 2000). Nonradioactive AEA, arachidonoyl trifluoromethyl ketone (ATMK), palmitoyl trifluoromethyl ketone (PTMK), oleoyl trifluoromethyl ketone (OTMK), arachidonoyl serinol, palmitoyl serinol, 2-linoleoylglycerol (2-LG), linoleoyl ethanolamine (LEA), arachidonic acid, URB597 and CAY10412 (5Z,8Z,11Z,14Z-eicosatetraenoic acid, 3-thienylmethyl ester, compound 6 of López-Rodríguez et al. (2001) were obtained from the Cayman Chemical Co. (Ann Arbor, MI, U.S.A.). Arachidonoyl glycine, palmitoylethanolamide (PEA), oleoylethanolamide (OEA) and noladin ether were obtained from TocrisCookson (Bristol, U.K.). Nonradioactive 2-OG and indomethacin were obtained from the Sigma Chemical Co. (St Louis, MO, U.S.A.). Fatty acid free bovine serum albumin was obtained either from Calbiochem-Novabiochem (La Jolla, CA, U.S.A.), or from the Sigma Chemical Co.
Assay of MAGL and FAAH
Cerebella previously obtained from adult Sprague–Dawley rats were thawed and homogenised at 4°C in sodium phosphate buffer (50 mM, pH 8) containing 0.32 M sucrose. Homogenates were then centrifuged at 100,000 × g for 60 min at 4°C. The supernatants (‘cytosol fractions') were collected and the pellets were suspended in sodium phosphate buffer (50 mM, pH 8) (‘membrane fractions'). The samples were stored frozen in aliquots at −70°C until used for assay. Protein concentration was determined by using the method described by Harrington (1990), with bovine serum albumin as standard.
Hydrolysis rates of [3H]AEA and [3H]2-OG were determined in the Umeå laboratory essentially as described by Omeir et al. (1995) and Dinh et al. (2002). Briefly, membrane or cytosol fractions were diluted to the appropriate assay protein concentrations in Tris-HCl buffer (10 mM, pH 7.2) containing 1 mM EDTA, unless otherwise stated. Aliquots (165 μl) were then added to glass tubes containing 10 μl of test compound. Blanks contained assay buffer instead of homogenate solution. Substrate (25 μl, final concentration 2 μM unless otherwise stated) was then added and the samples were incubated for 10 min at 37°C. Reactions were stopped by adding 400 μl chloroform : methanol (1/1 v v−1), vortex mixing the tubes two times and placing them on ice. The phases were then separated by centrifugation (10 min, 2500 r.p.m.). Aliquots (200 μl) of the methanol/buffer phase were taken and measured for tritium content by liquid scintillation spectroscopy with quench correction. In one series of experiments with [3H]AEA, parallel tubes were assayed as described above, but where the extraction process used active charcoal (80+320 μl of 0.5 M HCl per tube) (Boldrup et al., 2004) rather than chloroform/methanol.
Assays for CB1 cannabinoid receptor activity
CB1 cannabinoid receptor activity of selected compounds was determined using a combination of in vitro and in vivo assays. In vitro radioligand-binding assays were undertaken using [3H]CP 55,940 as ligand and membranes from CHO cells expressing CB1 receptors as described in detail previously (Pertwee et al., 2000). In vivo measures of central cannabimimetic activity were undertaken in the Richmond laboratory using male ICR mice (Harlan Laboratories, Dublin, VA, U.S.A.) and behavioural tests measuring spontaneous activity, rectal temperature, and nociceptive threshold (tail flick tests) (for details, see Martin et al., 1999; Pertwee et al., 2000).
Analysis of data
For the MAGL and FAAH assays, the pooled data for each test compound, expressed as % of control activity containing the same carrier concentration, were analysed using the built-in equation ‘sigmoidal dose–response (variable slope)' of the GraphPad Prism computer programme (GraphPad Software Inc., San Diego, CA, U.S.A.). The equation, as used here, fits observed % of control value=minimum % value+(100−% minimum value)/ (1 + 10(pX−pI50)nH). pI50 represents −log10 IC50 for the inhibitable component (i.e. 100−% minimum value), nH the Hill slope, and pX the −log10 concentration of the test compound. The ‘top' (i.e. uninhibited) values were fixed at 100% and the minimum % value were either fixed at 0% (defined by the programme as the simpler model) or determined by the programme (defined as the alternative model). When the 95% confidence limits of the minimum % values straddled zero, the simpler model was used as default to avoid artefactual results owing to unrealistic negative minimum values. When the 95% confidence limits of the minimum % values were both positive, the two models were compared by the programme using Akaike's information criterion, which determines which model is more likely to be correct. The model chosen by this analysis was then used.
Km and Vmax values were calculated using the direct linear plot analysis of Eisenthal & Cornish-Bowden (1974) as implemented by the Enzyme Kinetics v 1.4 software package, Trinity Software (Campton, NH, U.S.A.).
Results
MAGL and FAAH assay characterisation
Initial experiments were undertaken to determine the optimal assay conditions for MAGL and FAAH. Protein-dependency curves were constructed for both substrates using either Tris or sodium phosphate (both pH 8.0) as assay buffers. Hydrolysis of AEA was linear with protein content over the range tested (0–8 μg assay−1 for membranes and 0–16 μg assay−1 for supernatant), and there was no significant difference in the initial rates for the two buffer systems. Thus, for the Tris buffer, initial rates (the slope of the regression line of the observed activities over these protein ranges) were 0.74±0.053 and 0.042±0.017 nmol mg protein−1 min−1 for membrane and cytosol fractions, respectively. 2-OG was metabolised at a faster rate in both fractions, and linearity was lost for the membrane fractions at >4 μg assay−1, but the initial rates were not significantly different for the two buffers (6.49±0.38 and 6.02±0.29 nmol mg−1 protein−1 min−1 for Tris and sodium phosphate buffers, respectively). The cytosolic fractions showed linearity up to the highest protein content tested (16 μg assay−1), and the activity was significantly greater in the Tris buffer than in the sodium phosphate buffer (2.04±0.045 vs 1.23±0.090 nmol mg−1 protein−1 min−1). Tris buffer was thus used for the experiments reported here. The pH optimum for 2-OG (and AEA) metabolism by the cytosol fractions was lower than for AEA metabolism by the membrane fractions (Figure 1).
Figure 1.

Specific activity of: (a) cytosol; (b) membrane preparations towards 2 μM AEA and 2-OG using Tris buffers of different pH. Note that the pH values shown in the abscissae are the pH of the buffer (determined at room temperature) rather than of the final assay mixture at 37°C. Data are means±s.e.m., n=3.
The effect of different solvents upon the rate of hydrolysis of AEA and 2-OG was examined at pH 7.2 in membrane and cytosol fractions, respectively. Acetonitrile (0.05–5% v v−1) and DMSO (0.05–2% v v−1) had no effect on the observed rates of metabolism. Similarly ethanol (0.05–5%) did not affect the rate of AEA metabolism (data not shown). However, ethanol produced a small increase in the rate of 2-OG metabolism by the cytosolic fractions, mean (n=2) values (% of control) of 113, 125, 129, 138 and 151 being seen for added ethanol concentrations of 0.2, 0.5, 1, 2 and 5% v v−1, respectively (above the 0.25% ethanol present as vehicle for the substrates alone). In all experiments, solvent carrier concentrations were kept constant throughout, and never exceeded 0.5% (above the 0.25% ethanol present for the substrates alone). Under such conditions, [3H]2-OG was metabolised by the cytosolic fractions with a median (n=3) Km value of 2.2 μM (data not shown). The solvent used for each compound is indicated in Tables 1 and 2.
Table 1.
Inhibition of FAAH and MAGL by acyl gycerols, ethanolamines and trifluromethylketones
| pI50 (IC50 μM) [% max inhibition] for | |||
|---|---|---|---|
| Membrane [3H]AEA | Cytosol [3H]2-OG | Selectivity | |
| 2-acyl glycerols | 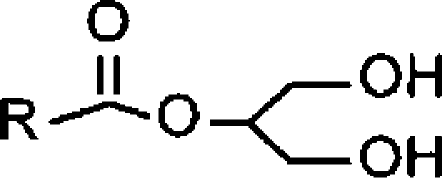 |
||
| 2-AG (C20:4)a | <4 (37% inhibition at 100 μM) | 4.88±0.05 (13) [77±4] | >7.6 (>4.2) |
| 2-OG (C18:1)a | 5.61±0.26 (2.5) [53±9] | 4.84±0.03 (15) [100] | 0.07 (2.5) |
| 2-LG (C18:2)a | 4.65±0.03 (22) [100] | 4.92±0.03 (12) [85±3] | 1.8 (1.3) |
| N-acyl ethanolamines | 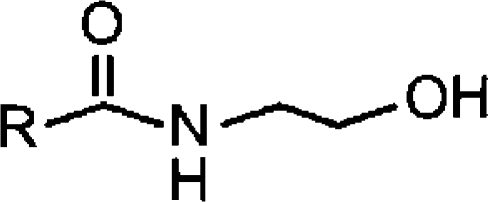 |
||
| AEA (C20:4)b | 5.89±0.12 (1.3)c [66±5] | 4.22±0.04 (60) [100] | 0.18 (0.12) |
| LEA (C18:2)b | 5.19±0.03 (6.5) [100] | 4.08±0.03 (84) [100] | 0.08 |
| PEA (C16:0)a | 4.38±0.27 (42) [100] | <4 (no inhibition at 100 μM) | <0.4 |
| Acyl trifluromethylketones | 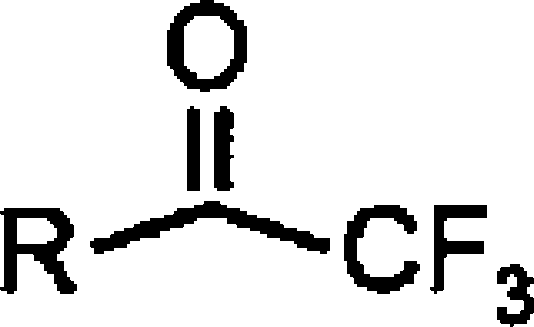 |
||
| ATMK (C20:4)b | 6.26±0.08 (0.55) [100] | 5.54±0.04 (2.9) [100] | 0.2 |
| OTMK (C18:1)b | 7.12±0.09 (0.076) [100] | 5.99±0.12 (1.0) [69±10] | 0.07 (0.03) |
| PTMK (C16:0)b | 7.14±0.09 (0.073) [100] | 5.11±0.09 (7.8) [100] | 0.009 |
Data are means±s.e.m. from analyses of data from three experiments. C20:4 refers to the number of carbon atoms and unsaturated bonds, respectively, in the acyl side chain (including the carbonyl group; thus PEA has an R of C15H29). Substrate concentrations were 2 μM. ‘Selectivity' is the ratio of pI50 values for the inhibitable components (in brackets are values assuming 100% inhibtion, when different): the higher the value, the more the MAGL selectivity.
Compounds were dissolved in aacetonitrile or bethanol.
Table 2.
Inhibition of FAAH and MAGL by analogues of 2-AG
| pI50 (IC50 μM) [% max inhibition] for | ||||
|---|---|---|---|---|
 |
Membrane [3H]AEA | Cytosol [3H]2-OG | Selectivity | |
| 2-AGa | 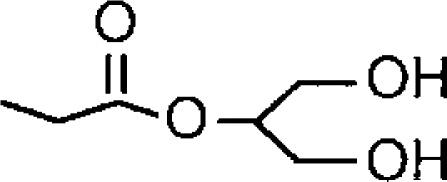 |
<4 (37% inhibition at 100 μM) | 4.88±0.05 (13) [77±4] | >7.6 (>4.2) |
| 1-AGa | 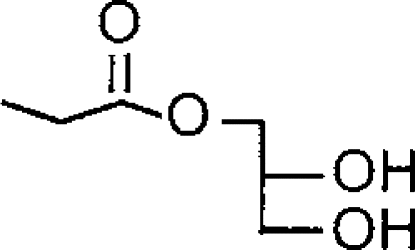 |
<4 (42% inhibition at 100 μM) | 4.77±0.05 (17) [100] | >5.9 |
| α-Me-1-AGa | 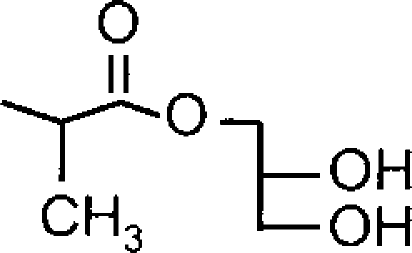 |
4.49±0.11 (33) [100] | 4.97±0.09 (11) [82±7] | 3.1 (1.9) |
| Arachidonoyl serinolb | 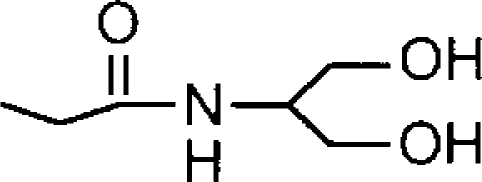 |
4.11±0.12 (78) [100] | 4.14±0.03 (73) [100] | 1.1 |
| O-1502a | 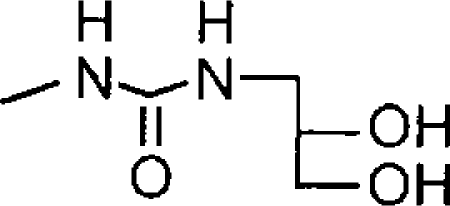 |
4.73±0.08 (19) [100] | <4 (39% inhibition at 100 μM) | <0.19 |
| Arachidonic acidb | 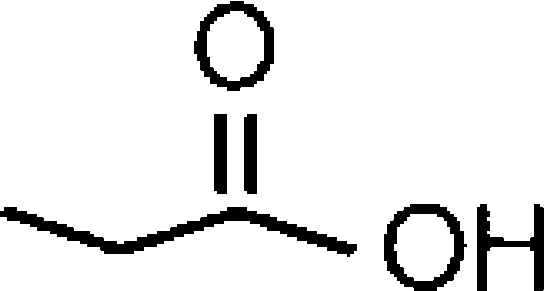 |
4.24±0.09 (58) [100] | 4.11±0.05 (78)d [100] | 0.74 |
| CAY-10402c | 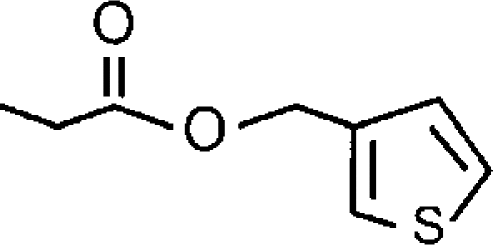 |
4.99±0.06 (10) [37±2] | <4 (14% inhibition at 100 μM)d | <0.10 (−e) |
| Noladin etherb | 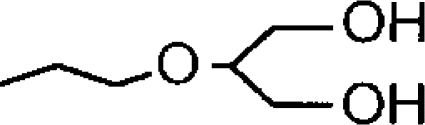 |
5.53±0.12 (3.0) [100] | 4.45±0.06 (36) [100%] | 0.08 |
| Arachidonoyl glycineb | 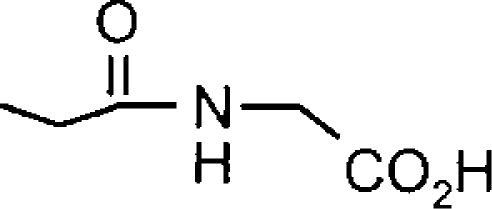 |
5.31±0.13 (4.9) [100] | <4 (42% inhibition at 100 μM) | <0.05 |
Data are means±s.e.m. from analyses of data from three experiments. The data for 2-AG are the same as in Table 1, and are included for reference purposes. ‘Selectivity'is the ratio of pI50 values for the inhibitable components (in brackets are values assuming 100% inhibtion, when different).
Compounds were dissolved in aacetonitrile, bethanol, cor provided dissolved in methyl acetate and diluted in acetonitrile.
The basal activities were rather high. In consequence, the 50 and 100 μM concentrations were retested at a lower (1 μg) protein concentration. For CAY10402, the % values were very similar, whereas arachidonic acid was slightly more potent in the second series. The pI50 and hence IC50 values are calculated from all data points, and therefore may be a slight underestimation of the potency of arachidonic acid.
pI50 value for both <4 when 100% inhibition used. The pH of the assay buffer was 7.2.
Validity of MAGL assay
Given that 2-AG (and presumably also 2-OG) can act as a substrate for FAAH (Goparaju et al., 1998), it is essential to demonstrate that the observed activity towards 2-OG in the cytosolic fractions is not owing to contamination by FAAH remaining in the cytosolic fraction. To this end, experiments were undertaken using URB597, which has been shown to be a potent (IC50 value 4.6 nM) inhibitor of rat brain FAAH but which does not affect the activity of rat brain MAGL (IC50 value >30 μM, Kathuria et al., 2003; see also Kim & Alger, 2004). Concentrations of 1 and 10 μM URB597 were tested and found completely to inhibit the hydrolysis of AEA by the membrane fractions without affecting the hydrolysis of 2-OG by the cytosolic fractions (Figure 2a). Similarly, palmitoylethanolamide and arachidonoylglycine, both of which are substrates for FAAH (Natarajan et al., 1984; Huang et al., 2001), inhibited the hydrolysis of AEA by the membrane fractions without affecting the hydrolysis of 2-OG by the cytosolic fractions (apart from the observed inhibition at 100 μM arachidonoylglycine) (Figure 2b and c). Finally, indomethacin, which is known to inhibit FAAH, particularly at low pH values (Fowler et al., 2003), did not affect 2-OG metabolism at concentrations ⩽50 μM (Figure 2d). Thus, the FAAH component of the cytosolic 2-OG metabolism is not at a level that will obfuscate interpretation of the data.
Figure 2.

Inhibition of cytosolic [3H]2-OG hydrolysis and membrane-bound [3H]AEA hydrolysis by: (a) URB597 and AEA; (b) PEA; (c) arachidonoyl glycine; (d) indomethacin. Data are means±s.e.m. (when not enclosed by the symbols), n=3.
In the experiments using URB597, AEA was also investigated. A concentration of 10 μM was without effect upon the cytosolic activity of 2-OG, whereas a concentration of 100 μM did reduce the activity (Figure 2c). Addition of URB597 had at best minor effects upon the observed inhibition produced by AEA. Further experiments indicated that AEA could produce a complete inhibition of 2-OG metabolism by the cytosolic fractions, although with a potency considerably lower than seen for inhibition of [3H]AEA metabolism by the membrane fractions (see Figure 3b).
Figure 3.

(a) Inhibition of membrane [3H]AEA hydrolysis by nonradioactive AEA, nonradioactive 2-OG and CAY-10402. (b) Inhibition of membrane [3H]AEA hydrolysis by nonradioactive AEA using either the standard assay (2.5 μg protein assay−1), the same conditions but either with an acid charcoal extraction, the addition of 0.2% glycerol and 0.02% Triton X-100 to the buffer, or the standard assay but with the assay buffer set to pH 9.0 (1.25 μg protein assay−1). The corresponding curve for the inhibition of cytosolic [3H]2-OG hydrolysis by AEA is also shown in the figure. (c) Inhibition of cytosolic [3H]2-OG hydrolysis by OTMK, α-Me-1-AG and 2-AG. (d) Inhibition of cytosolic [3H]2-OG hydrolysis by2-linoleoyl glycerol and O-2204. Data are means±s.e.m., n=3, unless enclosed by the symbols. Note that in (b), the error bars for the data using the charcoal extraction and for an assay pH of 9.0 are not shown for reasons of clarity. The curves shown were calculated as described in Methods assuming either complete inhibition (i.e. bottom value set to 0, dotted lines) or incomplete inhibition (i.e. bottom value determined by the computer programme to be significantly greater than 0, continuous lines in panels a, c and d). In all cases for panels a, c and d, the statistical analysis built into the programme (Akaike's information criterion, which determines which model is more likely to be correct) selected the curves showing incomplete inhibition to be the preferred curves.
Inhibition of FAAH and MAGL by 2-AG and related compounds
With the exception of the trifluoromethyl ketones, test compounds were tested at nine different concentrations over a range from 0.2 to 100 μM. For most cases, analysis of the data indicated that a curve ranging from 0 to 100% adequately fitted the data. However, in some cases, a curve with a residual activity fitted the data significantly better than a curve ranging from 0 to 100%. Such residual activity has previously been seen in this assay with fatty acid amide homologues and analogues and AEA as substrate (see e.g. Vandevoorde et al., 2003) and presumably reflects a solubility issue for these very lipophilic compounds. In Figure 2, curves are shown for such compounds assuming either complete inhibition (dotted lines) or residual inhibitions (continuous lines). In the case of the three compounds and AEA as substrate (Figure 2a), the differences in the lines are at first sight obvious.
The data with AEA as inhibitor are worth further comment, since it could reasonably be expected that the compound should inhibit its own metabolism. One explanation for the results, that the blank values are artefactually low, can be discounted, since the experiments were run together with indomethacin, which was found completely to inhibit the metabolism of [3H]AEA (Figure 2d). Inspection of the data, however, revealed that the two highest concentrations of AEA gave less inhibition of [3H]AEA metabolism than the 20 μM concentration, suggesting that perhaps during these experiments, the highest concentrations of AEA had not stayed in solution and thereby not interacted with the enzyme. Indeed, if the two highest concentrations were excluded, the residual activity was not significantly different from zero (pI50 value 5.36±0.09, IC50 value 4.3 μM). In this respect, it should be noted that in the experiments shown in Figure 2a, which were undertaken at a later date, an essentially complete inhibition of FAAH is seen with 100 μM AEA.
In order to shed more light on this anomaly, additional experiments were undertaken using membrane fractions, [3H]AEA, and different assay conditions: (a) the ‘standard' assay at pH 7.2; (b) the ‘standard' assay but using a different product extraction procedure which gives lower blank values (Boldrup et al., 2004); (c) addition of glycerol and Triton X-100 to the buffer; and (d) assay at pH 9.0 using the standard assay. In all these cases, the residual activity was not significantly different from zero (Figure 3b). The pI50 values (with IC50 values given in parentheses) were 5.34±0.04 (4.5 μM), 5.36±0.03 (4.4 μM), 4.93±0.25 (12 μM) and 5.26±0.08 (5.5 μM) for conditions (a), (b), (c) and (d), respectively. For comparative purposes, the inhibition curve for cytosolic 2-OG metabolism (which was run concomitantly with the AEA curve shown in Figure 3a) is shown in Figure 3b: the difference in potency for inhibition by AEA of membrane [3H]AEA and cytosol [3H]2-OG metabolism is very clear. 2-OG (100 μM) was also tested in these experiments. The % of control activity seen with 100 μM 2-OG vs membrane-bound [3H]AEA metabolism was 13±6, 7±2, 6±6 and –4±8 for conditions (a), (b), (c) and (d), respectively (means±s.e.m., n=3).
With respect to the compounds showing residual activities with 2-OG as substrate, the comparative curves assuming residual activity and 100% inhibition are almost superimposable (Figure 3c and d). This raises the issue as to whether in these cases the pI50 value calculated for the inhibitable component of the curves overestimates the true potency. In consequence, we have presented the results of these compounds as pI50 values for the inhibitable portion (so as to comply with the statistics), but calculated selectivity ratios for FAAH : MAGL for both analyses, in order to give a selectivity range, the lower (with respect to MAGL) selectivity value being considered as the more conservative estimate.
The data for the analogues and homologues of 2-AG and AEA are shown in Tables 1 and 2. 2-AG showed a >4-fold selectivity for MAGL over FAAH. Replacement of the arachidonoyl chain of 2-AG with a linoleoyl or oleoyl chain did not affect the observed potency towards inhibition of 2-OG metabolism, whereas these compounds interacted more potently with FAAH than 2-AG. For AEA and its shorter chain homologues (linoleoyl and palmitoylethanolamine), the compounds showed the expected FAAH selectivity, inhibiting AEA metabolism by acting as alternative substrates (Natarajan et al., 1984; Schmid et al., 1985; Maurelli et al., 1995) (Table 1). In contrast PTMK (palmitoyltrifluoromethylketone) was able to inhibit MAGL activity, albeit less potently than the arachidonoyl and oleoyl analogues, and considerably less potently than its ability to inhibit FAAH (Table 1).
With respect to the head group analogues of 2-AG (Table 2), the 1-AG analogue interacted with MAGL and FAAH with the same potency as 2-AG. Introduction of an α-methyl group into the 1-AG did not affect the potency towards MAGL, but increased the potency towards FAAH. Replacement of the COO group of 2-AG with a CONH group (arachidonoyl serinol) reduced the potency towards MAGL, so that the selectivity for this enzyme was lost. A further introduction of an –NH group (O-1502) resulted in an FAAH selective compound. Palmitoyl serinol only weakly interacted with FAAH and MAGL: thus, 20, 50 and 100 μM palmitoyl serinol produced 16±7, 18±6 and 20±11% inhibition, respectively, of membrane-bound AEA metabolism, and 6±0.6, 12±0.7 and 18±4% inhibition, respectively, of cytosolic 2-OG metabolism (means±s.e.m., n=3). Noladin ether, which has an ether linkage in place of the ester linkage was considerably FAAH selective, as was arachidonoyl glycine. Replacement of the glycerol group to give CAY10402 resulted in a weak inhibition of MAGL, whereas arachidonic acid had potencies towards both enzymes similar to arachidonoyl serinol.
Inhibition of MAGL and FAAH by O-2203 and O-2204
Two other compounds were tested, O-2203 and O-2204 (structures shown in Figure 4). These compounds (both dissolved in acetonitrile) inhibited FAAH and MAGL to similar extents (Figure 4a and b). Thus, for O-2203, the pI50 values towards membrane [3H]AEA and cytosol [3H]2-OG were 4.08±0.15 (IC50 83 μM) and 4.04±0.05 (IC50 90 μM), respectively, giving a selectivity ratio of 0.9. O-2204 inhibited membrane [3H]AEA hydrolysis with a pI50 value of 4.46±0.19 (IC50 35 μM). With respect to 2-OG hydrolysis, the compound did not produce complete inhibition (see Figure 3d), but for the inhibitable component (75±6%), the pI50 value was 4.85±0.10 (14 μM) giving a selectivity ratio of 2.5 (1.1 assuming 100 % inhibition in both cases).
Figure 4.

Inhibition of cytosolic [3H]2-OG and membrane-bound [3H]AEA hydrolysis by (a) O-2203 and (b) O-2204. The data for O-2204 and cytosolic [3H]2-OG is the same as that shown in Figure 3c, and is repeated here for comparative purposes. Data are means±s.e.m. (when not enclosed by the symbols), n=3.
Interaction of 2-AG, 1-AG, α-Me-1-AG, O-2203 and O-2204 with cannabinoid CB1 receptors
The ability of O-2203 and O-2204 to interact with CB1 receptors was investigated in behavioural and radioligand-binding assays. 2-AG and 1-AG were used as positive controls. As expected, 2-AG inhibited the binding of [3H]CP 55,940 to CB1 receptors expressed on CHO cells with a submicromolar affinity, and produced behavioural effects (inhibition of spontaneous activity, reduction of rectal temperature, thermal antinociception) consistent with central CB1 receptor activation (Table 3). 1-AG was also active in this respect, with the exception of the tail flick test (Table 3). This is in contrast to the study of van der Stelt et al. (2002) who reported a Ki value of >1000 nM for inhibition of [3H]CP 55,940 binding to rat forebrain CB1 receptors by this compound. However, Sugiura et al. (1999) reported that 1-AG showed good agonist efficacy towards CB1 receptors in NC108-15 cells and that a response was seen at submicromolar concentrations of this compound. The affinity towards CB1 receptors was reduced by an order of magnitude when an α-methyl group was inserted into 1-AG, and this compound did not show overt behavioural effects associated with central cannabinoid receptor activation (ED50 value >30 mg kg−1 i.v.) (Table 3). A similar result was seen for O-2203 and O-2204 (Table 3). Thus, α-Me-1-AG, O-2203 and O-2204 inhibit both cytosolic 2-OG and membrane AEA hydrolysis without having overt effects upon central CB1 receptors.
Table 3.
Cannabinoid receptor activities of O-2203 and related compounds
| ED50 (mg kg−1 i.v.) | ||||
|---|---|---|---|---|
| Compound | Ki (CHO-CB1) (nM) | Spontaneous activity | Rectal temperature | Tail flick latency |
| 2-AG | 538±17 | 19.4 | 7.9 | 4.8 |
| 1-AG | 241±99 | 12.0 | 10.0 | >30 |
| α-Me-1-AG | 1821±650 | >30 | >30 | >30 |
| O-2203 | 3710±910 | >30 | >30 | ND |
| O-2204 | 3240±489 | >30 | >30 | ND |
Discussion
In their recent mini review in this journal, De Petrocellis et al. (2004) listed a number of milestones that need to be reached to push the endocannabinoid field forward. One of these was ‘to develop selective and potent inhibitors of endocannabinoid biosynthesis and of 2-AG degradation that can be used in vivo'. With respect to 2-AG degradation, this aim can only be achieved when the pharmacology of MAGL is more completely understood. The present study, where the abilities of a series of 2-AG analogues to inhibit the hydrolysis of AEA and 2-OG have been investigated, is thus intended to provide a better understanding of the pharmacology of MAGL.
One particular confounding factor, at least for the early studies of 2-AG and 2-OG metabolism, is the ability of FAAH to interact with these substrates (see Introduction). Thus, for example, the early data of Ikeda et al. (1977) who characterised the hydrolysis of 2-OG and lower homologues by an enzyme purified from rat liver microsomes might in fact be an early FAAH study rather than a characterisation of MAGL, not least in view of its estimated molecular weight (62 kDa) and observed pH optimum value of 8.5. Later studies on membrane-bound enzymes are clearer: thus, for example, Chau & Tai (1988) demonstrated that 2-OG and 2-AG were metabolised by human platelet microsomal fractions with a pH optimum of ∼7 (although an activity with a higher pH optimum can be discerned from the figures), i.e. consistent with MAGL rather than FAAH, and showed in addition that the metabolism was not sensitive to inhibition by 100 μM indomethacin at pH 7.0, in contrast to the situation for rat brain FAAH (Fowler et al., 2003). The differential sensitivity to indomethacin has been confirmed here.
Although MAGL predominates in the metabolism of 2-AG in cerebellar membranes, as adjudged by its lack of sensitivity to standard FAAH inhibitors (Saario et al., 2004), we have elected to use the cytosolic rather than membrane-bound 2-OG metabolising activity as our source of MAGL in order to minimise any risk for contamination with the membrane-bound FAAH. Our data on the pH sensitivities for cytosolic 2-OG and membrane-bound AEA metabolism are entirely consistent with the selective assay of MAGL and FAAH, respectively (Tornqvist & Belfrage, 1976; Schmid et al., 1985) as are the differential sensitivities to inhibition by ATMK, URB597 and PEA (the latter acting towards FAAH as an alternate substrate) (Bisogno et al., 1997; Goparaju et al., 1999; Dinh et al., 2002; Kathuria et al., 2003; Saario et al., 2004). Similarly, the lack of difference in affinities of 2-AG and 1-AG as inhibitors of cytosolic 2-OG metabolism are entirely consistent with the known abilities of these regioisomers, and their oleoyl homologues, to act as substrates for MAGL (Tornqvist & Belfrage, 1976; Sakurada & Noma, 1981; Saario et al., 2004).
Three notable differences between the present study and the literature are, however, apparent: (a) the Km for 2-OG as substrate is relatively low; (b) AEA interacts, albeit with a low affinity, with MAGL; and (c) 2-AG is a rather weak inhibitor of FAAH. With respect to (a), our Km value of 2.2 μM is considerably lower than the Km values of 200 and 300 μM reported for the rat adipose tissue MAGL (soluble fraction) and mouse MAGL expressed in a baculovirus system, respectively (Tornqvist & Belfrage, 1976; Karlsson et al., 2000). However, the extremely lipophilic nature of 2-OG makes comparisons between different assays undertaken in different laboratories somewhat difficult – indeed, reported Km values for AEA as a substrate for FAAH also vary from 0.8 to 180 μM (see Fowler et al., 2001).
With respect to (b), Dinh et al. (2002) reported that MAGL cloned from rat brain and expressed in HeLa cells did not show appreciable AEA metabolism under the assay conditions used. Bisogno et al. (1997) found that 2-AG hydrolysis by cytosolic fractions from N18TG2 cells was only inhibited by 8% by 100 μM AEA. However, given that the AEA-sensitive component of the 2-OG metabolism seen here was not sensitive to inhibition by URB597, a reasonable interpretation of these data is that AEA can interact with MAGL, although with a much lower affinity than with FAAH, and not as a substrate. This conclusion (which can also be made for linoleoyl ethanolamine) raises the question as to whether the cytosolic AEA metabolism seen in the present study represents metabolism catalysed by FAAH or by MAGL. FAAH is a membrane-bound enzyme, but it is possible for homogenisation procedures to produce membrane micelles that do not sediment following centrifugation for 100,000 g × 60 min (see e.g. Fowler et al., 1980). The pH sensitivity of the cytosolic AEA metabolism is consistent with MAGL. However, subsequent studies comparing the sensitivities of cytosolic and pellet AEA metabolism to inhibition by PTMK, AEA, URB597 and arachidonoyl glycine were inconclusive, owing to the very low level (and hence poor activity : blank ratios) of the cytosolic AEA (data not shown). Thus, further work is necessary in determining the enzyme(s) responsible for the cytosolic AEA hydrolysis.
With respect to (c) it is well established that 2-AG can be metabolised by FAAH, and Goparaju et al. (1998) reported a Km value of ∼6 μM for the hydrolysis of 2-OG by membranes from COS-7 cells transfected with rat FAAH. It is possible that membrane-bound MAGL will remove some of the 2-AG (and 1-AG) during the assay and thereby give an artefactually low potency towards the FAAH. This might explain why α-Me-1-AG is a better inhibitor of FAAH than 1-AG. Ki values for inhibition by 2-AG and 1-AG of AEA metabolism in U937 cells of 5 and 3 μM, respectively, have been reported by van der Stelt et al. (2002). Similarly, 2-AG can inhibit FAAH in intact CHP100 cells with an IC50 (after a 10 min incubation) of ∼10 μM (Maccarrone et al., 2000). However, effects in intact cells are hard to compare with those in cell-free cytosol fractions not only owing to the possibility of intracellular accumulation of the inhibitor, but also since other effects of the compounds may be involved. Thus, at least a component of the inhibition of FAAH by 2-AG in the CHP100 cells is secondary to activation of the lipoxygenase pathway (Maccarrone et al., 2000). In addition, the problems of comparing data between laboratories are evident here: thus, for example, in the same experiments AEA was a poor inhibitor (Ki>10 μM) of FAAH activity in the U937 cells (van der Stelt et al., 2002). Such variations in potency between laboratories towards FAAH have also been seen with other arachidonoyl-based compounds (Fowler et al., 2004). This underlines the importance of the present approach, namely to compare effects upon FAAH and MAGL in parallel assays.
The main part of the study has been the characterisation of the affinities of a series of 2-AG analogues upon MAGL compared with FAAH. A number of observations can be made:
The shorter homologues of 2-AG, which retain at least one saturated bond in the acyl side chain, retain their affinity for MAGL. This was also seen when the arachidonoyl side chain of ATMK was replaced by an oleoyl side chain. Interestingly, the palmitoyl analogue (PTMK) was also able to inhibit 2-OG metabolism, suggesting that a saturated acyl side chain does not automatically result in a loss of affinity for MAGL.
The admittedly moderate selectivity towards MAGL for the arachidonoylglycerol regioisomers is lost either when the glycerol group is removed (arachidonic acid, a result consistent with the literature (Goparaju et al., 1999)) or replaced with a 3-thienylmethyl group (CAY10402); when either an α-methyl group is inserted (owing to an increased affinity for FAAH); or when the COO group is replaced by a CONH group to give arachidonoyl serinol (owing to a reduced affinity towards MAGL and a possible increased affinity for FAAH). This shift in selectivity is further accentuated when the terminal CH2 group of the arachidonoyl side chain is replaced by an NH group (O-1502). Whether or not this compound retains the ability of 2-AG to act as a substrate for FAAH is unclear. However, analogues of O-1502, albeit with simple aliphatic chains rather than the glycerol molecule, do not appear to be substrates for FAAH (as assessed by the lack of phenylmethylsulphonyl fluoride-dependence upon their affinities for CB1 receptors in radioligand-binding studies in vitro), but do interact with CB1 receptors to produce the expected behavioural responses (Ng et al., 1999). In this respect, O-1502 was found weakly to interact with CB1 receptors in vitro (Ki value=1749±136 nM, data not shown).
Replacement of the COO group of 2-AG with a simple ether linkage (noladin ether) greatly increases the affinity for FAAH with a corresponding small reduction in the affinity for MAGL. The high affnity of noladin ether for FAAH is an interesting property of this compound given that it is not metabolised by this enzyme (Fezza et al., 2002). This raises the possibility that this proposed endocannabinoid (Hanus et al., 2001; but see Oka et al., 2003) may not only have actions of its own, but also potentiate the actions of AEA in vivo by preventing its breakdown.
In conclusion, the present study has provided much needed basic pharmacological data for the interaction of MAGL with 2-AG (and AEA) analogues. Although this study did not identify an MAGL-selective compound, three compounds (α-Me-1-AG, O-2203 and O-2204) that inhibited FAAH and MAGL to the same extent were identified. These compounds in addition do not interact with central CB1 receptors, which is a prerequisite for their use in vivo. The potencies of the compounds are admittedly not very high, particularly when compared with FAAH inhibitors like URB597 (Kathuria et al., 2003). However, phenylmethylsulphonyl fluoride, which can be used as an FAAH inhibitor in vivo (Compton & Martin, 1997) has an in vitro potency towards rat brain forebrain FAAH of the same order of magnitude (12.9 μM, Hillard et al., 1995) as that of α-Me-1-AG for MAGL. Similarly, arachidonoylserotonin, another potentially useful FAAH inhibitor, inhibits AEA hydrolysis by RBL 2H3 cell membranes with an IC50 value of 5.6 μM (Bisogno et al., 1998).
Hopefully, it will be possible to build upon the data presented in the present study in the search for selective MAGL compounds. In this respect, an unsubstituted 1-glycerol head group seems to be important (although a recent abstract has reported arachidonoyl ethylene glycol as an inhibitor of MAGL (IC50 value 25 μM, FAAH>25 μM) (Cascio et al., 2004), hence future work could entail the synthesis of analogs with (i) variation in the length of the acyl side chain and (ii) substitution of different groups at the terminal carbon of the chain. An important parallel development would also be the identification of agents selectively inhibiting the biosynthesis of AEA and 2-AG. The recent cloning of key synthetic enzymes for these endocannabinoids (Bisogno et al., 2003; Okamoto et al., 2004) should help the development of screening models for such compound identification.
Acknowledgments
The authors are grateful to Britt Jacobsson for expert technical assistance with the FAAH and MAGL assays. This study was supported by grants from the Swedish Research Council (Grant no. 12158, medicine), Konung Gustav V's and Drottning Victorias Foundation, Stiftelsen för Gamla Tjänarinnor, Gun and Bertil Stohne's Foundation, and the Research Funds of the Medical Faculty, Umeå University (C.F., G.T.), and from the National Institute on Drug Abuse (Grant no. DA-09789) (B.M., R.R., R.G.P.).
Abbreviations
- 1-AG
1-arachidonoylglycerol
- 2-AG
2-arachidonoylglycerol
- AEA
arachidonoylethanolamide (anandamide)
- ATMK
arachidonoyltrifluoromethyl ketone
- CAY-10402
5Z,8Z,11Z,14Z-eicosatetraenoic acid, 3-thienylmethyl ester
- CP 55,940
(−)-cis-3-[2-hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol
- FAAH
fatty acid amide hydrolase
- 2-LG
2-linoleoylglycerol
- LEA
linoleoylethanolamide
- MAGL
monoacylglycerol lipase
- α-Me-1-AG
α-methyl-1-arachidonoylglycerol (α-methyl-1-AG)
- O-1502
1-nor-arachidonoyl-3-(2′3′-dihydroxypropyl)urea
- O-2203
1-(20-cyano-16,16-dimethyl-eicosa-5,8,11,14-tetraenoyl) glycerol)
- O-2204
2-(20-hydroxy-16,16-dimethyl-eicosa-5,8,11,14-tetraenoyl) glycerol)
- 2-OG
2-oleoylglycerol
- OTMK
oleoyltrifluoromethyl ketone
- PEA
palmitoylethanolamide
- PTMK
palmitoyltrifluoromethyl ketone
- URB597
3′-carbamoyl-biphenyl-3-yl-cyclohexylcarbamate
- VDM11
N-(4-hydroxy-2-methylphenyl) arachidonoyl amide
References
- BIFULCO M., DI MARZO V. Targeting the endocannabinoid system in cancer therapy: A call for further research. Nat. Med. 2002;8:547–550. doi: 10.1038/nm0602-547. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., HOWELL F., WILLIAMS G., MINASSI A., CASCIO M.G., LIGRESTI A., MATIAS I., SCHIANO-MORIELLO A., PAUL P., WILLIAMS E.-J., GANGADHARAN U., HOBBS C., DI MARZO V., DOHERTY P. Cloning of the first sn1-DAG lipases points to the spatial and temporal regulation of endocannabinoid signaling in the brain. J. Cell Biol. 2003;163:463–468. doi: 10.1083/jcb.200305129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BISOGNO T., MELCK D., DE PETROCELLIS L., BOBROV M.Y., GRETSKAYA N.M., BEZUGLOV V.V., SITACHITTA N., GERWICK W.H., DI MARZO V. Arachidonoylserotonin and other novel inhibitors of fatty acid amide hydrolase. Biochem. Biophys. Res. Comm. 1998;248:515–522. doi: 10.1006/bbrc.1998.8874. [DOI] [PubMed] [Google Scholar]
- BISOGNO T., SEPE N., MELCK D., MAURELLI S., DE PETROCELLIS L., DI MARZO V. Biosynthesis, release and degradation of the novel endogenous cannabimimetic metabolite 2-arachidonoylglycerol in mouse neuroblastoma cells. Biochem. J. 1997;322:671–677. doi: 10.1042/bj3220671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOLDRUP L., WILSON S.J., BARBIER A.J., FOWLER C.J. A simple stopped assay for fatty acid amide hydrolase avoiding the use of a chloroform extraction phase. J. Biochem. Biophys. Methods. 2004;60:171–177. doi: 10.1016/j.jbbm.2004.04.020. [DOI] [PubMed] [Google Scholar]
- CASCIO M.G., BISOGNO T., MATIAS I., DE PETROCELLIS L., ORLANDO P., DI MARZO V.Enzymes for 2-AG biosynthesis and metabolism in cell lines, and their pharmacological inhibition 20041102004 Symposium on the Cannabinoids, International Cannabinoid Research Society, Berlington, Vermont, U.S.A., p
- CHAU L.-Y., TAI H.-H. Monoglyceride and diglyceride lipases from human platelet microsomes. Biochim. Biophys. Acta. 1988;963:436–444. doi: 10.1016/0005-2760(88)90312-8. [DOI] [PubMed] [Google Scholar]
- CLEMENT A.B., HAWKINS E.G., LICHTMAN A.H., CRAVATT B.F. Increased seizure susceptibility and proconvulsant activity of anandamide in mice lacking fatty acid amide hydrolase. J. Neurosci. 2003;23:3916–3923. doi: 10.1523/JNEUROSCI.23-09-03916.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- COMPTON D.R., MARTIN B.R. The effect of the enzyme inhibitor phenylmethylsulfonyl fluoride on the pharmacological effect of anandamide in the mouse model of cannabimimetic activity. J. Pharmacol. Exp. Ther. 1997;283:1138–1143. [PubMed] [Google Scholar]
- CRAVATT B.F., DEMAREST K., PATRICELLI M.P., BRACEY M.H., GIANG D.K., MARTIN B.R., LICHTMAN A.H. Supersensitivity to anandamide and enhanced endogenous cannabinoid signaling in mice lacking fatty acid amide hydrolase. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9371–9376. doi: 10.1073/pnas.161191698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CRAVATT B.F., GIANG D.K., MAYFIELD S.P., BOGER D.L., LERNER R.A., GILULA N.B. Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature. 1996;384:83–87. doi: 10.1038/384083a0. [DOI] [PubMed] [Google Scholar]
- CRAVATT B.F., LICHTMAN A.H. The enzymatic inactivation of the fatty acid amide class of signaling lipids. Chem. Phys. Lipids. 2002;121:135–148. doi: 10.1016/s0009-3084(02)00147-0. [DOI] [PubMed] [Google Scholar]
- DASSE O., MAHADEVAN A., HAN L., MARTIN B.R., DIMARZO V., RAZDAN R.K. The synthesis of N-vanillyl-arachidonoyl-amide (arvanil) and its analogs: an improved procedure for the synthesis of the key synthon methyl 14-hydroxy-(all-cis)-5,8,11,-tetradecatrienonate. Tetrahedron. 2000;56:9195–9202. [Google Scholar]
- DE PETROCELLIS L., CASCIO M.G., DI MARZO V. The endocannabinoid system: a general view and latest additions. Br. J. Pharmacol. 2004;141:765–774. doi: 10.1038/sj.bjp.0705666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEUTSCH D.G., CHIN S.A. Enzymatic synthesis and degradation of anandamide, a cannabinoid receptor agonist. Biochem. Pharmacol. 1993;46:791–796. doi: 10.1016/0006-2952(93)90486-g. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., BISOGNO T., DE PETROCELLIS L., MELCK D., ORLANDO P., WAGNER J.A., KUNOS G. Biosynthesis and inactivation of the endocannabinoid 2-arachidonoylglycerol in circulating and tumoral macrophages. Eur. J. Biochem. 1999;264:258–267. doi: 10.1046/j.1432-1327.1999.00631.x. [DOI] [PubMed] [Google Scholar]
- DI MARZO V., BISOGNO T., SUGIURA T., MELCK D., DE PETROCELLIS L. The novel endogenous cannabinoid 2-arachidonoylglycerol is inactivated by neuronal- and basophil-like cells: connections with anandamide. Biochem. J. 1998;331:15–19. doi: 10.1042/bj3310015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DINH T.P., CARPENTER D., LESLIE F.M., FREUND T.F., KATONA I., SENSI S.L., KATHURIA S., PIOMELLI D. Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc. Natl. Acad. Sci. U.S.A. 2002;99:10819–10824. doi: 10.1073/pnas.152334899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EISENTHAL R., CORNISH-BOWDEN A. The direct linear plot. A new graphical procedure for estimating enzyme kinetic parameters. Biochem. J. 1974;139:715–720. doi: 10.1042/bj1390715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FEZZA F., BISOGNO T., MINASSI A., APPENDINO G., MECHOULAM R., DI MARZO V. Noladin ether, a putative novel endocannabinoid: inactivation mechanisms and a sensitive method for its quantification in rat tissues. FEBS Lett. 2002;513:294–298. doi: 10.1016/s0014-5793(02)02341-4. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J. Plant-derived, synthetic and endogenous cannabinoids as neuroprotective agents. Non-psychoactive cannabinoids, ‘entourage' compounds and inhibitors of N-acyl ethanolamine breakdown as therapeutic strategies to avoid pyschotropic effects. Brain Res. Rev. 2003;41:26–43. doi: 10.1016/s0165-0173(02)00218-7. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J. Metabolism of the endocannabinoids anandamide and 2-arachidonoyl glycerol, a review, with emphasis on the pharmacology of fatty acid amide hydrolase, a possible target for the treatment of neurodegenerative diseases and pain. Curr. Med. Chem. – Central Nervous System Agents. 2004;4:161–174. [Google Scholar]
- FOWLER C.J., CALLINGHAM B.A., O'CONNOR M.D.L., MATTHEWS E.K. The effect of sonication upon monoamine oxidase-A and -B in the rat liver. Biochem. Pharmacol. 1980;29:1185–1188. doi: 10.1016/0006-2952(80)90415-3. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J., HOLT S., TIGER G. Acidic non-steroidal anti-inflammatory drugs inhibit rat brain fatty acid amide hydrolase in a pH-dependent manner. J. Enzyme Inhib. Med. Chem. 2003;18:55–58. doi: 10.1080/1475636021000049726. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J., JONSSON K.-O., TIGER G. Fatty acid amide hydrolase: biochemistry, pharmacology and therapeutic possibilities for an enzyme hydrolyzing anandamide, 2-arachidonoylglycerol, palmitoylethanolamide and oleamide. Biochem. Pharmacol. 2001;62:517–526. doi: 10.1016/s0006-2952(01)00712-2. [DOI] [PubMed] [Google Scholar]
- FOWLER C.J., TIGER G., LIGRESTI A., LÓPEZ-RODRÍGUEZ M.L., DI MARZO V. Selective inhibition of anandamide cellular uptake versus enzymatic hydrolysis – a difficult issue to handle. Eur. J. Pharmacol. 2004;492:1–11. doi: 10.1016/j.ejphar.2004.03.048. [DOI] [PubMed] [Google Scholar]
- GERDEMAN G.L., LOVINGER D.M. Emerging roles for endocannabinoids in long-term synaptic plasticity. Br. J. Pharmacol. 2003;140:781–789. doi: 10.1038/sj.bjp.0705466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOPARAJU S.K., UEDA N., TANIGUCHIM K., YAMAMOTO S. Enzymes of porcine brain hydrolyzing 2-arachidonoylglycerol, an endogenous ligand of cannabinoid receptors. Biochem. Pharmacol. 1999;57:417–423. doi: 10.1016/s0006-2952(98)00314-1. [DOI] [PubMed] [Google Scholar]
- GOPARAJU S.K., UEDA N., YAMAGUCHI H., YAMAMOTO S. Anandamide amidohydrolase reacting with 2-arachidonoylglycerol, another cannabinoid receptor ligand. FEBS Lett. 1998;422:69–73. doi: 10.1016/s0014-5793(97)01603-7. [DOI] [PubMed] [Google Scholar]
- HAN L., RAZDAN R.K. Total synthesis of 2-arachidonylglycerol (2-Ara-Gl) Tetrahedron Lett. 1999;40:1631–1634. [Google Scholar]
- HANUS L., ABU-LAFI S., FRIDE E., BREUER A., VOGEL Z., SHALEV D.E., KUSTANOVICH I., MECHOULAM R. 2-Arachidonyl glyceryl ether, an endogenous agonist of the cannabinoid CB1 receptor. Proc. Natl. Acad. Sci. U.S.A. 2001;98:3662–3665. doi: 10.1073/pnas.061029898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARRINGTON C.R. Lowry protein assay containing sodium dodecyl sulfate in microtiter plates for protein determination on fractions from brain tissue. Analyt. Biochem. 1990;186:285–287. doi: 10.1016/0003-2697(90)90081-j. [DOI] [PubMed] [Google Scholar]
- HILLARD C., WILKISON D., EDGEMOND W., CAMPBELL W. Characterization of the kinetics and distribution of N-arachidonylethanolamine (anandamide) hydrolysis by rat brain. Biochim. Biophys. Acta. 1995;1257:249–256. doi: 10.1016/0005-2760(95)00087-s. [DOI] [PubMed] [Google Scholar]
- HO S.-Y., DELGADO L., STORCH J. Monoacylglycerol metabolism in human intestinal Caco-2 cells. Evidence for metabolic compartmentation and hydrolysis. J. Biol. Chem. 2002;277:1816–1823. doi: 10.1074/jbc.M108027200. [DOI] [PubMed] [Google Scholar]
- HUANG S.M., BISOGNO T., PETROS T.J., CHANG S.Y., ZAVITSANOS P.A., ZIPKIN R.E., SIVAKUMAR R., COOP A., MAEDA D.Y., DE PETROCELLIS L., BURSTEIN S., DI MARZO V., WALKER J.M. Identification of a new class of molecules, the arachidonoyl amino acids, and characterization of one member that inhibits pain. J. Biol. Chem. 2001;276:42639–42644. doi: 10.1074/jbc.M107351200. [DOI] [PubMed] [Google Scholar]
- IKEDA Y., OKAMURA K., FUJII S. Purification and characterisation of rat liver microsomal monoacylglycerol lipase in comparison to the other esterases. Biochim. Biophys. Acta. 1977;488:128–139. [PubMed] [Google Scholar]
- JÁRAI Z., WAGNER J.A., GOPARAJU S.K., WANG L., RAZDAN R.K., SUGIURA T., ZIMMER A.M., BONNER T.I., ZIMMER A., KUNOS G. Cardiovascular effects of 2-arachidonoyl glycerol in anesthetized mice. Hypertension. 2000;35:679–684. doi: 10.1161/01.hyp.35.2.679. [DOI] [PubMed] [Google Scholar]
- KARLSSON M., CONTRERAS J.A., HELLMAN U., TORNQVIST H., HOLM C. cDNA cloning, tissue distribution, and identification of the catalytic triad of monoglyceride lipase. Evolutionary relationship to esterases, lysophospholipases, and haloperoxidases. J. Biol. Chem. 1997;272:27218–27223. doi: 10.1074/jbc.272.43.27218. [DOI] [PubMed] [Google Scholar]
- KARLSSON M., REUE K., XIA Y.-R., LUSIS A.J., LANGIN D., TORNQVIST H., HOLM C. Exon–intron organization and chromosomal localization of the mouse monoglyceride lipase gene. Gene. 2001;272:11–18. doi: 10.1016/s0378-1119(01)00559-5. [DOI] [PubMed] [Google Scholar]
- KARLSSON M., TORNQVIST H., HOLM C. Expression, purification, and characterization of histidine-tagged mouse monoglyceride lipase from baculovirus-infected insect cells. Prot. Expr. Purif. 2000;18:286–292. doi: 10.1006/prep.1999.1194. [DOI] [PubMed] [Google Scholar]
- KATHURIA S., GAETANI S., FEGLEY D., VALIÑO F., DURANTI A., TONTINI A., MOR M., TARZIA G., LA RANA G., CALIGNANO A., GIUSTINO A., TATTOLI M., PALMERY M., CUOMO V., PIOMELLI D. Modulation of anxiety through blockade of anandamide hydrolysis. Nat. Med. 2003;9:76–81. doi: 10.1038/nm803. [DOI] [PubMed] [Google Scholar]
- KIM J., ALGER B. Inhibition of cyclooxygenase-2 potentiates retrograde endocannabinoid effects in hippocampus. Nat. Neurosci. 2004;7:697–698. doi: 10.1038/nn1262. [DOI] [PubMed] [Google Scholar]
- LICHTMAN A.H., HAWKINS E.G., GRIFFIN G., CRAVATT B.F. Pharmacological activity of fatty acid amides is regulated, but not mediated, by fatty acid amide hydrolase in vivo. J. Pharmacol. Exp. Ther. 2002;302:73–79. doi: 10.1124/jpet.302.1.73. [DOI] [PubMed] [Google Scholar]
- LIGRESTI A., BISOGNO T., MATIAS I., DE PETROCELLIS L., CASCIO M.G., CONSENZA V., D'ARGENIO G., SCAGLIONE G., BIFULCO M., SORRENTINI I., DI MARZO V. Possible endocannabinoid control of colorectal cancer growth. Gastroenterology. 2003;125:677–687. doi: 10.1016/s0016-5085(03)00881-3. [DOI] [PubMed] [Google Scholar]
- LÓPEZ-RODRÍGUEZ M.L., VISO A., ORTEGA-GUTIÉRREZ S., LASTRES-BECKER I., GONZÁLEZ S., FERNÁNDEZ-RUIZ J., RAMOS J.A. Design, synthesis and biological evaluation of novel arachidonic acid derivatives as highly potent and selective endocannabinoid transporter inhibitors. J. Med. Chem. 2001;44:4505–4508. doi: 10.1021/jm015545y. [DOI] [PubMed] [Google Scholar]
- MACCARRONE M., SALVATI S., BARI M., FINAZZI-AGRÒ A. Anandamide and 2-arachidonoylglycerol inhibit fatty acid amide hydrolase by activating the lipoxygenase pathway of the arachidonate cascade. Biochem. Biophys. Res. Commun. 2000;278:576–583. doi: 10.1006/bbrc.2000.3869. [DOI] [PubMed] [Google Scholar]
- MARTIN B.R., JEFFERSON R., WINCKLER R., WILEY J.L., HUFFMAN J.W., CROCKER P.J., SAHA B., RAZDAN R.K. Manipulation of the tetrahydrocannabinol side chain delineates agonists, partial agonists, and antagonists. J. Pharmacol. Exp. Ther. 1999;290:1065–1079. [PubMed] [Google Scholar]
- MAURELLI S., BISOGNO T., DE PETROCELLIS L., LUCCIA A.D., MARINO G., DI MARZO V. Two novel classes of neuroactive fatty acid amides are substrates for mouse neuroblastoma ‘anandamide amidohydrolase'. FEBS Lett. 1995;377:82–86. doi: 10.1016/0014-5793(95)01311-3. [DOI] [PubMed] [Google Scholar]
- NATARAJAN V., SCHMID P.C., REDDY P.V., SCHMID H.H.O. Catabolism of N-acylethanolamine phospholipids by dog brain preparations. J. Neurochem. 1984;42:1613–1619. doi: 10.1111/j.1471-4159.1984.tb12750.x. [DOI] [PubMed] [Google Scholar]
- NG E.W., AUNG M.M., ABOOD M.E., MARTIN B.R., RAZDAN R.K. Unique analogues of anandamide: arachidonoyl ethers and carbamates and norarachidonoyl carbamates and ureas. J. Med. Chem. 1999;42:1975–1981. doi: 10.1021/jm980711w. [DOI] [PubMed] [Google Scholar]
- OKA S., TSUCHIE A., TOKUMURA A., MURAMATSU M., SUHARA Y., TAKAYAMA H., WAKU K., SUGIURA T. Ether-linked analogue of 2-arachidonoylglycerol (noladin ether) was not detected in the brains of various mammalian species. J. Neurochem. 2003;85:1374–1381. doi: 10.1046/j.1471-4159.2003.01804.x. [DOI] [PubMed] [Google Scholar]
- OKAMOTO Y., MORISHITA J., TSUBOI K., TONAI T., UEDA N. Molecular characterization of a phospholipase D generating anandamide and its congeners. J. Biol. Chem. 2004;279:5298–5305. doi: 10.1074/jbc.M306642200. [DOI] [PubMed] [Google Scholar]
- OMEIR R.L., CHIN S., HONG Y., AHERN D.G., DEUTSCH D.G. Arachidonoyl ethanolamide-[1,2-14C] as a substrate for anandamide amidase. Life Sci. 1995;56:1999–2005. doi: 10.1016/0024-3205(95)00181-5. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G. Cannabinoid receptors and pain. Progr. Neurobiol. 2001;63:569–611. doi: 10.1016/s0301-0082(00)00031-9. [DOI] [PubMed] [Google Scholar]
- PERTWEE R.G., GIBSON T.M., STEVENSON L.A., ROSS R.A., BANNER W.K., SAHA B., RAZDAN R.K., MARTIN B.R. O-1057, a potent water-soluble cannabinoid receptor agonist with antinociceptive properties. Br. J. Pharmacol. 2000;129:1577–1584. doi: 10.1038/sj.bjp.0703245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SAARIO S.M., SAVINAINEN J.R., LAITINEN J.T., JÄRVINEN T., NIEMI R. Monoglyceride lipase-like enzymatic acitivity is responsible for hydrolysis of 2-arachidonoylglycerol in rat cerebellar membranes. Biochem. Pharmacol. 2004;67:1381–1387. doi: 10.1016/j.bcp.2003.12.003. [DOI] [PubMed] [Google Scholar]
- SAKURADA T., NOMA A. Subcellular localization and some properties of monoacylglycerol lipase in rat adipocytes. J. Biochem. 1981;90:1413–1419. doi: 10.1093/oxfordjournals.jbchem.a133607. [DOI] [PubMed] [Google Scholar]
- SCHMID P.C., ZUZARTE-AUGUSTIN M.L., SCHMID H.H.O. Properties of rat liver N-acylethanolamine amidohydrolase. J. Biol. Chem. 1985;260:14145–14149. [PubMed] [Google Scholar]
- SOMMA-DELPÉRO C., VALETTE A., LEPETIT-THÉVENIN J., NOBILI O., BOYER J., VÉRINE A. Purification and properties of a monoacylglycerol lipase in human erythrocytes. Biochem. J. 1995;312:519–525. doi: 10.1042/bj3120519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SUGIURA T., KODAKA T., NAKANE S., MIYASHITA T., KONDA S., SUHARA Y., TAKAYAMA H., WAKU K., SEKI C., BABA N., ISHIMA Y. Evidence that the cannabinoid CB1 receptor is a 2-arachidonoylglycerol receptor. Structure–activity relationship of 2-arachidonoylglycerol, ether-linked analogues, and related compounds. J. Biol. Chem. 1999;274:2794–2801. doi: 10.1074/jbc.274.5.2794. [DOI] [PubMed] [Google Scholar]
- TORNQVIST H., BELFRAGE P. Purification and some properties of a monoacylglycerol-hydrolysing enzyme of rat adipose tissue. J. Biol. Chem. 1976;251:813–819. [PubMed] [Google Scholar]
- VAN DER STELT M., VAN KUIK J.A., BARI M., VAN ZADELHOFF G., LEEFLANG B.R., VELDINK G.A., FINAZZI-AGRÒ A., VLIEGENTHART J.F.G., MACCARRONE M. Oxygenated metabolites of anandamide and 2-arachidonoylglycerol: conformational analysis and interaction with cannabinoid receptors, membrane transporter, and fatty acid amide hydrolase. J. Med. Chem. 2002;45:3709–3720. doi: 10.1021/jm020818q. [DOI] [PubMed] [Google Scholar]
- VANDEVOORDE S., JONSSON K.-O., FOWLER C.J., LAMBERT D.M. Modifications of the ethanolamine head in N-palmitoyl-ethanolamine: synthesis and evaluation of new agents interfering with the metabolism of anandamide. J. Med. Chem. 2003;46:1440–1448. doi: 10.1021/jm0209679. [DOI] [PubMed] [Google Scholar]
- WILEY J.L., DEWEY M.A., JEFFERSON R.G., WINCKLER R.L., BRIDGEN D.T., WILLOUGHBY K.A., MARTIN B.R. Influence of phenylmethylsulfonyl fluoride on anandamide brain levels and pharmacological effects. Life Sci. 2000;67:1573–1583. doi: 10.1016/s0024-3205(00)00749-9. [DOI] [PubMed] [Google Scholar]