

Abstract
Quinacrine was reported to have a marked in vitro antiprion action in mouse neuroblastoma cells. On compassionate grounds, quinacrine was administered to Creutzfeldt–Jakob disease patients, despite the absence of preclinical in vivo studies to evaluate efficacy. Quinacrine failed to provide therapeutic benefit. The aim of the study was to investigate possible pharmacokinetic and/or pharmacodynamic explanations for the discrepancy between the proven action of quinacrine in vitro and its lack of clinical efficacy.
We conducted in vitro experiments reproducing the culture conditions in which antiprion effects had been previously observed and recalculated the EC50 by determining the actual extracellular (120 nM) and intracellular (6713 nM) quinacrine neuroblastoma concentrations with the reported quinacrine EC50 (300 nM).
A randomized clinical trial in scrapie-affected ewes confirmed the absence of therapeutic benefit of quinacrine. The in vivo quinacrine exposure was evaluated in a pharmacokinetic investigation in healthy ewes. Cerebrospinal fluid concentrations (<10.6 and 55 nM after administration of therapeutic and toxic quinacrine doses, respectively) were much lower than the quinacrine extracellular neuroblastoma concentrations corresponding to the reported EC50. The total brain tissue concentrations (3556 nM) obtained after a repeated therapeutic dosage regimen were within the range of the intracellular neuroblastoma quinacrine concentrations.
In conclusion, in order to avoid in vivo trials for which failure can be predicted, the measurement in vitro of the antiprion EC50 in both intra- and extracellular biophases should be determined. It can then be established if these in vitro antiprion concentrations are achievable in vivo.
Keywords: Prion, therapeutic, quinacrine, pharmacokinetics, neuroblastoma, Creutzfeldt–Jakob disease (CJD), scrapie
Introduction
Prion accumulation is considered to be a key event in the pathophysiology of Creutzfeldt–Jakob disease (CJD) (Bruce et al., 1989; Jendroska et al., 1991), and cellular models for prion disorders have been used to screen the efficacy of many inhibitors of abnormal form of the PrPC protein (PrPSc) formation. Thus, a 6-day treatment with the anti-malarial drug quinacrine or with a phenothiazide derivative chlorpromazine cured a mouse neuroblastoma cell line (ScN2a) that was chronically infected with prions (Doh-Ura et al., 2000; Korth et al., 2001). The median effective concentration (‘EC50') for quinacrine and chlorpromazine (i.e. the nominal concentration of the test solution producing half-maximal inhibition of PrPSc formation) reported in this study were 300 nM (142 ng ml−1) and 3000 nM, respectively. Based on a review of the literature, it was anticipated that this effective in vitro concentration of quinacrine would be attainable in the in vivo biophase (not precisely identified at present). However, to date all patients with CJD treated with quinacrine at the dosage usually recommended in man for other conditions (approximately 300 mg per day) (Shannon et al., 1944; Goodman & Gilman, 1960) died without evidence of any retardation in disease progression (Furukawa et al., 2002; Follette, 2003).
The initial hypothesis was that the lack of quinacrine efficacy resulted from an inability to treat patients with a dosage regimen that would produce the same biophase concentration in vivo as in vitro. Scrapie is a naturally occurring disease of sheep, which provides a relevant model for testing this hypothesis, as scrapie is a prion disease for which the pathophysiological mechanisms of infection are likely to be similar to those occurring spontaneously in CJD.
A second hypothesis was that the reported in vitro EC50 (300 nM) was not the actual in vitro biophase EC50. Indeed, the in vitro EC50 for quinacrine reported by Korth et al. (2001) was only the nominal (calculated) concentration of the quinacrine solution added to the culture medium, which enabled half the maximum effect on prion replication in neuroblastoma cell lines, to be obtained with repeated administration in a volume of 4 ml for 2–7 days. It is not established that this culture medium ‘EC50' corresponds to the true EC50 in the in vitro biophase. Indeed, under both in vitro and in vivo conditions, the drug has to diffuse to the biophase and it can be removed and/or trapped by the neuroblastoma cellular organelles. Thus, it cannot be assumed that the concentration in the in vitro biophase is equal to the nominal concentration of the test solution added to the culture to obtain half quinacrine efficacy. Therefore, the in vitro EC50 should be determined experimentally from measured in vitro drug concentrations. This is particularly important when the in vitro effect of interest develops over several days.
The objectives of the present study were: (i) to determine if quinacrine together with chlorpromazine is effective in a controlled clinical trial in a naturally occurring prion disease of sheep; (ii) to recalculate the in vitro EC50 of quinacrine by establishing its disposition when added to the culture medium of N2a cells in vitro at the concentration equivalent to the reported ‘EC50'; (iii) to establish the in vivo exposure to quinacrine after the administration of therapeutic and toxic dosage regimens; and (iv) to compare the quinacrine concentrations in the N2a cell model with those obtained experimentally in the central nervous system (CNS) of ewes treated with quinacrine.
Methods
Animal procedures
All procedures involving animals were performed in accordance with French legal requirements regarding the protection of laboratory animals and with the authorization for animal experimentation no. 001889 of the French Ministry of Agriculture. The ewes used in experiments 1 and 3 were maintained under natural photoperiod conditions and they received daily rations of concentrate. Hay and water were given ad libitum.
Clinical trial
Experiment 1 was designed to evaluate the clinical efficacy of quinacrine in a randomized clinical trial in naturally infected scrapie ewes. The trial was carried out in 23 Manech red-faced ewes with naturally occurring scrapie. The scrapie diagnosis was established from clinical signs of pruritus, behavioural changes, tremor and locomotion incoordination, and confirmed by histopathology on brain samples after necropsy as described previously (Schelcher et al., 1999). The ewes were ranked in order of severity of clinical signs, pairs of ewes with similar clinical signs were formed and, within each pair, individual ewes were randomly allocated to treated or untreated (placebo) groups. At the time of inclusion in the clinical trial, the mean body weights (±s.d.) of 11 treated and 12 untreated scrapie ewes were 41.6±6.4 and 41.7±6.6 kg, respectively. The 11 ewes in the treated group received 150 mg (317 μmol) in toto of quinacrine dihydrochloride and 100 mg (281 μmol) chlorpromazine daily by injection into the gluteal muscle, for 7 days in the case of six ewes and for a nominal period of 30 days (except 1 day per week) for five ewes. The volumes of quinacrine and chlorpromazine solutions intramuscularly (i.m.) administered were 5 and 4 ml, respectively. Chlorpromazine was administered with quinacrine as recommended for human therapy of CJD (Korth et al., 2001). The 12 ewes in the untreated group received the equivalent volume of vehicle. The dosage regimen of quinacrine administered to the ewes was consistent with the dose used in humans (300 mg daily; Shannon et al., 1944). Blood samples were collected by venipuncture (jugular vein) from scrapie-infected ewes once each week during the time course of the disease. These ewes were killed as soon as they manifested clinical signs of irreversible recumbency.
Quinacrine disposition in N2a cells
Experiment 2 was designed: (i) to recalculate a posteriori the EC50 of quinacrine by determining both intra- and extracellular concentrations obtained when quinacrine was added to the culture medium of N2a cells at a nominal concentration equivalent to the in vitro EC50 reported by Korth et al. (2001); and (ii) to determine the relationship between intra- and extracellular quinacrine concentrations when increasing quinacrine amounts were added to the culture medium of N2a cells.
The mouse neuroblastoma cell line (N2a) was stably transfected as previously described (Lehmann & Harris, 1995). Transfected cells were plated at a density of 40,000 cm−2 into 25 cm2 flasks of 5 ml of OptiMEM (GIBCO, BRL, Cergy Pontoise, France) containing 10% foetal calf serum, and penicillin–streptomycin. These culture conditions were almost the same as those previously used (Korth et al., 2001), except that the cells were split at day 4 using 0.05 (w v−1) trypsin-EDTA (GIBCO, BRL) and were not infected with scrapie. The medium was changed every 48 h together with quinacrine, except on the 7th day of culture when the medium was collected after 24 h. The viability of neuroblastoma cells was assessed by counting in satellite flasks. The media from the final 24 or 48 h of culture were stored at −20°C until assayed for quinacrine content. At the end of the treatment period, cells were washed four times with isotonic saline solution, scraped and resuspended in 1 ml of deionized water and stored at −20°C until assayed. Intracellular concentrations of quinacrine were assayed after sonication of cells.
In a first series of in vitro experiments reproducing the culture conditions in which antiprion effects were observed (Korth et al., 2001), the time courses of intra- and extracellular quinacrine concentrations were measured over a period of 2–7 days in the presence of a nominal quinacrine concentration of 300 nM.
In a second set of experiments, the N2a cells were cultured in the presence of different concentrations of quinacrine (from 0 to 850 nM) for 2 days. At the end of the culture period, the quinacrine concentrations in the media and in the cells were determined.
In vivo quinacrine disposition
The objectives in Experiment 3 were: (i) to determine the overall in vivo quinacrine exposure in the animal model to enable comparison to exposure in man, and (ii) to compare the concentrations of quinacrine in the CNS of healthy ewes, treated with either a therapeutic dose or a toxic dose of quinacrine, with quinacrine concentrations that were effective in vitro.
The in vivo disposition of quinacrine was investigated in seven healthy Lacaune ewes, under conditions reproducing the therapeutic dosage regimen of quinacrine in the clinical trial. The ewes received 8 daily (day-0 to -7) i.m. injections of a therapeutic dose of 3 mg kg−1 day−1 of quinacrine. The daily injections were administered in turn in the gluteal, vastus lateris, brachiocephalicus or longissimus dorsi muscles, and in the right or left sides in an 8 × 8 Latin square design. One ewe has to be excluded from the experiment for an unrelated reason.
On day-0 and -7, blood samples were serially collected by direct venipuncture (jugular vein) within the hour preceding quinacrine i.m. administration and 10, 30, 60, 90, and 120 min after quinacrine administration, then at 2-h intervals until 10 h post-administration and finally at 23–24 h post-administration. From day-2 to -6, a blood sample was collected daily within the hour preceding quinacrine i.m. administration to determine trough plasma concentration. At 24 h after the final injection of quinacrine (day-8), cerebrospinal fluid (CSF) was sampled from the cisterna magna of four ewes. These ewes were then euthanized with intravenous (i.v.) pentobarbitone and exsanguinated. The brains were immediately removed and homogenized at 4°C in 15% (w v−1) deionized water and stored at −20°C until assayed.
Jugular venous blood samples were collected from the three remaining ewes daily until day-15, then every 2 or 3 days until day-21 and on day-28, when plasma quinacrine concentrations were no longer detectable. On day-28, CSF was sampled from the cisterna magna (two ewes) or from the lombosacral space (one ewe). The ewes were immediately euthanized and the brains homogenized at 4°C in 30% (w v−1) deionized water and stored at −20°C until assayed.
A second series of experiments was performed using three healthy ewes to determine CNS exposure to quinacrine over a wide range of quinacrine doses. Two ewes received an i.m. injection of a single therapeutic dose of 150 mg in toto of quinacrine and one ewe received a slow 5.4 h i.v. quinacrine infusion of a toxic dose of 2600 mg in toto. This i.v. dose was injected into the right jugular vein. At 24 h after the administration of quinacrine, a blood sample was taken from the left jugular vein via an indwelling catheter and CSF was sampled from the cisterna magna of all three ewes. The ewes were immediately euthanized and the brains homogenized at 4°C in 15% (w v−1) deionized water and stored at −20°C until assayed.
Sampling
Blood samples (5 ml) were collected in heparinized tubes and centrifuged for 10 min at 3000 × g. The plasma was removed and stored at −20°C until assayed. CSF was collected from anaesthetized animals (sodium pentobarbitone, Nesdonal®, Merial, Lyon, France) by puncture of the cisterna magna or the lombosacral space with a 20-gauge needle. A volume of 1–9 ml of CSF was gently withdrawn and centrifuged for 10 min at 1500 × g to remove cells and stored at −20°C until assayed.
Quinacrine assay
Quinacrine concentrations were determined using a validated high-performance liquid chromatography (HPLC) method in which the internal standard and all biological samples were extracted by liquid/liquid extraction. The HPLC apparatus consisted of a pump system equipped with an automatic injector and a variable-wavelength fluorescence monitor. The separation was achieved by reverse phase column (Inertsil ODS3 column, 3 μm, 150 × 4.0 mm2). The column was equilibrated at a flow rate of 0.3 ml min−1, with a mobile phase consisting of methanol : 25 mM citrate buffer (pH=4.0) (60 : 40, v v−1) containing 0.1 mM benzamidine. As far as possible, polypropylene was used for collecting, storing and assaying samples. The adsorption of drug on to materials during the assay was minimized by including a competing hydrophobic molecule, benzamidine in the mobile phase. The fluorescence detector was set at 300 nm (excitation) and 495 nm (emission). The sample volumes used in the assay were 200 μl for the plasma and 100 μl for the other biological samples. Quinacrine was extracted from biological samples with 1 ml (3 ml for the plasma samples) of 1,2 dichloroethane and 100 μl of 0.2 M sodium hydroxide and 100 μl of 277 nM ethodin as internal standard. Dichloroethane extracts of the matrices were evaporated under nitrogen at 40°C and resuspended in 100 μl DL-lactic acid (0.85%) before injection of a volume of 50 μl on to the column. The mean recoveries of quinacrine from the culture media and from the plasma were 85 and 50%, respectively. The quinacrine assay was performed accurately and reproducibly in the range of 21–254 nM. Within- and between-day precision was less than 15%. The validated quantification limit of the assay was 5.3 nM for the plasma and 10.6 nM for the CSF and culture media.
Pharmacokinetic analysis
Both compartmental and statistical moment approaches were used for the pharmacokinetic analysis of quinacrine concentrations, using WinNonlin 4.0 (Pharsight Corporation, Mountain View, CA, U.S.A.). The maximum concentration (Cmax) and time to maximum concentration (Tmax) were determined directly from plasma concentrations obtained after the first quinacrine i.m. administration, for each animal. The area under the curve (AUC0−24 h) for plasma quinacrine concentrations was calculated from t=0 to 24 h, after the first and eighth quinacrine administrations using the arithmetic trapezoidal rule. The AUC0–inf and the area under the first moment curve (AUMC) for quinacrine plasma concentrations after the first administration were calculated using the linear trapezoidal rule with extrapolation to infinity. The mean residence time (MRT, h) of quinacrine for a single administration was calculated using the equation
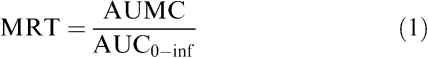 |
where AUMC is the area under the moment curve observed after i.m. administration of quinacrine.
The plasma concentration versus time curves after the eight i.m. quinacrine injections at the dose rate of 3 mg kg−1 day−1 were fitted with a polyexponential equation to assess possible dose and time dependencies of quinacrine disposition. The parameters were estimated by nonlinear regression. The number of exponents was determined by application of the Akaike's Information Criterion (Yamaoka et al., 1978). The data points were weighted with the inverse of the squared fitted value. The goodness of fit of the described model was assessed using least-squares criteria. A triexponential equation describing a bicompartmental open model with first-order absorption was selected
where C(t) represents the plasma quinacrine concentration at time t; Yi (nM) the coefficient of the ith exponential term; λ1 and λ2 the first-order rate constants of the initial and terminal phases; and K01(h−1) the first-order absorption rate constant.
The plasma half-life for the terminal phase was calculated using the equation
 |
with λ2 as defined above.
The apparent steady-state volume of distribution (Vss/F) was obtained with equation (4)
 |
where F is the relative bioavailability and Vc/F is the apparent volume of the central compartment, K12 is the first-order rate constant between central and peripheral compartments and K21 is the first-order rate constant between peripheral and central compartments.
The apparent plasma clearance (Cl/F) was obtained with equation (5)
with Vc/F as defined above and K10 the rate constant of quinacrine elimination.
Materials
Drug solutions were freshly prepared for in vitro and in vivo use. Quinacrine dihydrochloride (molecular weight: 472.9 Da), chlorpromazine–HCl, ethodin, benzamidine and 1,2 dichlororethane were obtained from Sigma-Aldrich (Saint Quentin Fallavier, France). Quinacrine was dissolved in saline to produce a concentration of 63.3 mM, except for the seven ewes in Experiment 3 for which quinacrine was dissolved in saline and dimethyl sulphoxide (50 : 50, v v−1). Chlorpromazine-HCl was dissolved at the concentration of 70.4 mM in vehicle containing 11.4 mM ascorbic acid, 5.3 mM sodium bisulphite, 7.9 mM sodium sulphite, 17.1 mM sodium chloride and 2% benzyl alcohol. Stock solutions of quinacrine were filtered throughout a 0.2 μm syringe filter for in vitro use.
Statistical analysis
Results are reported as mean±s.d. (or median). Statistical analyses were performed using SYSTAT 8.0 (SPSS Inc., Chicago, IL, U.S.A.). In Experiment 1, the median delay between inclusion in the clinical trial and death was determined for both treated and control groups. The percentage of ewes that died from the beginning of treatment was compared for the two treated and untreated groups with the log-rank test for equality of survival (Kaplan–Meier test).
Results
Experiment 1
The median survival time of untreated scrapie-affected control ewes (36 days, range 13–72 days, n=12) did not differ from that of ewes treated for 7 days (45 days, range 9–90 days, n=6) or for a nominal period of 30 days (22 days, range 6–91 days, n=5, Figure 1). Plasma quinacrine concentrations of the five treated diseased ewes varied from 47 to 721 nM (22–341 ng ml−1) 7 or 9 days after the beginning of the 7-day quinacrine treatment.
Figure 1.

Survival curves for scrapie-infected ewes (days after commencing a combined quinacrine and chlorpromazine treatment). There was no significant difference in survival between the two groups (11 treated ewes versus 12 untreated ewes).
Experiment 2
The time course of intra- and extracellular quinacrine concentrations was determined during a 2- to 7-day period of N2a cell culture in the absence and presence of a nominal quinacrine concentration of 300 nM (Figure 2). A 34±8% loss of quinacrine occurred after the dilution and filtration of a standard quinacrine solution of 21 μM through a 0.2 μm filter and 16±3% loss was due to adsorption to the culture disk. The quinacrine concentrations measured in the culture media remained relatively constant during the period of culture (mean: 120 nM; range: 100–140 nM) and were approximately 60% lower than the nominal concentration of the added solution.
Figure 2.

Distribution of quinacrine between extracellular and intracellular compartments in N2a neuroblastoma cells during the 7-day culture. Cells were cultured in the absence or presence of a nominal quinacrine concentration of 300 nM. At every change of medium, the concentration of quinacrine remaining in the medium and present in cells after 24–48 h of culture was determined. Values represent the mean±s.d. of an experiment performed in duplicate. Note that the scales of quinacrine concentration differ for the cells (left ordinate) and the culture media (right ordinate).
The mean intracellular quinacrine concentrations were calculated from an estimated 50 μl volume of subconfluent cells (i.e. a layer of 25 cm2 and 20 μm depth) to range from 2057 to 6713 nM (973–3175 ng ml−1) during the 7 days of culture. The ratio between the intra- and extracellular concentrations of quinacrine varied between 18 and 58 and tended to remain constant between day-2 and -6 of culture.
In the second in vitro experiment, the measured extracellular and intracellular quinacrine concentrations increased linearly with the added quinacrine concentrations (slope 0.43 and 13.8, for intra- and extracellular concentrations, respectively; R2=0.99, coefficient of determination, Figure 3). For a nominal medium concentration of 300 nM (i.e. the reported EC50), the intra- and extracellular quinacrine concentrations calculated from the regression line were 3761 nM (1779 ng ml−1) and 120 nM (57 ng ml−1), respectively. The ratio between the intra- and extracellular concentrations of quinacrine varied between 25 and 47.
Figure 3.

Distribution of quinacrine between extracellular and intracellular compartments in N2a neuroblastoma cells as a function of the quinacrine concentration, added to the culture medium. The N2a cells were cultured in the presence of seven concentrations of quinacrine (from 0 to 850 nM) for 2 days. At the end of the culture period, quinacrine concentration in the media and cells was determined. Values represent the mean±s.d. of an experiment performed in triplicate. Note that the scales of quinacrine concentration differ for the cells (left ordinate) and the culture media (right ordinate).
Experiment 3
The mean plasma quinacrine concentration versus time profile after the first i.m. quinacrine injection at the dose rate of 3 mg kg−1 is presented in Figure 4. The mean (±s.d.) quinacrine AUC0−24 h after the first i.m. injection was 898±593 nM h (425±280 ng ml−1 h), giving an average plasma quinacrine concentration of 37.4±24.7 nM (17.7±11.7 ng ml−1) over the first 24 h. The quinacrine MRT for a single dose was 13.43±5.90 h. The mean plasma maximum quinacrine concentration was 189±152 nM (89±72 ng ml−1) and mean time to maximal plasma concentration was 0.79±0.39 h.
Figure 4.

Semilogarithmic plot of mean plasma quinacrine concentration (nM) versus time (h) after a single i.m. quinacrine injection at the dose rate of 3 mg kg−1 in seven ewes.
After the eighth quinacrine administration, the mean (±s.d.) AUC0–24 h was 2958±1918 nM h (1399±907 ng ml−1 h), giving an average plasma quinacrine concentration of 123±80 nM (58±38 ng ml−1) and indicating an accumulation ratio of approximately 3 between the first and the eighth injections.
Figure 5 illustrates the time courses of observed and fitted plasma quinacrine concentrations for a representative healthy ewe for an 8-day i.m. quinacrine treatment of 3 mg kg−1 day−1. Visual inspection of Figure 5 indicates that plasma quinacrine concentrations increased almost linearly from day-0 to -6 and tended to remain constant from day-6 to -8. Table 1 presents the mean values of the quinacrine pharmacokinetic parameters as obtained by fitting the plasma quinacrine concentrations after an 8-day i.m. quinacrine treatment. After the final injection, the plasma concentration decreased, with a terminal half-life (mean±s.d.) of 52±11 h, to a value similar to the quantification limit of the assay at day-16. The apparent quinacrine clearance (Cl/F) was 3.04±0.70 l h−1 kg–1. The apparent steady-state volume of distribution (Vss/F) was 185±42 l kg–1.
Figure 5.

Observed (point) and fitted (line) plasma quinacrine concentrations (nM) in a representative ewe during 8 days of i.m. quinacrine administration at the dose rate of 3 mg kg−1 day−1 and for 5 further days after termination of dosing. Horizontal bar indicates the period of the quinacrine treatment. The good fitting supports the linearity (dose and time) of quinacrine disposition in our experimental conditions.
Table 1.
Pharmacokinetic parameters (mean±s.d.) describing the plasma disposition of quinacrine after an 8-day i.m. quinacrine treatment at the dose of 3 mg kg−1 day−1 in three ewes
| Parameters (units) | Mean±s.d. |
|---|---|
| K10 (h−1) | 0.069±0.021 |
| K12 (h−1) | 0.216±0.079 |
| K21 (h−1) | 0.069±0.012 |
| K01 (h−1) | 10.9±6.9 |
| t1/2 (h) | 52±11 |
| Vss/F (l kg−1) | 185±42 |
| Cl/F (l kg−1 h−1) | 3.04±0.70 |
K01 is the apparent absorption rate constant; K10 is the rate constant of quinacrine elimination; K12 is the first-order rate constant between central and peripheral compartments; and K21 is the first-order rate constant between peripheral and central compartments. t1/2 is the plasma half-life. Cl/F is the apparent clearance and Vss/F is the apparent steady-state volume of distribution.
At 20 days after the 8-day i.m. quinacrine treatment, quinacrine concentrations in the plasma, brain tissue and CSF were below the limit of quantification of the assay, except in the nervous tissue of one ewe, for which the value obtained was 63 nM.
Quinacrine concentrations were much higher in the nervous system than in plasma and CSF (Table 2). The ratios of the mean brain tissue concentration to mean plasma quinacrine concentration were 76, 53 and 24, 24 h after a single therapeutic quinacrine dose, repeated therapeutic doses and a toxic dose, respectively. The brain tissue/CSF ratio was 979 after the toxic dose of quinacrine and more than 335 after the chronic therapeutic dosage regimen.
Table 2.
Quinacrine concentrations (nM, mean±s.d.) in plasma, CSF and brain tissue in ewes for different conditions
| Quinacrine concentrations (nM) | ||||
|---|---|---|---|---|
| Dosage regimen | Delay after the (last) dose | Plasma | CSF | Brain |
| 150 mg i.m. in toto (n=2) | 24 h | 13.7±4.5 | <10.6a | 1043±199 |
| 2600 mg i.v. infusion (n=1) | 24 h | 2233 | 55 | 53 810 |
| 3 mg kg−1 i.m. daily for 8 days (n=4) | 24 h | 66.6±46.7 | <10.6a | 3556±965 |
| 3 mg kg−1 i.m. daily for 8 days (n=3) | 28 days | <5.3a | <10.6a | <35a (n=2) and 63 nM for one ewe |
Below the level of quantification of the assay.
Discussion
The results of this investigation failed to show any therapeutic benefit of a combined quinacrine and chlorpromazine treatment regimen in a controlled clinical trial in naturally infected scrapie ewes. Using 12 animals per arm, the power of this trial was theoretically sufficient to demonstrate a significant decrease in mortality rate of 24% for a unilateral risk of 5% and a power of 80%. This is satisfactory for this type of exploratory trial designed to demonstrate the existence or not of drug efficacy. This negative result is consistent with observations made in a model of mouse-adapted CJD (Collins et al., 2002) and in humans (Furukawa et al., 2002; Follette, 2003). However, the two mortality curves in our investigation were not parallel. Therefore, it cannot be totally excluded that treatment accelerated the death in the most affected ewes, while slowing disease progression in the less affected ewes.
In order to avoid the generation of debatable data with the in vitro test system, a second aim of our study was to determine why an antiprion drug (quinacrine) that was active in vitro was not clinically effective when administered to human patients and sheep. It was hypothesized that one explanation for the lack of clinical efficacy is the impossibility of treating patients with a quinacrine dosage regimen that enables similar quinacrine biophase concentrations to be achieved in vivo as in vitro. A second possible explanation is that while the reported quinacrine in vitro EC50 (300 nM) is the medium concentration required to obtain an effect on a neuroblastoma test system, this may not be the concentration required in the target biophase in vivo for clinical efficacy. The in vitro quinacrine disposition study showed first that the nonspecific quinacrine loss was approximately 50%, in agreement with the potential of quinacrine to be adsorbed on to surfaces (Björkman & Elisson, 1987). Secondly, the study demonstrated that quinacrine was relatively stable over 7 days of N2a cell culture. In addition, it is estimated that approximately 25% of the quinacrine loss is attributable to uptake of drug by the cells.
In the presence of a nominal concentration of quinacrine equivalent to the reported in vitro quinacrine EC50 (300 nM), the intracellular quinacrine concentrations (2057–6713 nM) attained values that were approximately 30 to 50 times higher than the extracellular levels (100–140 nM). This ratio is not very different to that which could be predicted from the pH partitioning hypothesis. Indeed, considering that quinacrine is a weak base with pKa=10.4 and assuming that the pH values of the culture medium, the neuroblastoma cytosol and the intralysosomal space are 7.4, 7.2 and 5, respectively, and that the lysosomial space represents about 5% of the cell volume, the predicted theoretical ratio of quinacrine concentrations between the intra- and extracellular spaces was about 13. Thus, the present experiment suggests that the antiprion in vitro EC50 may lie between 2000 and 7000 nM if the biophase is intracellular, but be only 120 nM if the biophase is extracellular. This extracellular concentration is almost identical to the free quinacrine concentration, as the binding to protein is very limited in the culture medium, whereas the intracellular concentration like the total tissue concentration comprises both free and bound quinacrine.
The effective quinacrine concentrations in the neuroblastoma cell model were compared to those obtained in the two putative biophases of the CNS, that is the brain tissue (representative of an intracellular biophase) and CSF (representative of an extracellular biophase) (De Lange & Danhof, 2002). The comparison was made under similar conditions to those prevailing in the sheep clinical trial. When quinacrine was administered at the recommended therapeutic dosage regimen (3 mg kg−1 day−1), plasma quinacrine concentrations increased progressively and attained an apparent steady-state level of approximately 120 nM after the eighth quinacrine administration. This is consistent with a quinacrine terminal half-life of 52 h. These results indicated that the ewes had been appropriately exposed during the clinical trial, despite considerable interindividual variability. Moreover, our data are consistent with findings in man. In humans, the recommended dosage regimen (800 mg per os the first day, then 100 mg three times daily) produces a similar quinacrine exposure, with plasma quinacrine concentrations in the range of 100–200 nM (Shannon et al., 1944; Nakajima et al., 2004). In addition, in sheep, the data indicate that the blood/plasma ratio of quinacrine concentrations was approximately 1.15 (unpublished observations) demonstrating that quinacrine is poorly accumulated in red blood cells as previously reported by Shannon et al. (1944).
Despite the variability in exposure to quinacrine, after both a single therapeutic and a toxic quinacrine dose, the relationship between the plasma and brain tissue quinacrine concentrations remained similar (range of brain tissue to plasma quinacrine concentrations ratio of 24–76). This confirms that plasma concentration is the driving concentration influencing drug distribution to and accumulation in the tissue compartment (Gibaldi & Perrier, 1982). Another fundamental tenet in pharmacokinetics is that it is the free drug concentration (and not the total drug concentration) which is the driving concentration for distribution. Therefore, it is the free drug plasma concentration which should be taken into account when considering drug efficacy and also when comparing in vitro and in vivo conditions.
The transport of (free) quinacrine across the blood–brain barrier was shown to involve both an influx system (organic cation transporter-like machinery) and an efflux system via the P-gp, which might restrict the entry of quinacrine into the brain (Dohgu et al., 2004). In the present experiment, the free drug concentration in vivo was not directly measured but can be estimated from the plasma/CSF concentrations ratio. Indeed, the CSF is an ultrafiltrate of plasma, virtually devoid of plasma protein and the drug concentration in the CSF represents the maximal value of the plasma free drug concentrations. The CSF quinacrine concentration was lower than the level of quantification of the analytical technique (10.6 nM) 24 h after repeated i.m. administration of 3 mg kg−1 day−1 of quinacrine, suggesting that the free quinacrine concentration was also less than 10.6 nM. After administration of a toxic quinacrine dose, quinacrine in CSF was measurable (55 nM) and the estimated plasma (total) to CSF (free) concentration ratio was 40, suggesting that the free quinacrine fraction in the plasma was greater than or equal to 2.5%. This latter value is of the same order as that reported for the free fraction in human plasma (Shannon et al., 1944; Goodman & Gilman, 1960), suggesting that the quinacrine concentrations in the CSF are similar to or slightly lower than the plasma free quinacrine concentrations. Considering this free fraction (2.5%), the estimated free plasma quinacrine concentration after a therapeutic dose of quinacrine (3 mg kg−1 day−1) for 8 days ranges from 0.5 to 3 nM, that is, much less than the reported in vitro EC50 (120 nM). Hence, if the biophase for antiprion activity is extracellular, that is, if the CSF quinacrine concentration is the relevant active quinacrine concentration, the current quinacrine dosage regimen will be wholly unable to achieve an in vivo therapeutic antiprion concentration.
On the other hand, it has been suggested that quinacrine interacts with the prion protein, PrPC within the endolysosomes (Doh-Ura et al., 2000), and that this interaction could prevent its conversion into the pathogenic form, PrPSc, in the endocytic pathway (Vogtherr et al., 2003). In this investigation, the total quinacrine concentration in the brain tissue was much higher (about 1000-fold) than the estimated CSF quinacrine concentrations (0.5–3 nM), attaining a concentration of 3556 nM after an 8-day treatment, that is, a tissue/CSF ratio much greater than the ratio of 13 predicted solely from equilibrium pH–pKa partition considerations, and assuming that the pH of CSF is equivalent to that of the culture medium (7.4). This finding is consistent with tissue trapping of the drug (Dubin et al., 1982) and with previous reports that quinacrine is concentrated in tissues, with only low concentrations in the CSF (Shannon et al., 1944). Lysosomal trapping of quinacrine (O'Neill et al., 1998) might account for most of the concentration of this drug in nervous tissue, but the extensive in vivo uptake of quinacrine by cells may also involve binding to other cell organelles or macromolecules. The lysosomes might be a privileged site of action for quinacrine, but this possibility does not exclude a plasma membrane biophase (Shyng et al., 1993).
Assuming that there is a lysosomal biophase, the total neuroblastoma and total brain tissue concentrations may be considered as relevant in relation to efficacy. As the total brain quinacrine concentration was of the same order as the in vitro measured EC50 (2000–7000 nM), attaining 3556 nM after a therapeutic treatment of quinacrine (3 mg kg−1 day−1), it can be reasonably assumed that the present dosage regimen sufficed to achieve an appropriate in vivo lysosomal quinacrine concentration. Consequently, it is likely that the lack of in vivo quinacrine efficacy in the clinical trial was of pharmacodynamic rather than pharmacokinetic origin. In agreement with this hypothesis, Barret et al. (2003) demonstrated that quinacrine could interact with PrPC to inhibit PrPSc formation in ScN2a cells, but was unable to affect the protease resistance of pre-existing PrPSc from brain homogenates of BSE-infected mice.
In conclusion, if the quinacrine biophase is located in the extracellular compartment (or intracellularly in the cytosol), the range of measured quinacrine concentrations required to obtain a 50% efficacy level in the neuroblastoma will clearly never be achieved in vivo, even with a toxic dose of quinacrine. In contrast, if the in vivo quinacrine biophase is lysosomal, appropriate quinacrine exposure can be achieved with a currently recommended therapeutic dosage regimen. In this circumstance, the lack of quinacrine clinical efficacy suggests that the in vitro quinacrine action of recovery of the neuroblastoma is not a relevant in vivo mechanism of action to obtain clinical improvement in treated patients.
Finally, from these experiments it can be recommended that in future investigations of putative antiprion drugs, the in vitro drug potency should be determined by measuring the actual in vitro drug concentration in the potential biophases. It should not be assumed that the in vitro biophase concentration is equal to the nominal concentration in the culture system. In vivo pharmacokinetic investigations are also required to predict whether a dosage regimen that has both antiprion effect and is safe will achieve an appropriate in vivo concentration in the possible biophases.
Acknowledgments
We thank S. Lehmann for providing the N2a cell clone, A. Chabert and G. Costes for technical assistance and P. Lees for comments on the manuscript. This study was supported by grants from the French National Institute for Agricultural Research (INRA), from GIS prion and from the General Direction for Education and Research (DGER) of the French Ministry of Agriculture.
Abbreviations
- AUC
area under the curve
- CJD
Creutzfeldt–Jakob disease
- Cl/F
apparent plasma clearance
- Cmax
maximum concentration
- MRT
mean residence time
- PrP
prion protein
- PrPC
normal cellular prion protein
- PrPSc
abnormal form of the PrPC protein
- ScN2a
scrapie-infected neuroblastoma
- Tmax
time to maximum concentration
- Vss/F
apparent steady-state volume of distribution
References
- BARRET A., TAGLIAVINI F., FORLONI G., BATE C., SALMONA M., COLOMBO L., DE LUIGI A., LIMIDO L., SUARDI L., ROSSI G., AUVRÉ F., ADJOU K.T., SALÈS N., WILLIAMS A., LASMÉZAS C., DESLYS J.P. Evaluation of quinacrine treatment for prion diseases. J. Virol. 2003;77:8462–8469. doi: 10.1128/JVI.77.15.8462-8469.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BJÖRKMAN S., ELISSON L.O. Determination of quinacrine (mepacrine) in plasma by high-performance liquid chromatography with fluorimetric detection. J. Chromatogr. 1987;420:341–348. doi: 10.1016/0378-4347(87)80189-5. [DOI] [PubMed] [Google Scholar]
- BRUCE M.E., MCBRIDE P.A., FARQUHAR C.F. Precise targeting of the pathology of the sialoglycoprotein, PrP, and vacuolar degeneration in mouse scrapie. Neurosci. Lett. 1989;102:1–6. doi: 10.1016/0304-3940(89)90298-x. [DOI] [PubMed] [Google Scholar]
- COLLINS S.J., LEWIS V., BRAZIER M., HILL A.F., FLETCHER A., MASTERS C.L. Quinacrine does not prolong survival in a murine Creutzfeldt–Jakob disease model. Ann. Neurol. 2002;52:503–506. doi: 10.1002/ana.10336. [DOI] [PubMed] [Google Scholar]
- DE LANGE E.C.M., DANHOF M. Considerations in the use of cerebrospinal fluid pharmacokinetics to predict brain target concentrations in the clinical setting: implications of the barriers between blood and brain. Clin. Pharmacokinet. 2002;41:691–703. doi: 10.2165/00003088-200241100-00001. [DOI] [PubMed] [Google Scholar]
- DOHGU S., YAMAUCHI A., TAKATA F., SAWADA Y., HIGUCHI S., NAITO M., TSURUO T., SHIRABE S., NIWA M., KATAMINE S., KATAOKA Y. Uptake and efflux of quinacrine, a candidate for the treatment of prion diseases, at the blood–brain barrier. Cell. Mol. Neurobiol. 2004;24:205–217. doi: 10.1023/B:CEMN.0000018617.21378.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOH-URA K., IWAKI T., CAUGHEY B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J. Virol. 2000;74:4894–4897. doi: 10.1128/jvi.74.10.4894-4897.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DUBIN N.H., BLAKE D.A., DIBLASI M.C., PARMLEY T.H., KING T.M. Pharmacokinetic studies on quinacrine following intrauterine administration to cynomolgus monkeys. Fertil. Steril. 1982;38:735–740. doi: 10.1016/s0015-0282(16)46703-8. [DOI] [PubMed] [Google Scholar]
- FOLLETTE P. New perspectives for prion therapeutics meeting. Prion disease treatment's early promise unravels. Science. 2003;299:191–192. doi: 10.1126/science.299.5604.191. [DOI] [PubMed] [Google Scholar]
- FURUKAWA H., TAKAHASHI M., NAKAJIMA M., YAMADA T. Prospects of the therapeutic approaches to Creutzfeldt–Jakob disease: a clinical trial of antimalarial, quinacrine. Nippon Rinsho. 2002;60:1649–1657. [PubMed] [Google Scholar]
- GIBALDI M., PERRIER D. Pharmacokinetics 1982New York: Marcel Dekker Inc; 2nd edn [Google Scholar]
- GOODMAN L.S., GILMAN A.Quinacrine The Pharmacological Basis of Therapeutics 1960New York: Macmillan; 1167–1173.2nd edn, ed. Goodman, L.S. & Gilman, A. pp. [Google Scholar]
- JENDROSKA K., HEINZEL F.P., TORCHIA M., STOWRING L., KRETZSCHMAR H.A., KON A., STERN A., PRUSINER S.B., DEARMOND S.J. Proteinase-resistant prion protein accumulation in Syrian hamster brain correlates with regional pathology and scrapie infectivity. Neurology. 1991;41:1482–1490. doi: 10.1212/wnl.41.9.1482. [DOI] [PubMed] [Google Scholar]
- KORTH C., MAY B.C.H., COHEN F.E., PRUSINER S.B. Acridine and phenothiazine derivatives as pharmacotherapeutics for prion disease. Proc. Natl. Acad. Sci. U.S.A. 2001;98:9836–9841. doi: 10.1073/pnas.161274798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEHMANN S., HARRIS D. A mutant prion protein displays an aberrant membrane association when expressed in cultured cells. J. Biol. Chem. 1995;270:24589–24597. doi: 10.1074/jbc.270.41.24589. [DOI] [PubMed] [Google Scholar]
- NAKAJIMA M., YAMADA T., KUSUHARA T., FURUKAWA H., TAKAHASHI M., YAMAUCHI A., KATAOKA Y. Results of quinacrine administration to patients with Creutzfeldt–Jakob disease. Dement. Geriatr. Cogn. Disord. 2004;17:158–163. doi: 10.1159/000076350. [DOI] [PubMed] [Google Scholar]
- O'NEILL P.M., BRAY P.G., HAWLEY S.R., WARD S.A., PARK B.K. 4-Aminoquinolines – past, present, and future: a chemical perspective. Pharmacol. Ther. 1998;77:29–58. doi: 10.1016/s0163-7258(97)00084-3. [DOI] [PubMed] [Google Scholar]
- SCHELCHER F., PICARD-HAGEN N., LAROUTE V., GAYRARD V., POPOT M.A., ANDRÉOLETTI O., TOUTAIN P.L. Corticoid concentrations are increased in the plasma and urine of ewes with naturally occurring scrapie. Endocrinology. 1999;140:2422–2425. doi: 10.1210/endo.140.5.6896. [DOI] [PubMed] [Google Scholar]
- SHANNON J.A., EARLE D.P., BRODIE B.B., TAGGART J.V., BERLINER R.W. The pharmacological basis for the rational use of atabrine in the treatment of malaria. J. Pharmacol. Exp. Ther. 1944;81:307–330. [Google Scholar]
- SHYNG S.L., HUBER M.T., HARRIS D.A. A prion protein cycles between the cell surface and an endocytic compartment in cultured neuroblastoma cells. J. Biol. Chem. 1993;268:15922–15928. [PubMed] [Google Scholar]
- VOGTHERR M., GRIMME S., ELSHORST B., JACOBS D.M., FIEBIG K., GRIESINGER C., ZAHN R. Antimalarial drug quinacrine binds to the C-terminal helix of cellular prion protein. J. Med. Chem. 2003;46:3563–3564. doi: 10.1021/jm034093h. [DOI] [PubMed] [Google Scholar]
- YAMAOKA K., NAKAGAWA T., UNO T. Application of Akaike's information criterion (AIC) in the evaluation of linear pharmacokinetic equations. J. Pharmacokinet. Biopharm. 1978;6:165–175. doi: 10.1007/BF01117450. [DOI] [PubMed] [Google Scholar]