

Abstract
Torsade de Pointes (TdP) is a well-described major risk associated with various kinds of drugs. However, prediction of this risk is still uncertain both in preclinical and clinical trials. We tested 45 reference compounds on the model of isolated canine Purkinje fibres. Of them, 22 are clearly associated and/or labelled with a risk of TdP, and 13 others are drugs with published clinical evidence of QT prolongation, with only one or two exceptional cases of TdP. The 10 remaining drugs are without reports of TdP and QT prolongation.
The relevance of different indicators such as APD90 increase, reverse use dependency, action potential triangulation or effect on Vmax was evaluated by comparison with available clinical data. Finally, a complex algorithm called TDPscreen™ and based on two subalgorithms corresponding to particular electrophysiological patterns was defined.
This latter algorithm enabled a clear separation of drugs into three groups: (A) drugs with numerous or several reports (>2 cases) of TdP, (B) drugs causing QT prolongation and/or TdP only, the latter at a very low frequency (⩽2 cases), (C) drugs without reports of TdP or QT prolongation.
The use of such an algorithm combined with a database accrued from reference compounds with available clinical data is suggested as a basis for testing new candidate drugs in the early stages of development for proarrhythmic risk prediction.
Keywords: Drug safety, proarrhythmic risk, Purkinje fibre, QT prolongation, Torsade de Pointes
Introduction
Drug-induced Torsade de Pointes (TdP) is a life-threatening form of polymorphic ventricular tachycardia, which appears in clinical use at a low or very low frequency and which is often associated with a QT prolongation. This arrhythmia is a well-described major risk for anti-arrhythmic drugs. This kind of deleterious effect has been described in other classes of drugs such as antihistaminics, psychotropics and antibiotics (Raehl et al., 1985). Several drugs such as terfenadine have been withdrawn from the market. Despite important progresses, prediction of this risk remains uncertain or problematic both in preclinical and clinical trials.
In preclinical trials, it is well established that use of only one approach is clearly not sufficient to assess properly the risk of TdP or QT prolongation. According to the most recent draft guidelines from the International Committee of Harmonisation (ICH S7B), a combination of different approaches based on both in vitro and in vivo methods is recommended. This suggestion emphasises the remark, pointing out the difficulty of thorough preclinical evaluation of this risk. Many in vivo preclinical approaches have been suggested, such as evaluation of QT prolongation by telemetry or in anaesthetised preparations. In the event of QT prolongation, these approaches are probably to be preferred because they enable evaluation of both the parent drug and metabolites, and furthermore enable the determination of a safety margin. Nevertheless, they may be limited for technical reasons, such as the need for QT correction, use of anaesthetics or drug-induced adverse effect, leading to a reduction in the sensitivity for detection of QT prolongation (Champeroux et al., 2000).
The blockade of a voltage-dependent potassium channel called hERG has been suggested as a common denominator for these agents. Assessment of this property is widely used mainly in preclinical screening processes. Nevertheless, retrospective analysis of the risk of TdP and the capability of inhibition of this channel demonstrates that some compounds already on the market can block this channel without inducing TdP, and even without inducing any QT prolongation. As a consequence of this observation, the main drawback is elimination of a large proportion of molecules in some chemical classes in the early preclinical development stages. In addition, use of in vitro conditions that do not exactly mimic the in vivo conditions may lead to false-negative results.
Evaluation of global electrophysiological effects in native cells embedded in its tissue by the microelectrode method is also widely used in parallel to the various methods described above. The main interest in this model is based on the possibility of characterisation of the electrophysiological profile of a drug in a native cardiac cell, not only on potassium currents, but also in the presence of all other currents. The main criticism of this model is linked to its advantages. Indeed, because of the presence of all native currents, interpretation of results can be difficult in case of effects of a drug on different currents simultaneously or according to the concentration. Clearly, the pathogenesis of TdP cannot be explained by a single blockade of the delayed rectifier potassium current IKR involving the hERG channel. Consequently, the native cell model is probably one of those which could enable determination of a common electrophysiological mechanism involving several ionic currents for drugs causing TdP.
In our laboratory, we have tested over several years many hundreds of molecules on the canine Purkinje fibre model in parallel to telemetry studies conducted in conscious dogs providing good correlation for QT prolongation assessment. Results obtained from 45 reference compounds have been analysed in detail. The relevance of different parameters has been evaluated by comparison with available clinical data. From this analysis, an algorithm was defined and is proposed for improved evaluation of the risk of TdP in early stages of preclinical development.
Methods
Preparations
Male Beagles dogs were anaesthetised with sodium pentobarbital (30 mg kg−1 i.v.). The heart was quickly removed and placed in an oxygenated and heparinised Tyrode's solution maintained at 4°C. Free-running Purkinje fibre bundles were dissected from the left ventricle of the heart. Subsequently, each Purkinje fibre was mounted in a 5 ml organ bath and was constantly irrigated with oxygenated (95% O2–5% CO2) Tyrode's solution (mM: NaCl 118, KCl 4, NaHCO3 27, MgCl2 1, NaH2PO4 1.8, CaCl2 1.8, Glucose 11) at a rate of 5 ml min−1. The bath temperature was maintained at 36.5±0.5°C and pH 7.3–7.5.
Action potential (AP) recordings
Bipolar stimulation electrodes were placed directly on the tissue in order to evoke an AP potential. Purkinje fibres were stimulated at normal rate (60 pulses per minute (ppm)) during an equilibration period of 30 min. Stimulation pulses had a duration of 2 ms, the amplitude was 1.5–2.0 times the diastolic threshold (i.e. approximately 2 V). Each Purkinje fibre was impaled with a conventional glass microelectrode filled with KCl 3 M and having a tip resistance of 10–30 MΩ. The Purkinje fibres were allowed to stabilise for 60 min at a stimulation rate of 60 ppm. At the end of this period of stabilisation, the sequence of perfusion was started. During each period of perfusion, preparations were stimulated at a normal rate of 60 ppm for the first 25 min, then at a low rate of 20 ppm for the last 5 min in order to reveal any possible reverse-use dependency. The recorded parameters were the amplitude of AP (APA; mV), the resting potential (RP; mV), the maximal rate of depolarisation (Vmax; V s−1) and AP durations at 50, 70 and 90% of repolarisation (APD50, APD70 and APD90, respectively). The experiments were performed over a period of 3 years and the parameters were measured using the DATAPAC (Biologic, France) and IOX (EMKA, France) data acquisition systems.
Each drug was perfused separately and directly into the organ bath. Substances were dissolved in an appropriate vehicle at concentrations 1000 times more concentrated than the final concentrations achieved in the organ bath, and then perfused at a rate of 5 μl min−1. Drugs were dissolved either in Tyrode or dimethylsulfoxide. The final concentration of these vehicles was never above 0.01%. Homogeneity of the perfusing solution containing the tested drug and Tyrode solution was ensured by utilisation of a mixing system just before the perfusion of tissues. This system was previously validated by checking the final concentration in the bath of a large number of test molecules (>20). The use of dimethylsulfoxide at 0.01% as vehicle was validated by testing five successive 30-min sequences of perfusion in five different preparations under the same experimental conditions as were used for the tested drugs.
Selection of drugs
Drugs were selected from the data set published by Redfern et al. (2003). This data set corresponds to a list of 100 drugs known or suspected to cause TdP and/or QT prolongation. They were selected on the criteria of availability of clinical data among all the five different categories defined by Redfern. Only drugs for which published clinical evidence of TdP and/or QT prolongation are available were selected. Some drugs not present in the Redfern classification and known to not cause TdP and QT prolongation were added in our database to test the robustness of algorithms. Initially, three groups were defined:
Group A: drugs with numerous or several reports (>2 cases) of TdP.
Group B: drugs causing QT prolongation and/or TdP only, the latter at a very low frequency (⩽2 cases).
Group C: drugs without reports of TdP or QT prolongation.
Most drugs were purchased from Sigma-Aldrich, France.
Algorithms
A minimum of three preparations obtained from different animals was used for evaluation of each drug. The lowest concentration was chosen so as not to induce any electrophysiological effect. In all cases, the highest concentration was defined as the highest soluble concentration, usually 10−5 M or up to 10−4 M in some cases. A logarithmic progression was followed, in most cases 10−8, 10−7, 10−6 and 10−5 M.
Data were analysed as the mean percentage of variation in relation to the control period of perfusion with the vehicle for each preparation. After analysis of data, several exclusion criteria or filters were defined to avoid biases due to nonspecific or exaggerated electrophysiological effects seen mainly at the highest effective concentrations: for example, a too large depolarisation of the resting membrane potential (loss of excitability). In this latter case, only the concentrations lower than those causing nonspecific or exaggerated effects were taken into account. These filters were common to all algorithms. No weighting linked to the concentration level was used.
Subsequently, proarrhythmic scores were determined using various algorithms. First, simple algorithms were evaluated, such as increase in APD90, decrease in Vmax, reverse use dependency (RUD) or AP triangulation. For most of the simple algorithms, proarrhythmic scores were determined from the mean percentage of variation of the considered parameter (e.g. APD90) in relation to the control period, for example,
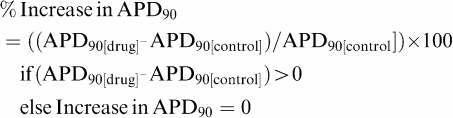 |
For some others such as RUD or triangulation, proarrhythmic scores were obtained from a difference of two parameters expressed in percentage of variation. As well, RUD was obtained as follows:
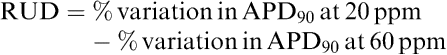 |
Likewise, triangulation in the delayed phase of repolarisation was calculated as follows:
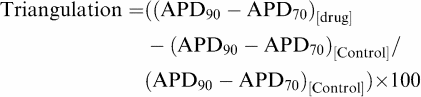 |
In total, more than 40 simple algorithms were tested. A proarrhythmic score was calculated for each concentration. Only the highest proarrhythmic score obtained at the most effective concentration was noted for each drug.
From this first analysis using only simple algorithms and on the basis of the results obtained from the first 20 compounds tested, a complex algorithm was defined and named TDPscreen™. This complex algorithm corresponds to the combination of two subalgorithms, giving each a high proarrhythmic score to the two main subgroups of torsadogenic compounds found from the database analysis:
Subgroup A1: compounds causing an exaggerated increase in APD90 combined with a high RUD.
Subgroup A2: compounds causing a triangulation of the delayed phase of repolarisation without shortening of APD90.
For the two subalgorithms and TDPscreen™, proarrhythmic scores are derived from a combination of simple algorithms and are thus expressed as percentage of variation with a cutoff value of 100. A computer program was built to run these algorithms, its name being protected by a trademark entitled TDPscreen™.
Results
Control
No relevant or noticeable effect was found at the end of five sequences of 30 min of perfusion, whatever the parameter. On average, the maximum deviation was less than 1% in the validation study, confirming that dimethylsulphoxide has no effect at a concentration of 0.01%, and showing that preparations were very stable throughout the whole period of recordings under the experimental conditions adopted.
APD90, RUD
Using this simple algorithm, most of the compounds causing QT prolongation and/or TdP in man have a score ranging between 10 and 80%. Nevertheless, there is no clear separation between compounds only causing QT prolongation and compounds causing TdP (Figure 1).
Figure 1.

Comparison of maximum increase in APD90 induced by drugs of groups A–C. Increase in APD90 induced by drugs expressed as a percentage of variation compared to control conditions. Most drugs causing TdP (group A) or QT prolongation only (group B) increase APD90. Some drugs causing TdP do not increase APD90 (terfenadine, thioridazine,…).
Approximately 20% of torsadogenic drugs have a typical pattern, that is, they induce an increase in APD as shown in Figure 2: dofetilide, sotalol, sparfloxacin and erythromycin. We put them in a subgroup called A1. They exhibit the highest score for the algorithm based on an increase in APD90. These increases in APD90 can be considered as exaggerated (more than 40%) when compared to those observed with most of the drugs only causing QT prolongation.
Figure 2.

Typical effects of dofetilide on AP of isolated canine Purkinje fibres. APs were triggered by field stimulation at 1 Hz in Tyrode (red line) and during the application of 1 μM dofetilide (blue line). Dofetilide and other drugs of the subgroup A1 (see Methods) cause an exacerbated (>40%) increase in AP duration.
However, some compounds inducing TdP in man do not increase APD90. Among these are some of the best-known drugs such as terfenadine, thioridazine, bepridil and astemizole. For these latter compounds, the common profile was an unchanged value of APD90, combined with a more or less marked triangulation (Figure 3). This typical pattern corresponds to approximately 80% of drugs causing TdP, and was put in a subgroup called A2. It is believed that, for torsadogenic drugs, the effects on APD90 are masked by a decrease in the plateau of the AP, related to a decrease in Vmax.
Figure 3.

Typical effects of terfenadine on AP of isolated canine Purkinje fibres. APs were triggered by field stimulation at 1 Hz in Tyrode (red line) and during the application of 10 μM terfenadine (blue line). Terfenadine and other drugs of the subgroup A2 (see Methods) cause a triangulation of the delayed phase of repolarisation, increase the difference APD90−APD70 and do not shorten the AP duration.
As expected, an RUD is found only for drugs causing an increase in APD90 (Figure 4); Therefore, this algorithm gives no more information than that based on APD90.
Figure 4.

Comparison of RUD on APD90 induced by drugs of groups A–C. RUD is calculated as the maximum difference in the percentages of increase in APD90 at low (20 ppm) and normal (60 ppm) stimulation rates. Torsadogenic drugs of the subgroup A1 exhibit the highest RUD (e.g. dofetilide, sotalol,…).
Except for erythromycin, all the other molecules of the first subgroup A1 of torsadogenic compounds causing exaggerated increases in APD90 exhibit a marked RUD.
In the second subgroup A2, two profiles of compounds may be defined. The first profile corresponds to compounds which do not cause any increase in APD90, combined with a more or less marked triangulation whatever the concentration: astemizole, terfenadine or thioridazine. These molecules exhibit little or no RUD.
The second profile in the subgroup A2 corresponds to molecules having generally dual effects depending on concentrations. In all cases, these latter compounds cause a reverse use-dependent increase in APD90 at low concentrations, followed by a more or less marked reduction in APD90 combined with a triangulation. This is related in most cases to a more or less large effect on Vmax at higher concentrations such as cisapride, droperidol, haloperidol and quinidine.
Vmax
A large proportion of compounds causing TdP and/or QT prolongation in humans have a more or less marked effect on Vmax (Figure 5), especially among the subgroup A2 of torsadogenic drugs. As expected, the most potent compounds are the class Ia antiarrhythmic drugs.
Figure 5.

Comparison of reduction in Vmax induced by drugs of groups A–C. Decrease in Vmax is expressed as a percentage of variation in relation to the control period. A large proportion of torsadogenic drugs of subgroup A2 causes a marked reduction in Vmax, suggesting an inhibition of the rapid sodium conductance. Drugs of subgroup A1 (see Methods) and group B have less marked effects on Vmax.
Triangulation APD90−APD70
A large proportion of compounds cause a more or less pronounced triangulation of the delayed phase of repolarisation, even among compounds with no reports of TdP or QT prolongation (Figure 6). Nevertheless, the highest proarrhythmic scores are found with compounds previously reported to incite TdP. Triangulation is related to the effects on currents involved in the plateau of the AP, such as the sodium or calcium currents. The difference APD90−APD70 was found to be better than the differences APD90−APD50 or APD90−APD30 to discriminate torsadogenic compounds from compounds only causing QT prolongation. This is probably because the difference APD90−APD70 is more dependent upon the IKR current involved in the final phase of repolarisation.
Figure 6.

Comparison of triangulation of the delayed phase of repolarisation induced by drugs of groups A–C. Triangulation is calculated from the maximum difference between APD90 and APD70 values and expressed as a percentage of variation in relation to control period. Torsadogenic drugs of subgroup A2 exhibit, in a general manner, a higher score than those found with drugs of the subgroup A1 or with drugs of group B. Some drugs of group C having calcium and/or sodium-inhibitory properties have a high score using this algorithm.
TDPscreen™
From the analysis of more than 40 different simple algorithms and, in particular, from the results of the previously mentioned algorithms, a more complex algorithm based on a combination of several simple algorithms and multiple conditions has been defined and is called the TDPscreen™. This algorithm takes advantage of its complexity to score drugs having any electrophysiological effect in order to highlight those being really proarrhythmics.
TDPscreen™ gives rise to a high proarrhythmic score for the two main subgroups of torsadogenic compounds in the database (Table 1):
Subgroup A1: Compounds causing an exaggerated increase in APD90 combined with a high RUD, for example, dofetilide, sotalol, sparfloxacin….
Subgroup A2: Compounds causing a triangulation of the delayed phase of repolarisation without shortening of APD90, for example, terfenadine, thioridazine….
Table 1.
Classification of tested molecules
| Groups | Subgroups | Molecules |
|---|---|---|
| A | A1 | Dofetilide, erythromycin, sotalol, sparfloxacin |
| A2 | Ajmaline, astemizole, bepridil,chlorpromazine, cisapride, disopyramide, droperidol, haloperidol, imipramine, maprotiline, pentamidine, pimozide, prenylamine, propafenone, quinidine, sertindole, terfenadine, thioridazine | |
| B | Azithromycin, chloroquine, ciprofloxacin, clarithromycin, clozapine, emetine, fluoxetine, ketoconazole, moxifloxacin, olanzapine, ondansetron, risperidone, verapamil | |
| C | Aspirine, captopril, codeine, diazepam, diltiazem, foscarnet, indomethaine, nicardipine, phenytoin, propranolol |
The first subalgorithm based on an exaggerated increase in APD90 and RUD was designed to differentiate the subgroup A1 (20%) of torsadogenic drugs, such as dofetilide, sotalol, sparfloxacin and erythromycin (Figure 7). The second subalgorithm based on triangulation of the delayed phase of repolarisation without shortening of APD90 was used to differentiate all the other drugs of the subgroup A2 (80%) of torsadogenic agents, in particular, drugs such as terfenadine (Figure 8), thioridazine and associated drugs.
Figure 7.

Comparison of proarrhythmic scores using the first subalgorithm for drugs of groups A–C. This subalgorithm differentiates torsadogenic drugs of subgroup A1, causing exacerbated increases in APD90 with a high RUD.
Figure 8.

Comparison of proarrhythmic scores using the second subalgorithm for drugs of groups A–C. This subalgorithm differentiates torsadogenic drugs of subgroup A2, causing triangulation of the delayed phase of repolarisation (APD90−APD70) without shortening in APD90.
TDPscreen™, being a combination of the two previously mentioned subalgorithms, clearly discriminates drugs triggering TdP, those causing only QT prolongation from drugs without reports of TdP or QT prolongation (Figure 9).
Figure 9.

Comparison of proarrhythmic scores obtained with TDPscreen™ for drugs of groups A–C. TDPscreen™ is a combination of the first and second subalgorithms used to differentiate the two subgroups of torsadogenic drugs A1 and A2. It clearly differentiates drugs causing TdP (group A) from those causing QT prolongation only or with exceptional reports of TdP (group B) and from drugs with no reports of TdP or QT prolongation (group C).
Discussion
Using single parameters such as triangulation of the AP or stimulation-dependent increase of APD90 (associated or not with RUD), it is not possible to distinguish compounds having proarrhythmic potential from those who do not have this property. This is why we built a complex algorithm based on the integration of several simple algorithms and called TdPScreen™.
The list of reference compounds tested on the model of isolated canine Purkinje fibres includes 22 molecules which are clearly associated and/or labelled with a risk of TdP (Redfern et al., 2003), which we named group A.
Among these 22 molecules, seven have been withdrawn from the market (astemizole, cisapride, droperidol, pimozide, prenylamine, sertindole and terfenadine (De Abajo & Rodriguez, 1999; Darpö, 2001; Haddad & Anderson, 2002). Six are antiarrhythmics, of which three are class Ia antiarrhythmics (ajmaline, disopyramide and quinidine), two are class III antiarrhythmic (dofetilide and D,L sotalol) and the last one is a class Ic antiarrhythmic, propafenone (Rosengarten & Brooks, 1987; Hii et al., 1991; Faber et al., 1994; Malik & Camm, 2001; Redfern et al., 2003).
Among the nine remaining drugs, all are non antiarrhythmic drugs with seven having been associated with numerous reports or a high risk of TdP, bepridil, that is, chlorpromazine, erythromycin, haloperidol, maprotiline, pentamidine and thioridazine and one with isolated documented cases of TdP, that is, sparfloxacin (Abinander, 1984; Singh, 1992; Glassman & Bigger, 2001; Redfern et al., 2003). Finally, one compound, imipramine, is associated with many fewer cases of TdP (Tzivoni et al., 1984), but is well documented for its proarrhythmic effects (Pacher et al., 1999). It is the only one in the group A for which the risk of TdP is not mentioned on the label. All these compounds show a score of 100 or close to 100 using TdPScreen™.
TDPscreen™ enables definition of a second group of 13 compounds named group B, with scores ranging from approximately 10 to 80. For six of them, there are published clinical data of QT prolongation but no reports of TdP (De Ponti et al., 2002; Warner & Hoffmann, 2002): clozapine, ketoconazole, moxifloxacin, olanzapine, ondansetron and risperidone. Five others have published clinical data of QT prolongation and reports of TdP at an extremely low frequency: azythromycin in one patient with congenital long QT syndrome (Arellano-Rodrigo et al., 2001), chloroquine in one patient following self medication (Demaziere et al., 1995), clarithromycin in two patients (Kamochi et al., 1999), ciprofloxacin (0.3 cases per 10 million prescriptions) (Frothingham, 2001) and fluoxetine in one elderly female patient (Redfern et al., 2003).
Verapamil and emetine are the only molecules not reported to cause TdP or QT prolongation in clinical use (De Ponti et al., 2002; Redfern et al., 2003). For verapamil, however, this calcium blocker is known to block IKR at relatively low concentrations (IC50: 0.14–0.83 μmol) (Chouabe et al., 2000) and cases of polymorphic ventricular tachycardia can be found in the literature (Shiraishi et al., 2002). Its intermediate score of 41 is thus not surprising and indeed seems logical. In the case of emetine, no data on the potassium currents involved in the ventricular repolarisation were found in the literature. Nevertheless, this amoebicide drug has been found to be cardiotoxic. Its cardiotoxicity was attributed to the inhibitory effects on sodium and calcium conductances (Lemmens-Gruber et al., 1996; 1997).
Interestingly, the scores obtained with some molecules in two therapeutic classes seem to fit with clinical and/or preclinical data in terms of potential for QT prolongation or TdP induction found in the literature. In the series of fluoroquinolone antibiotics, the following classification, based on their ability to increase the QT, is described from preclinical data (Webster et al., 2002): sparfloxacin>moxifloxacin>ciprofloxacin. We found respective scores of 82, 75 and 14 for these molecules. Likewise, in the series of macrolide antibiotics, the following classification, based on their ability to induce TdP, was suggested (Milberg et al., 2002): erythromycin>clarithromycin>azithromycin, while we found scores of 100, 57 and 17, respectively.
These observations suggest that the algorithm used could indicate the potential for QT prolongation when scores arise between 10 and 80. Above these values, it was preferred to attribute a cutoff value corresponding to the top score of 100 without trying to discriminate between compounds in terms of potential for TdP induction, since it is clear that all compounds exhibiting a very high score higher or close to 100 are torsadogenic.
Finally, a third group named C can be defined corresponding to drugs with no reports of TdP and QT prolongation (De Ponti et al., 2002): aspirin, captopril, codeine, diltiazem, foscarnet, nicardipine and propranolol. Among these compounds, one molecule was devoid of any electrophysiological effect: aspirin. Testing of a larger number of molecules with no electrophysiological effect contributes little to evaluation of algorithms based on changes of electrophysiological parameters, so no further molecules with this profile were tested. Captopril, codeine, indomethacine and foscarnet caused some various and minor changes in AP, in particular at the highest concentrations.
On the contrary, the five other molecules tested have marked electrophysiological effects. Diltiazem and nicardipine caused triangulation of AP due to the calcium-blocking property. At high concentrations, diltiazem was also found to decrease Vmax, suggesting that there is also an effect on the early depolarising sodium current. The effects of diazepam on the AP were very close to those of nicardipine. Indeed, diazepam was found to have calcium-blocking properties on cardiac cells (Nonaka et al., 1997). Propranolol and phenytoin were also found to cause a concentration-dependent triangulation that seems to be related to an effect on the depolarising sodium current, as suggested by the decrease in Vmax caused by these compounds. For these five molecules, the triangulation of APs was always associated with a shortening of the AP. This is the main difference when compared with the subgroup A2 of torsadogenic drugs, which cause triangulation. TDPscreen™ takes into account this pattern, enabling differentiation of drugs having effects on sodium and/or calcium conductances without effect on IKR.
Results obtained with TDPscreen™ demonstrate that it is possible to establish a clear link between clinical data from marketed drugs, or those withdrawn from the market, and their electrophysiological pattern. This also enables differentiation of drugs that cause only QT prolongation or TdP only under exceptional conditions. These correlations are not due to chance because a large number of molecules have been tested, approximately half of which are known to produce TdP. In addition, almost all of them have electrophysiological effects and are known to block the delayed rectifier potassium current IKR (Redfern et al., 2003).
The isolated canine Purkinje fibre model is probably the only one which can enable establishment of such a link within the same experiment. Indeed, this model exhibits a good equilibrium between the most important currents, especially INa and IKR. The TDPscreen™ algorithm is unlikely to be transposable to other models commonly in use, such as the model of papillary muscle of guinea-pigs in which the sensitivity to an inhibitor of sodium conductance is lower, or to the isolated Purkinje fibres of rabbits which are extremely sensitive to IKR blockers.
The need to use two subalgorithms, one discriminating drugs causing an exaggerated increase in AP and the other one based on triangulation without shortening in AP duration, suggests that torsadogenic drugs indeed have different electrophysiological patterns.
This strongly argues in favour of the use of models enabling evaluation of effects on multiple ion channels during the preclinical evaluation steps and not just models that are restricted to IKR.
It is well known that a common feature of drugs causing TdP is the blockade of IKR. Likewise, it is also clearly established that this pattern is alone not sufficient for induction of TdP and that other factors are involved, although presently they remain unclear. The two subgroups A1 and A2 of torsadogenic drugs on which TDPscreen™ is based confirm this. For the most numerous subgroup A2 of torsadogenic drugs, which cause triangulation without shortening in AP, an inhibition of the early depolarising sodium current was observed in many cases. By itself, an inhibition of the sodium current can be considered as antiarrhythmic. However, triangulation without shortening of the AP is expected to be torsadogenic because EADs are more easily induced under these conditions. This argues in favour of a role for inhibition of this current in the pathogenesis of TdP for this subgroup.
For the other less numerous subgroup A1 of torsadogenic drugs, an exaggerated increase in AP duration was found. The term ‘exaggerated' or ‘exacerbated' is used because the magnitude of increase is clearly larger than those observed with drugs known to induce only QT prolongation. This difference also argues in favour of additional electrophysiological properties, which could explain these abnormal increases in AP duration. The most likely hypothesis could be an inhibition of ITO, IKS and/or IK1 potassium current inhibition, in addition to IKR inhibition.
The characterisation of two subgroups among torsadogenic drugs supports the hypothesis based on a predominance of mechanisms of re-entry in genesis of TdP, in which an increase in heterogeneity of AP duration between different cellular types might play a crucial role (Antzelevitch et al., 1999a).
Indeed, it has been clearly demonstrated that some cells present in the mid-myocardium, called ‘M' cells (Antzelevitch et al., 1999b), are particularly sensitive to IKR blockers when compared to adjacent cells like epicardial or endocardial cells. Mechanisms of re-entry loops between these different kinds of cardiac cells are likely to be due to the persistence of depolarised cells (‘M' cells) adjacent to repolarised cells (endocardial or epicardial cells), which have passed their refractory period.
On the other hand, any mechanism that could enhance the heterogeneity of AP duration among these different cell types could be a source of re-entry and could initiate TdP. Such mechanisms are present for both torsadogenic drug subgroups A1 and A2 tested in our study.
For the subgroup A2 of drugs causing triangulation, the source of heterogeneity could be related to the effect seen on the early depolarising sodium current of most of the molecules, combined with their effect on IKR. This kind of electrophysiological pattern is expected to cause shortening of AP duration in some cardiac cells (e.g. endocardium, epicardium), few change in AP duration in others such as Purkinje fibres and prolongation in yet other cells, including ‘M' cells in which the IKR current is predominant. For the subgroup A1 of drugs causing an exaggerated increase in AP duration, the source of heterogeneity is certainly different and likely to be related to mechanisms that could prolong cell depolarisation, such as an ITo, IKS and/or IK1 inhibition. In this latter case, this kind of electrophysiological pattern is expected to maintain a depolarised state in some types of cells such as Purkinje fibres or ‘M' cells and to have little effect in the others such as endocardial and epicardial cells.
The interest in the canine Purkinje fibre model is certainly also linked to the similar sensitivity to IKR blockers between ‘M' cells and Purkinje cells, as suggested by the pharmacological responses on duration of the AP that we obtained on Purkinje cells with D,L sotalol, erythromycin and quinidine, when compared to those described on ‘M' cells with the same molecules (Antzelevitch et al., 1999b).
It must be pointed out that the TDPscreen™ algorithm does not involve any weighting of data according to the concentration level. This indicates that the most important point is the pattern of the electrophysiological effects of a drug rather than the concentration level at which its effects are seen in the model.
A methodological approach close to TDPscreen™ is suggested by Hondeghem & Hoffmann (2003) with the SCREENIT model in isolated female rabbit heart. The algorithms of this model give a high importance to three factors: triangulation, RUD and instability of APs.
Two of these factors, triangulation and RUD, are also used in the TDPscreen™ algorithm, but the instability factor is not taken into account here. Instability of AP duration is a major factor in the SCREENIT model and is probably the consequence of interaction with multiple cardiac ion channels, the latter factor being more important in the TDPscreen™ model. The concepts of instability and heterogeneity of AP duration are certainly very close and thus play a crucial role in the pathogenesis of TdP, as suggested by these two models, SCREENIT and TDPscreen™.
To conclude, TDPscreen™ confirms that the propensity of a compound to cause TDP is linked to common and particular electrophysiological properties in addition to hERG inhibition. It strongly argues in favour of a global evaluation of electrophysiological effects on integrated models such as on cardiac AP in preclinical trials. TDPscreen™ clearly enables a rapid classification of any new drug candidate between three simply defined groups and should be very helpful for prediction of the risk of TdP in Man when testing new drugs in the early stages of development, and in the absence of any clinical data.
Abbreviations
- AP
action potential
- APA
amplitude of action potential
- APD
action potential duration
- EAD
early after depolarisation
- RP
resting potential
- RUD
reverse use dependency
- TdP
Torsade de Pointes
References
- ABINANDER E.G. QT prolongation and torsade de pointes ventricular tachycardia produced by maprotiline. Am. J. Cardiol. 1984;53:654. doi: 10.1016/0002-9149(84)90065-1. [DOI] [PubMed] [Google Scholar]
- ANTZELEVITCH C., SICOURI S., LITOVSKY S.H., LUKAS A., KRISHNAN S.C., DI DIEGO J.M., GINTANT G.A., LIU D.W. Heterogeneity within the ventricular wall: electrophysiology and pharmacology of epicardial, endocardial and M cells. Circ. Res. 1999a;21:45–61. doi: 10.1161/01.res.69.6.1427. [DOI] [PubMed] [Google Scholar]
- ANTZELEVITCH C., SHIMIZU W., YAN G.X., SICOURI S., WEISSENBURGER J., NESTERENKO V.V., BURASHNIKOV A., DI DIEGO J., SAFFITZ J., THOMAS G.P. The M cell: its contribution to the ECG and to normal and abnormal electrical function of the heart. J. Cardiovasc. Electrophysiol. 1999b;10:1124–1152. doi: 10.1111/j.1540-8167.1999.tb00287.x. [DOI] [PubMed] [Google Scholar]
- ARELLANO-RODRIGO E., GARCIA A., MONT L., ROQUE M. Torsade de Pointes and cardiorespiratory arrest induced by azithromycin in a patient with congenital long QT syndrome. Med. Clin. (Barc.). 2001;117:118–119. doi: 10.1016/s0025-7753(01)72036-2. [DOI] [PubMed] [Google Scholar]
- CHAMPEROUX P., MARTEL E., VANNIER C., BLANC V., LEGUENNEC J.Y., FOWLER J., RICHARD S. The preclinical assessment of the risk for QT interval prolongation. Therapie. 2000;55:101–109. [PubMed] [Google Scholar]
- CHOUABE C., DRICI M.-D., ROMEY G., BARHANIN J. Effects of calcium channel blockers on cloned cardiac K+ channels IKR and IKS. Therapie. 2000;55:195–202. [PubMed] [Google Scholar]
- DARPÖ B. Spectrum of drugs prolonging QT interval and the incidence of torsade de pointes. Eur. Heart J. Suppl. 2001;3:K70–K80. [Google Scholar]
- DE ABAJO F.J., RODRIGUEZ L.A.G. Risk of ventricular arrhythmias associated with nonsedating antihistamine drugs. Br. J. Clin. Pharmacol. 1999;47:307–313. doi: 10.1046/j.1365-2125.1999.00885.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DEMAZIERE J., FOURCADE J.M., BUSSEUIL C.T., ADELEINE P., MEYER S.M., SAISSY J.M. The hazards of chloroquine self prescription in west Africa. J. Toxicol. Clin. Toxicol. 1995;33:369–370. doi: 10.3109/15563659509028925. [DOI] [PubMed] [Google Scholar]
- DE PONTI F., POLUZZI E., CAVALLI A., RECANATINI M., MONTANARO N. Safety of non-antiarrhythmic drugs that prolong the QT interval or induce torsade de pointes: an overview. Drug Saf. 2002;25:263–286. doi: 10.2165/00002018-200225040-00004. [DOI] [PubMed] [Google Scholar]
- FABER T.S., ZEHENDER M., JUST H. Drug-induced torsade de pointes. Incidence, management and prevention. Drug Saf. 1994;11:463–476. doi: 10.2165/00002018-199411060-00007. [DOI] [PubMed] [Google Scholar]
- FROTHINGHAM R. Rates of torsade de pointes associated with ciprofloxacin, ofloxacin, levofloxacin, gatifoloxacin, and moxifloxacin. Pharmacotherapy. 2001;21:1468–1472. doi: 10.1592/phco.21.20.1468.34482. [DOI] [PubMed] [Google Scholar]
- GLASSMAN A.H., BIGGER J.T. Antipsychotic drugs: prolonged QTc interval, torsade de pointes, and sudden death. Am. J. Psychiatry. 2001;158:1774–1782. doi: 10.1176/appi.ajp.158.11.1774. [DOI] [PubMed] [Google Scholar]
- HADDAD P.M., ANDERSON I.M. Antipsychotic-related QTc prolongation, torsade de pointes and sudden death. Drugs. 2002;62:1649–1671. doi: 10.2165/00003495-200262110-00006. [DOI] [PubMed] [Google Scholar]
- HII J.T., WYSE D.G., GILLIS A.M., COHEN J.M., MITCHELL L.B. Propafenone-induced torsade de pointes: cross-reactivity with quinidine. Pacing Clin. Electrophysiol. 1991;14:1568–1570. doi: 10.1111/j.1540-8159.1991.tb02729.x. [DOI] [PubMed] [Google Scholar]
- HONDEGHEM L.M., HOFFMANN P. Blinded test in isolated female rabbit heart reliably identifies action potential duration prolongation and proarrhythmic drugs: importance of triangulation, reverse use dependency and instability. J. Cardiovasc. Pharmacol. 2003;41:14–24. doi: 10.1097/00005344-200301000-00003. [DOI] [PubMed] [Google Scholar]
- KAMOCHI H., NII T., EGUCHI K., MORI T., YAMAMOTO A., SHIMODA K., IBARAKI K. Clarithromycin associated with torsade de pointes. Jpn. Circ. J. 1999;63:421–422. doi: 10.1253/jcj.63.421. [DOI] [PubMed] [Google Scholar]
- LEMMENS-GRUBER R., KARKHANEH A., STUDENIK C., HEISTRACHER P. Cardiotoxicity of emetine dihydrochloride by calcium channel blockade in isolated preparations and ventricular myocytes of guinea pig hearts. Br. J. Pharmacol. 1996;117:377–383. doi: 10.1111/j.1476-5381.1996.tb15202.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEMMENS-GRUBER R., STUDENIK C., KARKHANEH A., HEISTRACHER P. Mechanism of sodium channel blockade in the cardiotoxic action of emetine dihydrochloride in isolated cardiac preparations and ventricular myocytes of guinea pigs. J. Cardiovasc. Pharmacol. 1997;30:554–561. doi: 10.1097/00005344-199711000-00004. [DOI] [PubMed] [Google Scholar]
- MALIK M., CAMM A.J. Evaluation of drug-induced QT interval prolongation: implications for drug approval and labeling. Drug Saf. 2001;24:323–351. doi: 10.2165/00002018-200124050-00001. [DOI] [PubMed] [Google Scholar]
- MILBERG P., ECKARDT L., BRUNS H.J., BIERTZ J., RAMTIN S., REINSCH N., FLEISCHER D., KIRCHHOF P., FABRITZ L., BREITHARDT G., HAVERKAMP W. Divergent proarrythmic potential of macrolide antibiotics despite similar QT prolongation: fast phase 3 repolarization prevents early afterdepolarizations and torsade de pointes. J. Pharmacol. Exp. Ther. 2002;1:218–225. doi: 10.1124/jpet.102.037911. [DOI] [PubMed] [Google Scholar]
- NONAKA A., KASHIMOTO S., IMAMURA M., FURUYA A., KUMAZAWA T. Mechanism of the negative inotropic effect of midazolam and diazepam in cultured foetal mouse cardiac myocytes. Eur. J. Anaesthesiol. 1997;14:481–487. doi: 10.1046/j.1365-2346.1997.00111.x. [DOI] [PubMed] [Google Scholar]
- PACHER P., UNGVARI Z., NANASI P.P., FURST S., KECSKEMETI V. Speculations on difference between tricyclic and selective serotonin reuptake inhibitor antipressants on their cardiac effects. Is there any. Curr. Med. Chem. 1999;6:469–480. [PubMed] [Google Scholar]
- RAEHL C.L., PATEL A.K., LEROY M. Drug induced torsade de pointes. Clin. Pharm. 1985;4:675–690. [PubMed] [Google Scholar]
- REDFERN W.S., CARLSSON L., DAVIS A.S., LYNCH W.G., MACKENZIE I., PALETHORPE S., SIEGL P.K., STRANG I., SULLIVAN A.T., WALLIS R., CAMM A.J., HAMMOND T.G. Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 2003;58:32–45. doi: 10.1016/s0008-6363(02)00846-5. [DOI] [PubMed] [Google Scholar]
- ROSENGARTEN M., BROOKS R. Torsade de pointes ventricular tachycardia in a hypothyroid patient treated with propafenone. Can. J. Cardiol. 1987;3:234–239. [PubMed] [Google Scholar]
- SHIRAISHI H., ISHIBASHI K., URAO N., HYOGO M., TSUKAMOTO M., KEIRA N., HIRASAKI S., SEO Y., SHIRAYAMA T., NAKAGAWA M. Two cases of polymorphic ventricular tachycardia induced by the administration of verapamil against paroxysmal supraventricular tachycardia. Intern. Med. 2002;41:445–448. doi: 10.2169/internalmedicine.41.445. [DOI] [PubMed] [Google Scholar]
- SINGH B.N. Safety profile of bepridil determined from clinical trials in chronic stable angina in the United States. Am. J. Cardiol. 1992;69:68D–74D. doi: 10.1016/0002-9149(92)90962-x. [DOI] [PubMed] [Google Scholar]
- TZIVONI D., KEREN A., COHEN A.M., LOEBEL H., ZAHAVI I., CHENZBRAUN A., STERN S. Magnesium therapy for torsade de pointes. Am. J. Cardiol. 1984;53:528–530. doi: 10.1016/0002-9149(84)90025-0. [DOI] [PubMed] [Google Scholar]
- WARNER B., HOFFMANN P. Investigation of the potential of clozapine to cause torsade de pointes. Adverse Drug React. Toxicol. Rev. 2002;21:189–203. doi: 10.1007/BF03256196. [DOI] [PubMed] [Google Scholar]
- WEBSTER R., LEISHMAN D., WALKER D. Towards a drug concentration effect relationship for QT prolongation and torsade de pointes. Initiation of antiarrhythmic drug therapy for patients with supraventricular tachycardia. Curr. Opin. Drug Disc. 2002;5:116–126. [PubMed] [Google Scholar]