

Abstract
Chronic haloperidol treatment has been associated with an increased incidence of glucose intolerance and type-II diabetes mellitus. We studied the effects of haloperidol on native ATP-sensitive potassium (KATP) channels in mouse pancreatic β cells and on cloned Kir6.2/SUR1 channels expressed in HEK293 cells.
The inhibitory effect of haloperidol on the KATP channel was not mediated via the D2 receptor signaling pathway, as both D2 agonists and antagonists blocked the channel.
KATP currents were studied using the patch-clamp technique in whole-cell and outside-out patch configurations. Addition of haloperidol to the extracellular solution inhibited the KATP conductance immediately, in a reversible and voltage-independent manner. Haloperidol did not block the channel when applied intracellularly in whole-cell recordings.
Haloperidol blocked cloned Kir6.2/SUR1 and Kir6.2ΔC36 KATP channels expressed in HEK cells. This suggests that the drug interacts with the Kir6.2 subunit of the channel.
The IC50 for inhibition of the KATP current by haloperidol was 1.6 μM in 2 mM extracellular K+ concentration ([K+]o) and increased to 23.9 μM in 150 mM [K+]o. The Hill coefficient was close to unity, suggesting that the binding of a single molecule of haloperidol is sufficient to close the channel.
Haloperidol block of KATP channels may contribute to the side effects of this drug when used therapeutically.
Keywords: Haloperidol, ATP-sensitive potassium channel, KATP channel, pancreatic β cell, diabetes mellitus, D2 receptor
Introduction
Haloperidol is frequently used in clinical practice to treat psychotic disorders and neurological diseases, and to control the symptoms of Tourette's syndrome (Kapur & Remington, 2001). Its pharmacological effect is believed to be due to the ability of the drug to block the dopamine signaling pathway in the central nervous system (for a review, see Seeman & Van Tol, 1994). It is well established that treatment with antipsychotic agents is often associated with excess body weight gain, abnormal glucose metabolism, and, in some cases, even diabetes mellitus (DM) (Baptista et al., 2002; Wirshing et al., 2002). A large-scale cohort study showed the relative risk of DM in haloperidol-treated patients to be three-fold greater than in an untreated control population (Buse et al., 2003). The exact mechanisms underlying the development of DM in haloperidol-treated patients are still unknown, but it has been proposed that the dopamine signaling pathway might be involved (Hägg et al., 1998). Haloperidol has been shown to directly inhibit various types of ion channels, including the G-protein-activated inwardly rectifying potassium channel (Kobayashi et al., 2000), the calcium-activated potassium channel (Akamine et al., 2002), HERG and HEAG potassium channels (Shuba et al., 2001; Gessner & Heinemann, 2003), and L-, N-, and P-type calcium channels (Galizzi et al., 1986; Sah & Bean, 1994).
The ATP-sensitive potassium (KATP) channel links the metabolic state to membrane electrical excitability in many cell types, including pancreatic β cells, heart, and neurons (Ashcroft & Rorsman, 1989; Carmeliet 1999; Chiou & How, 2001). In pancreatic β cells, the KATP channel couples elevation in the blood glucose level to insulin secretion (Ashcroft & Rorsman, 1990; Miki et al., 1998). Closure of this channel at elevated glucose leads to membrane depolarization, opening of voltage-gated calcium channels, and a rise in intracellular calcium that triggers insulin secretion (for a review, see Kanno et al., 2002).
The KATP channel is a heterotetrameric complex, composed of four Kir6.2 and four sulfonylurea receptor (SUR) subunits. Kir6.2 is an inwardly rectifying K+ channel that serves as a common pore-forming subunit of the KATP channel in most tissues except vascular smooth muscle (Inagaki et al., 1995). It possesses inhibitory binding sites for ATP and drugs such as phentolamine (Proks & Ashcroft, 1997; Tucker et al., 1997). SUR is a regulatory protein that confers sensitivity to MgADP and drugs such as K-channel openers and sulfonylureas (which block the channel) (for a review, see Aguilar-Bryan et al., 1998). There are two SUR genes, which exhibit different tissue distributions: SUR1 is found in β cells, gut L cells, and some neurons, SUR2A in cardiac and skeletal muscle and SUR2B in smooth muscle and neurons. Different SURs confer different sensitivities to drugs and nucleotides (Reimann et al., 2000) Both Kir6.2 and SUR1 are required for correct trafficking of the KATP channel to the plasma membrane; however, deletion of the last 26 residues of Kir6.2 (Kir6.2ΔC26) removes an endoplasmic reticulum retention signal and enables Kir6.2ΔC26 to reach the surface membrane in the absence of SUR (Tucker et al., 1997; Zerangue et al., 1999). This construct therefore provides a useful tool for examining the subunit of the KATP channel that is targeted by a drug.
In this study, we investigated the effects of haloperidol on native KATP channels in mouse pancreatic β cells and cloned β cell (Kir6.2/SUR1) KATP channels expressed in HEK293 cells. We found that haloperidol blocks KATP channels by binding to an external site on Kir6.2, and that this effect is modulated by extracellular K+ concentration ([K+]o).
Methods
Isolation of islet cells
All animal studies were conducted according to the National Institutes of Health's Guidelines for Care and Use of Experimental Animals and were approved by the Committee on Animal Care and Use of the local institution and state. Adult male NMRI mice were killed by cervical dislocation. Liberase (0.3 mg ml−1) (Roche, U.S.A.) was dissolved in Hank's buffer salt solution (Invitrogen, U.S.A.) and injected into pancreas via the bile duct. The pancreas was then removed and digested for 20–30 min at 37°C. Islets were first enriched by Ficoll gradient centrifugation (Amersham, Sweden) and then hand picked. Isolated islets were shaken in CMRL-1066 medium (Invitrogen, U.S.A.) plus 2 mM EGTA, then triturated into single cells. Cells were plated onto poly-L-ornithine-coated coverslips and cultured in CMRL-1066 medium supplemented with 10% fetal bovine serum (Invitrogen, U.S.A.), 100 U ml−1 penicillin G, and 0.1 mg ml−1 streptomycin in a humidified atmosphere of 5% CO2/95% O2 at 37°C. Cultured cells were used within 4 days.
Cloned channel expression
Mouse Kir6.2 (Genbank D50581; Bond et al., 1995; Inagaki et al., 1995) and rat SUR1 (Genbank L40624; Aguilar-Bryan et al., 1995) cDNAs were cloned into the pcDNA3 plasmid. A truncated form of Kir6.2 (Kir6.2ΔC36), which lacks the C-terminal 36 amino acids and forms functional channels in the absence of SUR, was prepared as described previously (Tucker et al., 1997).
HEK293 cells were cultured in DMEM (Sigma) containing 10% FBS (Life Technologies, Paisley, Scotland), 3 mM glucose, and 2 mM glutamine at 37°C in a humidified atmosphere of 5% CO2/95% O2 at 37°C. Cells were plated on poly L-ornithine-coated glass coverslips and transiently transfected with either 0.2 μg of the pcDNA3 containing Kir6.2 construct and 0.8 μg of pcDNA3 containing rat SUR1 construct or 1 μg pcDNA3 containing Kir6.2ΔC36 by using FuGENE 6 (Roche Biochemicals) according to the manufacturers' instructions. Cells were used 2–4 days after transfection.
Electrophysiology
KATP currents were recorded using the whole-cell patch-clamp configuration by an EPC9 and EPC10 amplifier (HEKA Electronik, Germany). Data were acquired at 20 kHz with PULSE8.6 software (HEKA Electronik). Pipettes were pulled from 1.5 mm borosilicate glass capillaries (Harvard Apparatus Ltd, U.K.) and their tips were heat polished using a microforge (MF830, Narishige, Japan). Pipette resistances were 2–4 MΩ in the standard intracellular solution. The series resistances of whole-cell configuration ranged between 5 and 20 MΩ. β Cells were identified by their larger size (>5 pF; Göpel et al., 2000) and their electrophysiological properties. In particular, they did not exhibit voltage-gated Na+ currents at a holding potential of −70 mV, but this could be activated in most cells by a conditioning pulse to −150 mV, as previously described (Göpel et al., 1999). Single-channel currents were recorded from outside-out patches at −70 mV and digitized at 10 kHz. The pipette resistances were 5–10 MΩ. All experiments were carried out at room temperature.
Solutions and chemicals
The extracellular solution contained (in mM): 150 NaCl, 10 HEPES, 2 KCl, 2 CaCl2, 1 MgCl2, pH 7.2. (adjusted with NaOH). The potassium concentration was varied by equimolar replacement of NaCl with KCl, and 150 mM [K+]o solution was used in single-channel recordings. The intracellular solution contained (in mM): 150 KCl, 10 HEPES, 1 MgCl2, 5 EGTA, pH 7.2 (adjusted with KOH). For whole-cell recordings in native β cells, Na2ATP (0.5 mM) was added to the intracellular solution to prevent rundown of KATP currents. The osmolarity of all solutions was 300±10 mOsm. The recording chamber had a volume of 2 ml and solutions containing chemicals were perfused focally by gravity.
L-741,626 and sulpiride were purchased from Tocris (Biotrend, Germany) and all other chemicals from Sigma (Sigma-Aldrich, Germany). However, different batches of haloperidol were purchased from both companies. Haloperidol, sulpiride, and L-741,626 were dissolved in DMSO, tolbutamide was dissolved in ethanol. The final concentrations of DMSO were less than 0.1%, which did not affect the KATP currents.
Data analysis
Concentration-dependent curves were fitted by the Hill equation:
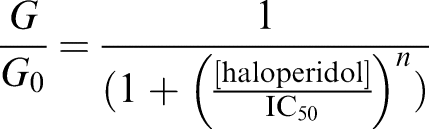 |
where G and G0 are the KATP conductance in the presence and absence of haloperidol, respectively, [haloperidol] is the concentration of haloperidol, IC50 is the half-maximal inhibitory concentration of haloperidol and n is the slope parameter (Hill coefficient). The conductance was measured as a slope conductance between −100 and −50 mV of a 100 ms voltage ramp, independently of whether the current flow was inward (as at high [K+]o) or outward (as at low [K+]o). This is justifiable as experiments at a given [K+]o showed that neither the extent nor rate of block varied with membrane potential or the direction of current flow. The control conductance was taken as the mean of that recorded in control solution before and after drug addition.
Curve fitting was carried out using PulseFit v8.65 (HEKA Electronik, Germany), Matview (Matlab extension, Wise Technologies, Slovenia), and SigmaPlot v8.0 (Jandel Scientific, U.S.A.). Single-channel data were analyzed using TAC v.4.1.5 (Bruxton, Seattle, WA, U.S.A.). Recordings were analyzed before and after haloperidol treatment and the average data were taken as the control. For measurements of open and closed times, events were detected using 50% threshold level method. The open time distribution was fitted by a Gaussian distribution, and the closed time distribution was best fitted by the sum of two Gaussian distributions. The burst duration was defined by two openings separated by an interval of <1 ms (approximately twice the mean short closed time).
Data are given as mean±s.e.m., and n indicates the number of cells analyzed. Statistical significance (P<0.05) was determined using Student's t-test or one-way ANOVA test.
Results
Haloperidol inhibition of KATP channels does not involve the D2 receptor
In whole-cell recordings on β cells, the KATP conductance was induced by dialysis with low [ATP]i (<0.5 mM) and quantified by measuring the slope conductance between −100 and −50 mV of a 100 ms voltage ramp. Addition of the D2 receptor antagonist haloperidol to the extracellular solution (2 mM [K+]o) significantly reduced the whole-cell KATP conductance, an effect which was reversible upon wash out of haloperidol (Figure 1a and b). The simplest explanation of this observation is that haloperidol acts as a direct channel blocker. However, it is also possible that the drug blocks a tonic activation of D2 receptors, caused by low levels of dopamine in the extracellular solution. To exclude this possibility, we tested the effect of other D2 receptor antagonists and agonists. The specific D2 antagonist L-741,626 also partially blocked the KATP current (Figure 1c); however, the antagonist sulpiride had no effect (Figure 1d). We next tested if the D2 receptor agonist quinpirole (20 μM) could increase the KATP current and reverse the effect of haloperidol. In contrast to our expectations, quinpirole partially inhibited the KATP conductance when applied alone, and it did not prevent the inhibitory effect of haloperidol (Figure 1e). All the drugs we tested have comparable affinities for the D2 receptor (IC50, 1–10 nM), but the extent of KATP channel block they caused was significantly different (Figure 1g). These results suggest that haloperidol inhibition is unlikely to involve the D2 receptor signaling pathway.
Figure 1.

Inhibition of KATP conductance by tolbutamide, D2 receptor agonist, and antagonists in pancreatic β cells (2 mM [K+]o). (a) Haloperidol block in a concentration-dependent manner, which can be reversed upon washout, is shown. The KATP currents were elicited by 100 ms voltage-ramp stimulation (−100 to −50 mV, 1 Hz) from holding potential −110 mV. (b–f) KATP current–voltage relation before (control) and after application of: (b) 100 μM haloperidol (HAL), (c) 10 μM L741-626 (L), (d) 100 μM sulpiride (SUL), (e) 20 μM quinpirole (QUIN), 20 μM quinpirole plus 100 μM haloperidol, and (f) 100 μM tolbutamide (TOL). (g) Comparison of mean KATP conductances in the presence of the drugs indicated normalized to control solution. Numbers of cells tested are indicated in parentheses. One-way ANOVA indicated a significant difference between 100 μM sulpiride and the other D2 antagonists (P<0.001). Haloperidol and quinpirole plus haloperidol were not significantly different (t-test, P=0.86).
Haloperidol binding site is on Kir6.2. subunit
To determine whether haloperidol binds to the Kir6.2 or SUR1 subunit of the KATP channel, we compared its effect on the cloned KATP channels Kir6.2/SUR1 and a C-terminal-deleted Kir6.2 construct, Kir6.2ΔC36, expressed in HEK293 cells. Figure 2 shows that the extent of haloperidol block was independent of whether Kir6.2 was expressed alone or in combination with SUR1. This result demonstrates that haloperidol block only requires Kir6.2. In addition, this experiment provides further support for the idea that the D2 signaling pathway is not involved, because HEK293 cells do not express endogenous D2 receptors (Senogles et al., 2004).
Figure 2.

Effect of haloperidol on wild-type and ΔC36 KATP channels expressed in HEK293 cells (2 mM [K+]o). (a) Sample traces of haloperidol inhibition of wild-type (Kir6.2/SUR1) (left) or Kir6.2ΔC36 (right) overexpressed in HEK cells. The whole-cell KATP currents were elicited as in Figure 1. (b). The concentration-dependent inhibition curves of haloperidol blockage of wild-type (right) and Kir6.2ΔC36 (left) KATP currents (n from 2 to 6 for each point). The lines are the best-fit curves of Hill equation with IC50=4.4 μM with n=1.4 for wild-type KATP conductances and IC50=1.7 μM with n=1.5 for Kir6.2ΔC36 KATP conductances.
Haloperidol-binding site is extracellular
To determine the location of the haloperidol-binding site, we used native β cells and dialyzed them intracellularly with 100 μM haloperidol, a concentration that blocked more than 90% of KATP current when applied extracellularly in 2 mM [K+]o (Figure 1). To prevent extracellular accumulation of haloperidol following diffusion across the plasma membrane, cells were constantly perfused with standard extracellular solution. However, the KATP current still developed following washout of ATP, and could only be blocked when haloperidol was applied extracellularly (Figure 3a and d). This suggests that haloperidol can only access its binding site from the extracellular side of the membrane. In contrast, when β cells were dialyzed with 10 μM glibenclamide, which is thought to act at an intracellular site (Ashcroft & Gribble, 1999), no discernible KATP current developed during 10 min of dialysis (Figure 3b and c).
Figure 3.

Effect of intracellular dialysis of channel blockers on the whole-cell KATP conductance in pancreatic β cells (2 mM [K+]o). Cells were dialyzed with intracellular solution plus 100 μM haloperidol (a) or 10 μM glibenclamide (b). Time zero indicates the time of establishment of the whole-cell configuration. Haloperidol (30 μM) or glibenclamide (1 μM) was applied to the extracellular solution as indicated by the bar in (a). (c) Peak KATP conductances during 10 min dialysis with intracellular haloperidol (100 μM; 2.78±0.65 nS, n=7) are significantly bigger than with glibenclamide (10 μM; 0.38±0.06 nS, n=5) (P<0.01, t-test). (d) Whole-cell KATP conductances in cells dialyzed with intracellular haloperidol (2.44±0.5 nS) still can be further blocked by extracellular haloperidol perfusion (30 μM; 0.19±0.06 nS) (n=5, P<0.01, paired t-test).
Voltage dependency of haloperidol blockage
It has been reported that haloperidol blocks other potassium channels, such as HERG, in a strongly voltage-dependent manner (Suessbrich et al., 1997). We examined this effect using cloned KATP channels expressed in HEK293 cells (which do not possess significant endogenous voltage-dependent currents), to avoid interference from voltage-gated channels in β cells. [K+]o was raised to 10 mM to increase the inward current and the cells were held at EK, the potassium equilibrium potential (−68 mV). As shown in Figure 4, currents elicited by either +50 mV (outward current) or −50 mV (inward current) pulses were blocked equally by 10 μM haloperidol, indicating that the block is not voltage dependent.
Figure 4.

Haloperidol block of KATP currents in HEK293 cells is voltage independent. (a) Effect of 10 μM haloperidol on Kir6.2/SUR1 KATP channels expressed in HEK cells. Currents (lower trace) were elicited by ±50 mV voltage steps from a holding potential of −68 mV (top trace), which is the calculated EK. (b) The time course of haloperidol block of both −50 mV (inward current) and +50 mV (outward current). (c) The inhibition by 10 μM haloperidol at +50 mV (67±14%) and −50 mV (67±13%) was not significantly different (n=3, P=0.82, paired t-test).
Haloperidol block is reduced by increasing [K+]o
In the previous section, we found that in 10 mM [K+]o, haloperidol only blocked the KATP current by 67% (Figure 4c), whereas approximately 80% of the current is blocked in standard extracellular solution which contains 2 mM [K+]o (Figure 1a). This implies that [K+]o might affect haloperidol binding. To explore this possibility, we constructed concentration-inhibition curves for haloperidol block of mouse β-cell KATP currents at different [K+]o. As Figure 4b shows, haloperidol block was not voltage dependent, did not depend on the direction of current flow, and exhibited comparable block kinetics at all potentials. The KATP conductance at different haloperidol concentrations was normalized to that in the absence of the drug, and is plotted against the haloperidol concentration in Figure 5a. The results clearly show that haloperidol block is reduced at higher [K+]o, the IC50 increasing from 1.67 to 23.88 μM when [K+]o was raised from 2 to 150 mM (Figure 5b). The Hill coefficient was close to unity (Figure 5c), suggesting that binding of a single molecule of drug was sufficient to block the KATP channel.
Figure 5.

[K+]o reduced the haloperidol effect on KATP conductances in mouse pancreatic β cells. (a). Haloperidol concentration-dependent inhibition curves were constructed at different [K+]o (see Results for details on whole-cell KATP conductance measurements). The number of cells tested at each [K+]o was between 2 and 12 (median=5). Dashed lines were fitted by the Hill equation. (b) IC50 values for haloperidol blockage determined from the concentration-dependent inhibition curves shown in (a). Calculated values were 1.67±0.25, 3.06±0.65, 8.97±0.95, 14.84±2.75, and 23.88±2.21 for 2, 10, 50, 100, 150 mM [K+]o, respectively. (c) Hill coefficients for haloperidol block determined from the concentration-dependent inhibition curves shown in (a). Calculated values were 0.72±0.07, 0.62±0.09, 0,84±0.07, 0.68±0.10, and 0.75±0.06 for 2, 10, 50, 100, 150 mM [K+]o, respectively.
Single-channel analysis
We also examined the effect of haloperidol on native β cell KATP currents at the single-channel level (Figure 6). Since haloperidol can only access to its binding site from the extracellular side (Figure 3), we used outside-out patch recordings. In ATP-free solution, KATP channel currents rundown exponentially with a time constant of 1–2 min, followed by a sustained phase of activity (Nichols et al., 1991). We measured the effect of haloperidol on the single-channel kinetics during this sustained phase. The results are summarized in Table 1. As seen in Figure 6, KATP channel openings occurred in bursts, which were separated by longer closed intervals. The short closed time is primarily determined by openings within a burst and the long closed time reflects the interburst intervals. Our long closed time is longer than previously published data (Proks & Ashcroft, 1997), which we attribute to KATP channel rundown. Neither the open time nor the short closed time was altered by haloperidol and the unitary current size was not affected. These data indicate that haloperidol does not act as an open channel blocker. However, the drug markedly decreased the channel open probability by prolonging the long closed time, which accounts for the reduced macroscopic whole-cell conductance.
Figure 6.

Effects of haloperidol on single-channel currents in pancreatic β cells (150 mM [K+]o). Single-channel currents recorded at −70 mV in (a) control or (b) in the presence of 10 μM haloperidol. The dashed line indicated the zero current level.
Table 1.
Outside-out analysis of haloperidol effect on KATP channels in pancreatic β cells
| 150 [K+]o (n=5) | Mean open time (ms) | Mean short closed time (ms) | Mean long closed time (ms) | Mean burst duration (ms) | Open probability (Po) | I (pA, at −70 mV) |
| Control | 1.62±0.12 | 0.58±0.02 | 109.7±17.3 | 11.1±4.5 | 0.20±0.13 | 4.2±0.2 |
| 10 μM haloperidol | 1.47±0.20 | 0.57±0.03 | *176.9±35.8 | 9.1±2.7 | *0.15±0.11 | 4.4±0.1 |
P<0.05, paired Student's t-test.
Discussion
In this study, we found that haloperidol blocks both native KATP channels in mouse pancreatic β cells and the cloned β-cell type of KATP channel (Kir6. 2/SUR1) expressed in HEK293 cells. The effect of the drug is voltage independent, rapidly reversible, is observed only when haloperidol is added to the external side of the membrane, and is modulated by [K+]o. The inhibitory effect is mediated via the Kir6.2 subunit of the KATP channel.
Haloperidol is a hydrophobic compound (Froemming et al., 1989) and can therefore freely diffuse through the plasma membrane. In our experiments, haloperidol was only able to block the channel when applied to the extracellular solution. A possible explanation for this finding is that when applied intracellularly, any haloperidol that crosses the membrane is rapidly diluted by the bulk extracellular solution before it has time to bind to its binding site.
Our experiments indicate that external potassium ions modulate channel inhibition by haloperidol. The mechanism of this effect was not explored but theoretically could involve a reduction in haloperidol binding (either by direct competition or via an allosteric effect), interference with the transduction mechanism by which haloperidol binding results in closure of the channel, or, if the drug interacts preferentially with a particular gating state, an effect of external potassium ions on gating. It has been shown that the binding of open channel blockers, including haloperidol, to voltage-gated potassium channels are reduced at higher [K+]o (Kuo, 1998; Jo et al., 2000; Shuba et al., 2001). The detailed mechanism is still not clear, but it has been proposed that external potassium ions compete with the open channel blocker imipramine for binding to the external pore region of the A-type potassium channel in rat hippocampal neurons (Kuo, 1998). Single-channel analysis of KATP channels showed that haloperidol is not an open channel blocker but [K+]o still can hinder its binding to the channel. It has also been confirmed by the X-ray crystallography that potassium ions are bound close to the external mouth of the pore (Zhou et al., 2001; Kuo et al., 2003), and it is possible that these potassium ions and channel blockers might compete for the same binding site. A similar interaction might explain how the elevated [K+]o reduces haloperidol block of KATP channels. It is noteworthy that many preclinical toxicological and pharmacological experiments on potassium channels have been carried out in high [K+]o solutions (e.g. Kobayashi et al., 2000), which may possibly lead to underestimation of the safety margin of these compounds as is the case for haloperidol (Redfern et al., 2003).
Our results indicate that the effect of haloperidol is mediated via Kir6.2, probably by direct binding of the drug to the protein. In this respect, haloperidol resembles the imidazoline phentolamine (Proks & Ashcroft, 1997), which also appears to interact directly with Kir6.2. Kir6.2 also serves as the pore-forming subunit of the KATP channel in other islet cells (α and δ cells; Göpel et al., 2000), in the L cells of the gut which secrete the insulin incretin GLP-1 (Gribble et al., 2003), in many neurons, and in cardiac and skeletal muscle (Inagaki et al., 1995). Muscle KATP channels are mostly closed under resting conditions and open only in response to ischemia. However, KATP channels in islet cells and L cells are involved in the release of hormones that modulate insulin secretion. Likewise, KATP channels in ventromedial hypothalamic neurons appear to be involved in the counter-regulatory response to glucose (Miki et al., 2001), and those of the substantia nigra may be involved in movement control (Roeper et al., 1990). Since Kir6.2 is the binding site for haloperidol, it might exert unspecific side effects by inhibiting different KATP channels in a wide range of tissues.
An important question is whether the inhibitory effect of haloperidol on the KATP channel is clinically relevant. There are several arguments against this possibility. First, the peak plasma concentration found in patients during treatment for schizophrenia is about 10–100 nM (Froemming et al., 1989), which is an order of magnitude lower than the IC50 for KATP channel inhibition. However, at least in brain, haloperidol rapidly accumulates to higher concentration (>200 nM; Kornhuber et al., 1999) and this could block a significant fraction of KATP channels. Second, not all D2 antagonists which precipitate DM block KATP channels with the same affinity. For example, sulpiride alters glucose homeostasis in rats (Baptista, 1999), but our studies indicate it has little inhibitory effect on KATP channels, even at high concentration. Third, studies have shown that haloperidol can either reduce insulin secretion (Hermansen, 1978) or have no effect (El-Denshary et al., 1982), but an increase in secretion (which is expected if the drug blocks KATP channels) has not been reported. It is possible that haloperidol blocks Ca2+ currents in β cells, as it does in other tissues, which leads to a reduction in insulin secretion, glucose intolerance, and diabetes. Further studies are needed to resolve this question.
Acknowledgments
We thank the Max-Planck Society (to SBY and MR), the Wellcome Trust, and the Royal Society (to PP and FMA) for support. FMA is the Royal Society GlaxoSmithKline Research Professor. SBY is a Ph.D. student of the International MD/PhD Program in the Neurosciences of the International Max Planck Research School. In addition, this work was supported by European Commission (GROWBETA, project QLG1-CT-2001-02233). We also thank M. Niebeling and H. Röhse for excellent technical assistance.
Abbreviations
- KATP
ATP-sensitive potassium channel
- Kir
inwardly rectifying potassium channel
- [K+]o
extracellular K+ concentration
- SUR
sulfonylurea receptor
References
- AGUILAR-BRYAN L., CLEMENT IV J.P., GONZALEZ G., KUNJILWAR K., BABENKO A., BRYAN J. Toward understanding the assembly and structure of KATP channels. Physiol. Rev. 1998;78:227–245. doi: 10.1152/physrev.1998.78.1.227. [DOI] [PubMed] [Google Scholar]
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT IV J.P., BOYD A.E., III, GONZALEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- AKAMINE T., NISHIMURA Y., ITO K., UJI Y., YAMAMOTO T. Effects of haloperidol on K+ currents in acutely isolated rat retinal ganglion cells. Invest. Ophthalmol. Vis. Sci. 2002;43:1257–1261. [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. ATP-sensitive K+ channels and insulin secretion: their role in health and disease. Diabetologia. 1999;42:903–919. doi: 10.1007/s001250051247. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. Electrophysiology of the pancreatic beta-cell. Prog. Biophys. Mol. Biol. 1989;54:87–143. doi: 10.1016/0079-6107(89)90013-8. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. ATP-sensitive K+ channels: a link between β-cell metabolism and insulin secretion Biochem. Soc. Trans. 1990;18:109–111. doi: 10.1042/bst0180109. [DOI] [PubMed] [Google Scholar]
- BAPTISTA T. Body weight gain induced by antipsychotic drugs: mechanisms and management. Acta. Psychiat. Scand. 1999;100:3–16. doi: 10.1111/j.1600-0447.1999.tb10908.x. [DOI] [PubMed] [Google Scholar]
- BAPTISTA T., KIN N.M., BEAULIEU S., DE BAPTISTA E.A. Obesity and related metabolic abnormalities during antipsychotic drug administration: mechanisms, management and research perspectives. Pharmacopsychiatry. 2002;35:205–219. doi: 10.1055/s-2002-36391. [DOI] [PubMed] [Google Scholar]
- BOND C.T., ÄMMÄLÄ C., ASHFIELD R., BLAIR T.A., GRIBBLE F., KHAN R.N., LEE K., PROKS P., ROWE I.C.M., SAKURA H., ASHFORD M.J., ADELMAN J.P., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding an inwardly-rectifying potassium channel expressed in pancreatic beta-cells and in the brain. FEBS Lett. 1995;367:61–66. doi: 10.1016/0014-5793(95)00497-w. [DOI] [PubMed] [Google Scholar]
- BUSE J.B., CAVAZZONI P., HORNBUCKLE K., HUTCHINS D., BREIER A ., JOVANOVIC L. A retrospective cohort study of diabetes mellitus and antipsychotic treatment in the United States. J. Clin. Epidemiol. 2003;56:164–170. doi: 10.1016/s0895-4356(02)00588-7. [DOI] [PubMed] [Google Scholar]
- CARMELIET E. Cardiac ionic currents and acute ischemia: from channels to arrhythmias. Physiol. Rev. 1999;79:917–1017. doi: 10.1152/physrev.1999.79.3.917. [DOI] [PubMed] [Google Scholar]
- CHIOU L.C., HOW C.H. ATP-sensitive K+ channels and cellular actions of morphine in periaqueductal gray slices of neonatal and adult rats. J. Pharmacol. Exp. Ther. 2001;298:493–500. [PubMed] [Google Scholar]
- EL-DENSHARY E.E., ISMAIL N.A., GAGERMAN E., SEHLIN J., TALJEDAL I.B. Bromocriptine and insulin secretion. Biosci. Rep. 1982;2:115–116. doi: 10.1007/BF01116177. [DOI] [PubMed] [Google Scholar]
- FROEMMING J.S., LAM Y.W., JANN M.W., DAVIS C.M. Pharmacokinetics of haloperidol. Clin. Pharmacokinet. 1989;17:396–423. doi: 10.2165/00003088-198917060-00004. [DOI] [PubMed] [Google Scholar]
- GALIZZI J.P., FOSSET M., ROMEY G., LADURON P., LAZDUNSKI M. Neuroleptics of the diphenylbutylpiperidine series are potent calcium channel inhibitors. Proc. Natl. Acad. Sci. U.S.A. 1986;83:7513–7517. doi: 10.1073/pnas.83.19.7513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GESSNER G., HEINEMANN S.H. Inhibition of hEAG1 and hERG1 potassium channels by clofilium and its tertiary analogue LY97241. Br. J. Pharmacol. 2003;138:161–171. doi: 10.1038/sj.bjp.0705025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÖPEL S., KANNO T., BARG S., GALVANOVSKIS J., RORSMAN P. Voltage-gated and resting membrane currents recorded from β-cells in intact mouse pancreatic islets. J. Physiol. 1999;521:717–728. doi: 10.1111/j.1469-7793.1999.00717.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GÖPEL S.O., Kanno T., BARG S., WENG X.G., GROMADA J., RORSMAN P. Regulation of glucagon release in mouse α-cells by KATP channels and inactivation of TTX-sensitive Na+ channels. J. Physiol. 2000;528:509–520. doi: 10.1111/j.1469-7793.2000.00509.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GRIBBLE F.M., WILLIAMS L., SIMPSON A.K., REIMANN F. A novel glucose-sensing mechanism contributing to glucagon-like peptide-1 secretion from the GLUTag cell line. Diabetes. 2003;52:1147–1154. doi: 10.2337/diabetes.52.5.1147. [DOI] [PubMed] [Google Scholar]
- HÄGG S., JOELSSON L., MJORNDAL T., SPIGSET O., OJA G., DAHLQVIST R. Prevalence of diabetes and impaired glucose tolerance in patients treated with clozapine compared with patients treated with conventional depot neuroleptic medications. J. Clin. Psychiatry. 1998;59:294–299. doi: 10.4088/jcp.v59n0604. [DOI] [PubMed] [Google Scholar]
- HERMANSEN K. Haloperidol, a dopaminergic antagonist: somatostatin-like inhibition of glucagon and insulin release from the isolated, perfused canine pancreas. Diabetologia. 1978;15:343–347. doi: 10.1007/BF03161000. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., TSUURA Y., NAMBA N., MASUDA K., GONOI T., HORIE M., SEINO Y., MIZUTA M., SEINO S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J. Biol. Chem. 1995;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- JO S.H., YOUM J.B., LEE C.O., EARM Y.E., HO W.K. Blockade of the HERG human cardiac K+ channel by the antidepressant drug amitriptyline. Br. J. Pharmacol. 2000;129:1474–1480. doi: 10.1038/sj.bjp.0703222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KANNO T., GOPEL S.O., RORSMAN P., WAKUI M. Cellular function in multicellular system for hormone-secretion: electrophysiological aspect of studies on alpha-, beta- and delta-cells of the pancreatic islet. Neurosci. Res. 2002;42:79–90. doi: 10.1016/s0168-0102(01)00318-2. [DOI] [PubMed] [Google Scholar]
- KAPUR S., REMINGTON G. Atypical antipsychotics: new directions and new challenges in the treatment of schizophrenia. Annu. Rev. Med. 2001;52:503–517. doi: 10.1146/annurev.med.52.1.503. [DOI] [PubMed] [Google Scholar]
- KOBAYASHI T., IKEDA K., KUMANISHI T. Inhibition by various antipsychotic drugs of the G-protein-activated inwardly rectifying K+ (GIRK) channels expressed in xenopus oocytes. Br. J. Pharmacol. 2000;129:1716–1722. doi: 10.1038/sj.bjp.0703224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KORNHUBER J., SCHULTZ A., WILTFANG J., MEINEKE I., GLEITER C.H., ZÖCHLING R., BOISSL K-W., LEBLHUBER F., RIEDERER P. Persistence of haloperidol in human brain tissue. Am. J. Psychiatry. 1999;156:885–890. doi: 10.1176/ajp.156.6.885. [DOI] [PubMed] [Google Scholar]
- KUO A., GULBIS J.M., ANTCLIFF J.F., RAHMAN T., LOWE E.D., ZIMMER J., CUTHBERTSON J., ASHCROFT F.M., EZAKI T., DOYLE D.A. Crystal structure of the potassium channel KirBac1.1 in the closed state. Science. 2003;300:1922–1926. doi: 10.1126/science.1085028. [DOI] [PubMed] [Google Scholar]
- KUO C.C. Imipramine inhibition of transient K+ current: an external open channel blocker preventing fast inactivation. Biophys. J. 1998;75:2845–2857. doi: 10.1016/S0006-3495(98)77727-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIKI T., LISS B., MINAMI K., SHIUCHI T., SARAYA A., KASHIMA Y., HORIUCHI M., ASHCROFT F.M., MINOKOSHI Y., ROEPER J., SEINO S. ATP-sensitive K+ channels in the hypothalamus are essential for the maintenance of glucose homeostasis. Nat. Neurosci. 2001;4:507–512. doi: 10.1038/87455. [DOI] [PubMed] [Google Scholar]
- MIKI T., NAGASHIMA K., TASHIRO F., KOTAKE K., YOSHITOMI H., TAMAMOTO A., GONOI T., IWANAGA T., MIYAZAKI J., SEINO S. Defective insulin secretion and enhanced insulin action in KATP channel-deficient mice. Proc. Natl. Acad. Sci. U.S.A. 1998;95:10402–10406. doi: 10.1073/pnas.95.18.10402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NICHOLS C.G., LEDERER W.J., CANNELL M.B. ATP dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophys. J. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PROKS P., ASHCROFT F.M. Phentolamine block of KATP channels is mediated by Kir6.2. Proc. Natl. Acad. Sci. U.S.A. 1997;94:11716–11720. doi: 10.1073/pnas.94.21.11716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- REDFERN W.S., CARLSSON L., DAVIS A.S., LYNCH W.G., MACKENZIE I., PALETHORPE S., SIEGL P.K., STRANG I., SULLIVAN A.T., WALLIS R., CAMM A.J., HAMMOND T.G. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- REIMANN F., GRIBBLE F.M., ASHCROFT F.M. Differential response of KATP channels containing SUR2A or SUR2B subunits to nucleotides and pinacidil. Mol. Pharmacol. 2000;58:1318–1325. doi: 10.1124/mol.58.6.1318. [DOI] [PubMed] [Google Scholar]
- ROEPER J., HAINSWORTH A.H., ASHCROFT F.M. Tolbutamide reverses membrane hyperpolarisation induced by activation of D2 receptors and GABAB receptors in isolated substantia nigra neurones. Pflügers. Arch. 1990;416:473–475. doi: 10.1007/BF00370758. [DOI] [PubMed] [Google Scholar]
- SAH D.W., BEAN B.P. Inhibition of P-type and N-type calcium channels by dopamine receptor antagonists. Mol. Pharmacol. 1994;45:84–92. [PubMed] [Google Scholar]
- SEEMAN P., VAN TOL H.H. Dopamine receptor pharmacology. Trends. Pharmacol. Sci. 1994;15:264–270. doi: 10.1016/0165-6147(94)90323-9. [DOI] [PubMed] [Google Scholar]
- SENOGLES S.E., HEIMERT T.L., ODIFE E.R., QUASNEY M.W. A region of the third intracellular loop of the short form of the D2 dopamine receptor dictates Gi coupling specificity. J. Biol. Chem. 2004;279:1601–1606. doi: 10.1074/jbc.M309792200. [DOI] [PubMed] [Google Scholar]
- SHUBA Y.M., DEGTIAR V.E., OSIPENKO V.N., NAIDENOV V.G., WOOSLEY R.L. Testosterone-mediated modulation of HERG blockade by proarrhythmic agents. Biochem. Pharmacol. 2001;62:41–49. doi: 10.1016/s0006-2952(01)00611-6. [DOI] [PubMed] [Google Scholar]
- SUESSBRICH H., SCHONHERR R., HEINEMANN S.H., ATTALI B., LANG F., BUSCH A.E. The inhibitory effect of the antipsychotic drug haloperidol on HERG potassium channels expressed in Xenopus oocytes. Br. J. Pharmacol. 1997;120:968–974. doi: 10.1038/sj.bjp.0700989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TUCKER S.J., GRIBBLE F.M., ZHAO C., TRAPP S., ASHCROFT F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- WIRSHING D.A., BOYD J.A., MENG L.R., BALLON J.S., MARDER S.R., WIRSHING W.C. The effects of novel antipsychotics on glucose and lipid levels. J. Clin. Psychiatry. 2002;63:856–865. doi: 10.4088/jcp.v63n1002. [DOI] [PubMed] [Google Scholar]
- ZERANGUE N., SCHWAPPACH B., JAN Y.N., JAN L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]
- ZHOU Y., MORAIS-CABRAL J.H., KAUFMAN A., MACKINNON R. Chemistry of ion coordination and hydration revealed by a K+ channel–Fab complex at 2.0 resolution. Nature. 2001;414:43–48. doi: 10.1038/35102009. [DOI] [PubMed] [Google Scholar]