

Abstract
β-Adrenoceptor antagonists (‘β-blockers') are one of the most widely used classes of drugs in cardiovascular medicine (hypertension, ischaemic heart disease and increasingly in heart failure) as well as in the management of anxiety, migraine and glaucoma. Where known, the mode of action in cardiovascular disease is from antagonism of endogenous catecholamine responses in the heart (mainly at β1-adrenoceptors), while the worrisome side effects of bronchospasm result from airway β2-adrenoceptor blockade. The aim of this study was to determine the selectivity of β-antagonists for the human β-adrenoceptor subtypes.
3H-CGP 12177 whole cell-binding studies were undertaken in CHO cell lines stably expressing either the human β1-, β2- or the β3-adrenoceptor in order to determine the affinity of ligands for each receptor subtype in the same cell background.
In this study, the selectivity of well-known subtype-selective ligands was clearly demonstrated: thus, the selective β1 antagonist CGP 20712A was 501-fold selective over β2 and 4169-fold selective over β3; the β2-selective antagonist ICI 118551 was 550- and 661-fold selective over β1 and β3, respectively, and the selective β3 compound CL 316243 was 10-fold selective over β2 and more than 129-fold selective over β1.
Those β2-adrenoceptor agonists used clinically for the treatment of asthma and COPD were β2 selective: 29-, 61- and 2818-fold for salbutamol, terbutaline and salmeterol over β1, respectively. There was little difference in the affinity of these ligands between β1 and β3 adrenoceptors.
The clinically used β-antagonists studied ranged from bisoprolol (14-fold β1-selective) to timolol (26-fold β2-selective). However, the majority showed little selectivity for the β1- over the β2-adrenoceptor, with many actually being more β2-selective.
This study shows that the β1/β2 selectivity of most clinically used β-blockers is poor in intact cells, and that some compounds that are traditionally classed as ‘β1-selective' actually have higher affinity for the β2-adrenoceptor. There is therefore considerable potential for developing more selective β-antagonists for clinical use and thereby reducing the side-effect profile of β-blockers.
Keywords: β-Adrenoceptor, β-blocker, β-agonist, β-antagonist, drug selectivity, whole cell binding
Introduction
β-Adrenoceptor antagonists (β-blockers) are one of the most widely used classes of drugs in clinical practice. Pronethalol and propranolol, the first β-blockers, were first shown to lower blood pressure and have beneficial effects in the management of angina (Black et al., 1965). By binding to cardiac β-adrenoceptors, these early β-blockers were able to block the binding (and therefore action) of the endogenous catecholamines adrenaline and noradrenaline, resulting in a reduction in the rate and force of cardiac contraction. As well as their major use in cardiovascular disease (hypertension, ischaemic heart disease and increasingly heart failure; Prichard, 1988; CIBIS-II, 1999; Heidenreich et al., 1999; Prichard et al., 2001; COMET, 2003), β-blockers are also used in the treatment of glaucoma, tremor, anxiety, migraine and hyperthyroidism (Peet & Yates, 1981; Feely & Peden, 1984; Uitti, 1998; Limmroth & Michel, 2001; Stamper et al., 2002).
However, while the main cardiovascular use of β-blockers is the antagonism of cardiac β-adrenoceptor responses in the heart (mainly β1-adrenoceptors), a main side effect is due to antagonism of β2-adrenoceptors in the airways, resulting in bronchospasm (Lewis & Lofthouse, 1993). Drugs that are more cardio-selective (or β1-selective) have therefore been developed, as these should offer a lower side-effect profile (Prichard, 1988). Initially, these drugs were screened for ‘selectivity' by observing changes in heart rate (taken to be β1-antagonism; Prichard, 1988), changes in contraction of atrial appendages (β1) or bronchial smooth muscle (β2) isolated from animals or humans (Harms, 1976). Although a good reflection of tissue-binding affinities, this is more difficult to extrapolate to specific receptor affinities. Firstly, these comparisons were made in different cell backgrounds and from different individuals, that is, atrial appendage from one individual and bronchus from another. Secondly, these measurements are subject to the changes involved in the disease process or those induced by any previous medication (Harms, 1976). For example, exposure to highly efficacious agonists can alter the β2-adrenoceptor such that its affinity for ligands is reduced 10-fold (Baker et al., 2003a). Thus, assays in which cells have had prior exposure to agonists many therefore give an underestimate of β2-adrenoceptor binding. Thirdly, many tissues express more than one receptor subtype, which may complicate the evaluation of ligand affinities.
Often, the affinity of antagonists is assessed by their ability to inhibit agonist responses. Although this allows direct comparisons across many ligands and receptors to be made, by definition, it also requires the presence of an agonist and this in itself may alter receptor binding (Baker et al., 2003a). Furthermore, many of the β-blockers have been shown to possess intrinsic efficacy of their own (e.g. Jasper et al., 1990; Azzi et al., 2003; Baker et al., 2003b, 2003c). As this can vary depending on the cellular response being measured (Azzi et al., 2003; Baker et al., 2003c), these properties provide additional complications to ligand affinity measurements from functional studies.
Thus, although estimated β-adrenoceptor-binding affinity legitimately varies between the tissues examined, to achieve a true reflection of the individual receptor affinities, affinity should ideally be measured in systems which guarantee the existence of only a single receptor subtype, in the same cell background and in the absence of agonists. The aim of the present study was therefore to determine the selectivity of a wide range of clinically used β-blockers for binding to recombinant human β1, β2 and β3 adrenoceptors stably expressed in living mammalian cells in the same cell background.
Methods
Materials
Cell culture reagents were from Sigma Chemicals (Poole, Dorset, U.K.), except foetal calf serum, which was from PAA Laboratories (Teddington, Middlesex, U.K.). White-sided view plates were from Costar and Microscint 20 scintillation fluid from Packard. 3H-CGP 12177 was from Amersham International (Buckinghamshire, U.K.). Betaxolol, practolol, pronethalol, ICI 215001, bisoprolol, salmeterol, CGP 12177, ICI 118551, ICI 89406, salbutamol, sotalol, timolol, CL 316243 and xamoterol were from Tocris Life Sciences (Avonmouth, U.K.). All other reagents were from Sigma Chemicals.
Cell lines
Mammalian cells (CHO-K1) stably expressing either the human β1-adrenoceptor (Baker et al., 2003b) or the human β2-adrenoceptor (Baker et al., 2002) were used. A further stable cell line was made by transfection of CHO-K1 cells with the human β3-adrenoceptor (DNA from Guthrie DNA Resource Centre) using Lipofectaime and Optimem according to the manufacturer's instructions. Transfected cells were selected for 3 weeks using resistance to neomycin (at 1 mg ml−1). A single clone was then isolated by dilution cloning.
Cell culture
All the CHO cells were grown in Dulbecco's modified Eagle's medium nutrient mix F12 (DMEM/F12) containing 10% foetal calf serum and 2 mM L-glutamine in a 37°C humidified 5% CO2 : 95% air atmosphere. The day prior to experimentation, the cells were seeded into white-sided, clear-bottomed 96-well view plates such that they were confluent for the following day's experiment.
3H-CGP 12177 whole cell binding
On the day of experimentation, the media was removed from each well of the 96-well view plate. The β-ligand under investigation (diluted in DMEM/F12 containing 2 mM L-glutamine only, that is, serum-free media) followed immediately by radioligand 3H-CGP 12177 (0.3–0.6 nM for β1- and β2-expressing cells and 6–20 nM for β3-expressing cells) were then added to each well. The cells were incubated for 1.5 h at 37°C. The cells were washed twice by the addition and removal of 200 μl phosphate-buffered saline. Microscint 20 (200 μl) was added to each well, a white base applied to the plate to convert the wells into white-sided/white-bottomed wells and the plates counted on a Topcount.
Data analysis
All data points on each binding curve were performed in triplicate and each 96-well plate also contained triplicate determinations of total and nonspecific binding. Nonspecific binding was determined in the presence of 100 nM CGP 20712A for the β1-adrenoceptor, 100 nM ICI 118551 for the β2-adrenoceptor and 100 μM CGP 12177 for the β3-adrenoceptor. In all cases where a KD value is stated, increasing concentrations of the competing ligand were used until the specific binding of 3H-CGP 12177 was completely inhibited. The following equation was then fitted to the data using Graphpad Prism 2.01 and the IC50 was then determined as the concentration required to inhibit 50% of the specific binding
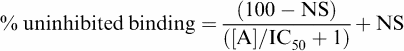 |
where [A] is the concentration of the ligand, IC50 is the concentration at which half of the specific binding of 3H-CGP 12177 has been inhibited and NS is the nonspecific binding.
From the IC50 value and the known concentration of radioligand 3H-CGP 12177, a KD (concentration at which half the receptors are bound by the competing ligand) value was calculated using the equation:
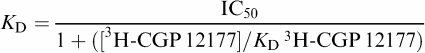 |
Results
The KD for 3H-CGP 12177 in the β1 cell line has been previously determined from saturation-binding experiments and was 0.42 nM, with a receptor expression level of 1146 fmol mg−1 protein (Baker et al., 2003b). The KD value for 3H-CGP 12177 in the β2 cell line was 0.17±0.01 nM (n=11) and receptor expression level 466 fmol mg−1 protein (Baker et al., 2002). For the β3 cells, saturation-binding experiments were performed and an estimate of the KD value for 3H-CGP 12177 of 97±27 nM (n=5) obtained. However, the maximum concentration of 3H-CGP 12177 that could be achieved (220 nM) was only two-fold over this KD value and the specific binding at this concentration was only 30.6% of total binding. When CGP 12177 was used to displace the binding of a lower concentration of 3H-CGP 12177, the KD value obtained from the above equation (which simplifies to KD=IC50−[3H-CGP 12177]) was 109.2±11.9 nM (log KD=−6.99±0.05; n=12) and the receptor expression level 789.7±130 fmol mg−1 protein. This value (109.2 nM) was used as the KD value for 3H-CGP 12177 in all subsequent calculations of KD values of the competing ligands at the β3-adrenoceptor. At the concentration of 3H-CGP 12177 used in the competition-binding experiments, specific binding represents 98.8, 93.2 and 50.0% of total binding for the β1, β2 and β3 cells, respectively. This concentration of 3H-CGP 12177 will not detect binding to the secondary non-catecholamine site of the β1-adrenoceptor (Baker et al., 2003b).
CGP 20712A, the selective β1-adrenoceptor antagonist, was found to have 501 times higher affinity for the β1- than the β2-adrenoceptor, and 4169 times higher affinity for the β1- than the β3-adrenoceptor (see Table 1). Likewise, the selective β2-adrenoceptor ligand ICI 118551 was 543-fold selective for the β2- over the β1-adrenoceptor and 661-fold selective for β2- over the β3-adrenoceptor. The β2-agonists used in the clinical management of asthma and COPD were β2-selective: 29-, 61- and 2818-fold for salbutamol, terbutaline and salmeterol over β1, with little β1/β3 selectivity.
Table 1.
Log KD values of β-blockers and β-agonists for binding to the human β1-, β2- and β3-adrenoceptors
| Log KD values | ||||||
|---|---|---|---|---|---|---|
| β1 | n | β2 | n | β3 | n | |
| β-ligands | ||||||
| CGP 20712A | −8.81±0.03 | 10 | −6.11±0.05 | 4 | −5.19±0.09 | 6 |
| ICI 89406 | −8.91±0.09 | 6 | −7.07±0.06 | 5 | −5.69±0.06 | 6 |
| Practolol | −6.14±0.05 | 4 | −4.99±0.07 | 4 | >−4 | 7 |
| Xamoterol | −7.22±0.04 | 5 | −6.07±0.08 | 5 | −4.45±0.07 | 8 |
| Bisoprolol | −7.83±0.04 | 4 | −6.70±0.05 | 4 | −5.67±0.10 | 6 |
| Betaxolol | −8.21±0.07 | 6 | −7.38±0.06 | 8 | −5.97±0.08 | 7 |
| Atenolol | −6.66±0.05 | 7 | −5.99±0.14 | 9 | −4.11±0.07 | 7 |
| ICI 215001 | −6.37±0.05 | 6 | −5.86±0.04 | 5 | −6.63±0.11 | 7 |
| Acebutolol | −6.46±0.03 | 9 | −6.08±0.07 | 6 | −4.41±0.12 | 7 |
| Metoprolol | −7.26±0.07 | 7 | −6.89±0.09 | 9 | −5.16±0.12 | 7 |
| CGP 12177 | −9.21±0.04 | 8 | −9.39±0.07 | 7 | −6.99±0.05 | 12 |
| Labetolol | −7.63±0.05 | 4 | −8.03±0.07 | 10 | −6.18±0.10 | 6 |
| Carvedilol | −8.75±0.09 | 7 | −9.40±0.08 | 5 | −8.30±0.11 | 6 |
| Pronethalol | −6.44±0.07 | 7 | −7.36±0.07 | 5 | −5.89±0.15 | 6 |
| Propranolol | −8.16±0.08 | 5 | −9.08±0.06 | 8 | −6.93±0.11 | 7 |
| Sotalol | −5.77±0.11 | 7 | −6.85±0.09 | 10 | −5.05±0.07 | 8 |
| CL 316243 | >−3 | 3 | −4.10±0.19 | 3 | −5.11±0.05 | 6 |
| Alprenolol | −7.83±0.06 | 9 | −9.04±0.07 | 8 | −6.93±0.07 | 7 |
| Bupranolol | −8.51±0.04 | 4 | −9.85±0.05 | 4 | −7.04±0.13 | 6 |
| Nadolol | −7.23±0.04 | 8 | −8.60±0.07 | 6 | −6.18±0.20 | 7 |
| Timolol | −8.27±0.08 | 5 | −9.68±0.02 | 4 | −6.80±0.11 | 7 |
| ICI 118551 | −6.52±0.02 | 4 | −9.26±0.03 | 12 | −6.44±0.16 | 6 |
| Clinically-used β-agonists | ||||||
| Salbutamol | −4.66±0.07 | 6 | −6.12±0.07 | 9 | −4.33±0.08 | 7 |
| Terbutaline | −3.82±0.07 | 5 | −5.62±0.06 | 9 | −3.90±0.10 | 7 |
| Salmeterol | −5.38±0.01 | 6 | −8.83±0.07 | 11 | −5.73±0.12 | 8 |
Values represent mean±s.e.m. of n separate experiments.
However, although the majority of clinically used β-blockers showed great variation in their ability to bind to the receptors (from nanomolar affinity for timolol to micromolar for atenolol, see Table 1, Figures 1 and 2), very little selectivity is seen for binding between the β1 and β2 adrenoceptors. Only CL 316243 was found to be β3 selective.
Figure 1.

Inhibition of 3H-CGP 12177 binding to whole cells by CGP 21712A in (a) CHO β1 cells, (b) CHO β2 cells and (c) CHO β3 cells. Bars represent total 3H-CGP 12177 binding and nonspecific binding was determined in the presence of (a) 100 nM CGP 20712A, (b) 100 nM ICI 118551 or (c) 100 μM CGP 12177. The concentrations of 3H-CGP 12177 present in each case are (a) 0.35, (b) 0.35 and (c) 13.2 nM. Data points are mean±s.e.m. of triplicate determinations. These single experiments are representative of (a) 10, (b) four and (c) six separate experiments. CGP 20712A shows high β1-selectivity.
Figure 2.

Inhibition of 3H-CGP 12177 binding to whole cells by sotalol in (a) CHO β1 cells, (b) CHO β2 cells and (c) CHO β3 cells. Bars represent total 3H-CGP 12177 binding and nonspecific binding was determined in the presence of (a) 100 nM CGP 20712A, (b) 100 nM ICI 118551 or (c) 100 μM CGP 12177. The concentrations of 3H-CGP 12177 present in each case are (a) 0.35, (b) 0.58 and (c) 6.9 nM. Data points are mean±s.e.m. of triplicate determinations. These single experiments are representatives of (a) seven, (b) 10 and (c) eight separate experiments. Sotalol has relatively little β-adrenoceptor selectivity.
Discussion
β-Adrenoceptor antagonists (β-blockers) are one of the most widely used classes of drugs in clinical practice and are currently used in the management of hypertension, ischaemic heart disease, heart failure, anxiety, tremor, migraine and glaucoma. This study suggests that many ligands previously considered to have β1-selectivity, for example, metoprolol and atenolol (Lewis & Lofthouse, 1993), have poor β1/β2 selectivity, while others that are often prescribed for cardiovascular, disorders, for example, carvedilol, sotalol and timolol actually have higher affinities for the β2-adrenoceptor (Table 2).
Table 2.
Selectivity ratios of the β-blockers for human β1-, β2- and β3-adrenoceptors, where a ratio of 1 demonstrates no selectivity for a given receptor subtype over another
| Selectivity ratios | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| β1 | vs | β2 | β2 | vs | β3 | β1 | vs | β3 | |
| β-ligands | |||||||||
| CGP 20712A | 501.2 | 8.3 | 4168.7 | ||||||
| ICI 89406 | 69.2 | 24.0 | 1659.6 | ||||||
| Practolol | >14.1 | >9.8 | >138.0 | ||||||
| Xamoterol | 14.1 | 41.7 | 588.8 | ||||||
| Bisoprolol | 13.5 | 10.7 | 144.5 | ||||||
| Betaxolol | 6.8 | 25.7 | 173.8 | ||||||
| Atenolol | 4.7 | 75.9 | 354.8 | ||||||
| ICI 215001 | 3.2 | 5.9 | 1.8 | ||||||
| Acebutolol | 2.4 | 46.8 | 112.2 | ||||||
| Metoprolol | 2.3 | 53.7 | 125.9 | ||||||
| CGP 12177 | 1.5 | 251.2 | 166.0 | ||||||
| Labetolol | 2.5 | 70.8 | 28.2 | ||||||
| Carvedilol | 4.5 | 12.6 | 2.8 | ||||||
| Pronethalol | 8.3 | 29.5 | 3.5 | ||||||
| Propranolol | 8.3 | 141.3 | 17.0 | ||||||
| Sotalol | 12.0 | 63.1 | 5.2 | ||||||
| CL 316243 | >12.6 | 10.2 | >128.8 | ||||||
| Alprenolol | 16.2 | 128.8 | 7.9 | ||||||
| Bupranolol | 21.9 | 645.7 | 29.5 | ||||||
| Nadolol | 23.4 | 263.0 | 11.2 | ||||||
| Timolol | 25.7 | 758.6 | 29.5 | ||||||
| ICI 118551 | 549.5 | 660.7 | 1.2 | ||||||
| Clinically-used β-agonists | |||||||||
| Salbutamol | 28.8 | 61.7 | 2.1 | ||||||
| Terbutaline | 63.1 | 52.5 | 1.2 | ||||||
| Salmeterol | 2818.4 | 1258.9 | 2.2 | ||||||
Thus, the affinity of CGP 20712A is 501-fold more at the β1- than β2-receptor.
Although the affinity of a great many β-adrenergic ligands has been assessed over the years, direct comparisons are often difficult to interpret as studies have been conducted in different tissues, from different species and by different methods. At times, species differences are very important, for example, ICI 118551 is not subtype specific in the pig (Liang & Mills, 2001). The methods of determining ligand affinity are also important. As mentioned above, measurements made in the presence of an agonist may alter ligand affinity measurements. There are also likely to be discrepancies in antagonist affinities at GPCRs in studies made in intact cells and those from membrane preparations. In intact cells, there is always endogenous GTP present. The GPCR–G-protein complex will therefore be dissociated and the GPCR will be in the low receptor–ligand affinity state. In membrane preparations in the absence of GTP, two site-binding affinities for agonists are seen as receptors also exist in a high-affinity G-protein-bound state (where the G-protein is unable to dissociate in the absence of GTP; Kobilka, 1992). Whole cell-binding studies would therefore be expected to yield measurements similar to the low-affinity sites normally detected in membranes in the presence of GTP. In keeping with this, only a single-site competition-binding curve was seen with the β-agonists in this study.
Despite these difficulties, a few studies do exist that have examined the selectivity in the same cell background. Smith & Teitler (1999) examined the selectivity of seven β-blockers using membranes from insect cells transfected with recombinant human β1- and β2-adrenoceptor receptors. The selectivity ratios obtained (for bisoprolol, betaxolol, atenolol, metoprolol, carvedilol, propranolol and ICI 118551) are very similar to those in this study and also suggest that carvedilol and propranolol are actually more β2-selective. Another study, assessing the β-adrenoceptor subtype selectivity of 12 antagonists at human β1, β2 and β3 adrenoceptors expressed stably in CHO cells, has very recently been published (Hoffmann et al., 2004). In this study, Hoffmann et al. (2004) used 125I-cyanopindolol binding to membranes to assess receptor selectivity. Although the authors did not detect any salbutamol or terbutaline β1/β2 selectivity, selectivity was demonstrated with ICI 118551 and CGP 20712A. Bisoprolol, metoprolol and atenolol were found to be more β1-selective in membrane preparations than reported here in intact cells, although, again, carvedilol and propranolol showed slight β2-selectivity in both systems. However, despite a few differences between studies, it is clear that, although highly selective β1 compounds do exist, the β-blockers currently available for clinical use do not show much selectivity between the β-adrenoceptors.
Thus, although in the clinical setting β-blockers are primarily used for their β1-antagonist effect, the majority actually appear to have rather poor β1/β2 selectivity (Table 2). However, despite this, β-blockers have been and continue to be a highly effective treatment for many cardiovascular disorders. The effectiveness of the drugs in man obviously depends on more than just receptor affinity. The pharmacokinetic profile of the drugs, the absorption, metabolism, tissue distribution and elimination of the drugs, as well as their longevity of action at the given receptors, also are important. Also, there are several different polymorphic variants of the β-adrenoceptors within the population and this may give rise to different drug affinities and actions both in the laboratory and in a clinical setting.
Finally, many ‘β-blockers' are not neutral antagonists, but have some agonist and inverse agonist actions of their own at the different β-adrenoceptors (Jasper et al., 1990; Chidiac et al., 1994; Bond et al., 1995; Azzi et al., 2001; 2003; Baker et al., 2003b, 2003c; Hoffmann et al., 2004). The contribution of this to their overall clinical effects is so far unknown. However, the clinical benefit of β-blockers in heart failure does not appear to be a class effect, nor is it completely explained by β1-antagonism (CIBIS-II, 1999; BEST, 2001; COMET, 2003). The agonist and inverse agonist effects of the different β-blockers may therefore explain some of the differences between drugs and their mode of action in conditions where β1-antagonism does not seem to be the whole explanation.
In conclusion, although selective β1-antagonism is the goal of most β-blocker treatment regimes, the majority of clinically used β-blockers have little selectivity for the human β1- over the human β2-adrenoceptor in intact living cells. Clearly, as more β1-adrenoceptor-selective antagonists do exist than those currently clinically available (e.g. CGP 20712A), there is considerable potential for developing more selective β-antagonists for clinical use and thereby reducing the side-effect profile of β-blockers.
Acknowledgments
J.G. Baker is a Wellcome Trust Clinical Scientist Fellow and thanks Professor S.J. Hill for his helpful comments in the preparation of this manuscript.
Abbreviations
- CGP 12177
(−)-4-(3-tert-butylamino-2-hydroxypropoxy)-benzimidazol-2-one
- CGP 20712A
2-hydroxy-5-(2-[{hydroxy-3-(4-[1-methyl-4-trifluoromethyl-2-imidazolyl]phenoxy)propyl}amino]ethoxy)benzamide
- CHO
Chinese hamster ovary
- CL 316243
disodium (R,R)-5-(2-[{2-(3-chlorophenyl)-2-hydroxyethyl}-amino]propyl)-1,3-benzodioxole-2,2,dicarboxylate
- COPD
chronic obstructive pulmonary disease
- ICI 118551
(−)-1-(2,3-[dihydro-7-methyl-1H-inden-4-yl]oxy)-3-([1-methylethyl]-amino)-2-butanol
- ICI 215001
(S)-4-[2-hydroxy-3-phenoxypropylaminoethoxy]phenoxyacetic acid hydrochloride
- ICI 89406
N-[2-[3-(2-cyanophenoxy)-2-hydroxypropylamino]ethyl]-N′-phenylurea
References
- AZZI M., CHAREST P.G., ANGERS S., ROSSEAU G., KOHOUT T., BOUVIER M., PINERYO G. Beta-arrestin-mediated activation of MAPK by inverse agonists reveals distinct active conformations for G protein-coupled receptors. Proc. Natl. Acad. Sci. U.S.A. 2003;100:11406–11411. doi: 10.1073/pnas.1936664100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AZZI M., PINERYO G., PONTIER S., PARENT S., ANSANAY H., BOUVIER M. Allosteric effects of G protein overexpression on the binding of β-adrenergic ligands with distinct inverse effacacies. Mol. Pharmacol. 2001;60:999–1007. doi: 10.1124/mol.60.5.999. [DOI] [PubMed] [Google Scholar]
- BAKER J.G., HALL I.P., HILL S.J. Pharmacological characterization of CGP 12177 at the human β2-adrenoceptor. Br. J. Pharmacol. 2002;137:400–408. doi: 10.1038/sj.bjp.0704855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BAKER J.G., HALL I.P., HILL S.J. Influence of agonist efficacy and receptor phosphorylation on antagonist affinity measurements: differences between second messenger and reporter gene responses. Mol. Pharmacol. 2003a;64:679–688. doi: 10.1124/mol.64.3.679. [DOI] [PubMed] [Google Scholar]
- BAKER J.G., HALL I.P., HILL S.J. Agonist actions of ‘β-blockers' provide evidence for two agonist activation sites or conformations of the human β1-adrenoceptor. Mol. Pharmacol. 2003b;63:1312–1321. doi: 10.1124/mol.63.6.1312. [DOI] [PubMed] [Google Scholar]
- BAKER J.G., HALL I.P., HILL S.J. Agonist and inverse agonist actions of β-blockers at the human β2-adrenoceptor provide evidence for agonist-directed signalling. Mol. Pharmacol. 2003c;64:1357–1369. doi: 10.1124/mol.64.6.1357. [DOI] [PubMed] [Google Scholar]
- Beta-blocker Evaluation of Survival Trial Investigators (BEST) A trial of the beta-blocker bucindolol in patients with advanced chronic heart failure. N. Engl. J. Med. 2001;344:1659–1667. doi: 10.1056/NEJM200105313442202. [DOI] [PubMed] [Google Scholar]
- BLACK J.W., DUNCAN W.A.M., SHANKS R.G. Comparison of some properties of pronethalol and propranolol. Br. J. Pharmacol. 1965;25:577–591. doi: 10.1111/j.1476-5381.1965.tb01782.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOND R.A., LEFF P., JOHNSON T.D., MILANO C.A., ROCKMAN H.A., MCMINN T.R., APPARSUNDARAM S., HYEK M.F., KENAKIN T., ALLEN L.F., LEFKOWITZ R.J. Physiological effects of inverse agonists in transgenic mice with myocardial overexpression of the β2-adrenoceptor. Nature. 1995;374:272–276. doi: 10.1038/374272a0. [DOI] [PubMed] [Google Scholar]
- CHIDIAC P., HEBERT T.E., VALIQUETTE M., DENNIS M., BOUVIER M. Inverse agonist activity of β-adrenergic antagonists. Mol. Pharmacol. 1994;45:490–499. [PubMed] [Google Scholar]
- CIBIS-II Investigators and Committees: The Cardiac Insufficiency Bisoprolol Study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13. [PubMed] [Google Scholar]
- COMET. POOLE-WILSON P.A., SWEDBERG K., CLELAND J.G., DI LENARDA A., HANRATH P., KOMAJDA M., LUBSEN J., LUTIGER B., METRA M., REMME W.J., TORP-PEDERSEN C., SCHERHAG A, SKENE A., CARVEDILOL OR METOPR Comparison of carvedilol and metoprolol on clinical outcomes in patients with chronic heart failure in the Carvedilol Or Metoprolol European Trial (COMET): randomised controlled trial. Lancet. 2003;362:7–13. doi: 10.1016/S0140-6736(03)13800-7. [DOI] [PubMed] [Google Scholar]
- FEELY J., PEDEN N. Use of beta-adrenoceptor blocking drugs in hyperthyroidism. Drugs. 1984;27:425–446. doi: 10.2165/00003495-198427050-00003. [DOI] [PubMed] [Google Scholar]
- HARMS H.H. Isoproterenol antagonism of cardioselective beta adrenergic receptor blocking agents: a comparative study of human and guinea-pig cardiac and bronchial beta adrenergic receptors. J. Pharmacol. Exp. Ther. 1976;199:329–335. [PubMed] [Google Scholar]
- HEIDENREICH P.A., MCDONALD K.M., HASTIE T., FADEL B., HAGAN V., LEE B.K., HLATKY M.A. Meta-analysis of trials comparing beta-blockers, calcium antagonists, and nitrates for stable angina. J. Am. Medic. Assoc. 1999;281:1927–1936. doi: 10.1001/jama.281.20.1927. [DOI] [PubMed] [Google Scholar]
- HOFFMANN C., LEITZ M.R., OBERDORF-MAASS S., LOHSE M.J., KLOTZ K.N. Comparative pharmacology of human β-adrenergic receptor subtypes – characterization of stably transfected receptor in CHO cells. Naunyn-Schmiedeberg's Arch. Pharmacol. 2004;369:151–159. doi: 10.1007/s00210-003-0860-y. [DOI] [PubMed] [Google Scholar]
- JASPER J.R., MICHEL M.C., INSEL P.A. Amplification of cyclic AMP generation reveals agonistic effects of certain beta-adrenergic antagonists. Mol. Pharmacol. 1990;37:44–49. [PubMed] [Google Scholar]
- KOBILKA B. Adrenergic receptors as models for G protein-coupled receptor. Annu. Rev. Neurosci. 1992;15:87–114. doi: 10.1146/annurev.ne.15.030192.000511. [DOI] [PubMed] [Google Scholar]
- LEWIS R.V., LOFTHOUSE C. Adverse reactions with beta-adrenoceptor blocking drugs. An update. Drug Saf. 1993;9:272–279. doi: 10.2165/00002018-199309040-00005. [DOI] [PubMed] [Google Scholar]
- LIANG W., MILLS S. Profile of ligand binding to the porcine β2-adrenergic receptor. J. Anim. Sci. 2001;79:877–883. doi: 10.2527/2001.794877x. [DOI] [PubMed] [Google Scholar]
- LIMMROTH V., MICHEL M.C. The prevention of migraine: a critical review with special emphasis of β-adrenergic blockers. Br. J. Clin. Pharmacol. 2001;52:237–243. doi: 10.1046/j.0306-5251.2001.01459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PEET M., YATES R.A. Beta-blockers in the treatment of neurological and psychiatric disorders. J. Clin. Hosp. Pharm. 1981;6:155–171. doi: 10.1111/j.1365-2710.1981.tb00988.x. [DOI] [PubMed] [Google Scholar]
- PRICHARD B.N. Beta-blockade therapy and cardiovascular disease. Past, present and future. Postgrad. Med. 1988;29:8–18. [PubMed] [Google Scholar]
- PRICHARD B.N., CRUICKSHANK J.M., GRAHAM B.R. Beta-adrenergic blocking drugs in the treatment of hypertension. Blood Press. 2001;10:366–386. doi: 10.1080/080370501753400665. [DOI] [PubMed] [Google Scholar]
- SMITH C., TEITLER M. Beta-blocker selectivity at cloned human beta1- and beta2-adrenoceptors. Cardiovasc. Drugs Ther. 1999;13:123–126. doi: 10.1023/a:1007784109255. [DOI] [PubMed] [Google Scholar]
- STAMPER R.L., WIGGINGTON S.A., HIGGINBOTHAM E.J. Primary drug treatment for glaucoma: beta-blocker versus other medications. Surv. Ophthalmol. 2002;47:63–67. doi: 10.1016/s0039-6257(01)00286-7. [DOI] [PubMed] [Google Scholar]
- UITTI R.J.Medical treatment of essential tremor and Parkinson's disease Geriatrics 19985346–48.53–57 [PubMed] [Google Scholar]