

Abstract
Prostaglandin E2 produced endothelium-independent relaxation of phenylephrine- and 5-HT-contracted piglet saphenous vein (PSV; pEC50=8.6±0.2; n=6).
The prostanoid EP4 receptor antagonist GW627368X (30–300 nM) produced parallel rightward displacement of PGE2 concentration–effect (E/[A]) curves (pKb=9.2±0.2; slope=1). Higher concentrations of GW627368X did not produce further rightward shifts, revealing the presence of non-EP4 prostanoid receptors.
In all, 18 other prostanoid receptor agonists relaxed PSV in a concentration-related manner. Relative potencies of agonists most sensitive to 10 μM GW627368X (and therefore predominantly activating EP4 receptors) correlated well with those at human recombinant EP4 receptors in human embryonic kidney (HEK-293) cells (r2=0.74).
In the presence of 10 μM GW627368X, the rank order of agonist relative potency matched that of the human recombinant EP2 receptor in Chinese hamster ovary cells (r2=0.72).
Iloprost, cicaprost and PGI2 relaxed PSV maximally and were antagonised by 10 μM GW627368X, demonstrating that they were full EP4 receptor agonists. Residual responses to these compounds in the presence of GW627368X suggested the presence of IP receptors.
BW245C relaxed PSV maximally (pEC50=6.8±0.1). In the presence of 10 μM GW627368X, BW245C produced biphasic E/[A] curves (phase one pEC50=6.6; α=24%; phase two pEC50=5.1; α=112%). Phase two was antagonised by the DP receptor antagonist BW A868C (1 μM), demonstrating that BW245C is an agonist at DP and EP4 receptors.
We conclude that PSV contains EP4, EP2, DP and IP receptors; IP receptor agonists are also porcine EP4 receptor agonists.
Keywords: Saphenous, venous pharmacology, prostanoid, receptor, EP2, EP4, DP, IP, GW627368X, EP4 antagonist
Introduction
Prostanoids are a group of lipid hormone mediators that are derived from C-20 fatty acids by the action of cyclooxygenases 1 and 2. They consist of the prostaglandins (PG) and the thromboxanes (Tx) and elicit a wide variety of biological responses through activation of G-protein-coupled receptors. The prostanoid receptor family consists of eight distinct rhodopsin-like receptor proteins, each being the product of an individual gene (Coleman et al., 1994a, 1994b; Narumiya et al., 1999). These have been termed the prostanoid DP, EP1, EP2, EP3, EP4, FP, IP and TP receptors, based on the natural prostanoid that displays the highest potency at each receptor. Thus, prostaglandin E2 (PGE2) is the prostaglandin that displays the highest potency at receptors of the EP type. With the recent identification of prostaglandin D2 as a potent agonist at the CRTH2 (chemottractant receptor homologous molecule of TH2 cells) receptor (Hirai et al., 2001), the total number of prostanoid receptor subtypes is now nine.
The prostanoid receptors may be grouped according to the G-proteins to which they preferentially couple. Receptors normally associated with smooth muscle relaxation (DP, EP2, EP4 and IP) couple via Gs or, in the case of DP, by some poorly defined mechanism, to elevation of intracellular cyclic adenosine monophosphate (cAMP) levels. EP3, FP and TP couple via both Gi and Gq to either reduce intracellular cAMP or elevate Ca2+. EP1 couples via an as yet unidentified G-protein to raise intracellular Ca2+.
Regulation of venous and arterial smooth muscle tone is a major role for prostanoid receptors across all mammalian species. Vasorelaxation leading to dilation of arteries and veins is mediated by DP (Giles & Leff, 1988; 1989; Liu et al., 1996), EP2 (Humbles et al., 1991), EP4 (Coleman et al., 1994a; Milne et al., 1995; Lydford et al., 1996a, 1996b; Rouaud et al., 1999; Jones et al., 2000) and IP (Oliva & Nicosia, 1987) receptors, while vasoconstriction is mediated by TP (for a review, see Smith et al., 1980), EP1 (Walch et al., 2001; Li et al., 2004) and EP3 (Qian et al., 1994; Jones et al., 1997) receptors. In almost all vascular preparations studied, a mixed population of relaxatory prostanoid receptors has been observed.
Piglet saphenous vein (PSV) contains prostanoid receptors coupled to smooth muscle relaxation. The identity of these receptors has become a matter of debate because of the lack of potent and truly selective antagonists for EP2 and EP4 receptors (Coleman et al., 1994a; Jones et al., 2000). Accurate identification of the receptor subtypes involved is of relevance to the development of models and techniques in coronary artery bypass graft surgery (Angelini et al., 1990) and to assay development in drug discovery. In order to clarify the identity of the receptors mediating vasorelaxation in PSV, we have used the selective EP4 receptor antagonist GW627368X (Giblin et al., 2002a, 2002b; Wilson et al., 2003; Figure 1; Table 1) to challenge a series of standard prostanoid agonists. The results have been correlated with agonist relative potencies generated in recombinant systems containing single receptor types (Wilson et al., 2004).
Figure 1.

The structure of GW627368X (Giblin et al., 2002a, 2002b; Wilson et al., 2003).
Table 1.
The affinity of GW627368X for cloned human and native porcine prostanoid receptors
| Binding (pKi) | Functional (pKb) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Receptor | hDP | hEP1 | hEP2 | hEP3 | hEP4 | hFP | hIP | hTP | hEP4 | pEP4 |
| Affinity | <5.0 | <5.1 | <5.1 | <5.2 | 7.1 | <5.1 | <5.3 | 6.9 | 7.9 | 9.2 |
Data taken from Giblin et al. (2002a, b) and Wilson et al. (2003).
Methods
Preparation of PSV sections
Large White piglets of either sex (3–6 days old) were obtained from a commercial breeder and humanely killed by a Schedule 1 method in accordance with U.K. legislation. The saphenous vein was removed from each hind leg of the animal and dissected free of connective and other adhering tissue. Where required, de-endothelialisation was achieved by gently passing a length of vein over a roughened 125 μM diameter silver wire five times. Unless otherwise stated, studies were performed using endothelium-intact tissue. Rings of tissue of 5 mm length were suspended between Tungsten wire hooks in 5 ml side-arm bubbling tissue baths for isometric force recording containing Krebs solution at 37°C, aerated with 95% O2+5% CO2. Krebs solution was of the following composition: NaCl 118 mM, NaHCO3 25 mM, KCl 4.8 mM, KH2PO4 1.2 mM, MgSO4 1.2 mM, glucose 11.1 mM, CaCl2 1.25 mM and contained indomethacin 3 μM to block endogenous prostaglandin synthesis and GR32191B 1 μM to block TP receptors. Tissue was initially collected into Krebs solution additionally supplemented with the monoamine oxidase inhibitor pargyline (500 μM), such that a 30 min exposure to this agent was achieved. Changes in force were detected using Grass FT03C force displacement transducers and recorded digitally on a MacLab data acquisition system running Chart v3.4.2 software (sampling frequency 0.66 Hz; AD Instruments, Hastings, U.K.).
Tissue bath experimental procedure
An initial force of 1 g was applied to each tissue ring for a period of 10 min, at the end of which the bathing solution in each bath was replaced. A force of 2 g was then applied for a period of 30 min followed by exposure to 80 mM KCl to establish the maximum level of force generated by each ring. This concentration of KCl had previously been shown to be maximally effective in these tissues (data not shown). Washout of vasoactive agents was achieved by four exchanges of bathing medium, after which basal tone was allowed to re-establish for 10 min prior to the addition of the EP4 receptor antagonist GW627368X or vehicle. In order to study the functional effects of prostanoids at relaxant receptors in whole tissues, tone must first be elevated with a suitable spasmogen. This was achieved by the addition of either 1 μM phenylephrine (PE) which has previously been shown to represent an EC80 concentration of this compound (data not shown), or 1 μM 5-hydroxytryptamine (5-HT). Pre-exposure of PSV to KCl (80 mM) stabilised subsequent contractions in response to PE (1 μM) and gave greater reproducibility (data not shown). Responses to PE or 5-HT were allowed to stabilise such that an overall antagonist incubation time of 60 min elapsed before the construction of agonist E/[A] curves. In order to maximise the number of experiments performed in tissues from each animal, a single agonist concentration–effect (E/[A]) curve was produced in each ring of tissue by the cumulative addition of compound at 0.5 log10 intervals. A representation of the experimental method is shown as Figure 2.
Figure 2.

Typical isometric force recording generated in rings of piglet saphenous vein treated as described in Methods. Substances were added to the bathing solution at the points indicated. Addition of PGE2 resulted in rapid tissue relaxation responses which were stably maintained. Inset: Mean PGE2 concentration–effect curve in rings of PE (1 μM) precontracted PSV. A four-parameter logistic equation was fitted to individual curve data as described in Methods to generate curve parameters as follows: pEC50=8.6±0.2, nH=1.9 (1.5–2.4), α=98±3%. Data are mean±s.d.; data for slope are geometric mean with 95% confidence intervals; n=6.
Data analysis
Data from tissue bath studies were extracted as grams tension and either normalised with respect to the KCl response (contractile responses) or to the PE/5-HT response (relaxant responses). Responses to prostanoid receptor agonists are therefore expressed as % of spasmogen-induced tone. A four-parameter logistic equation of the form
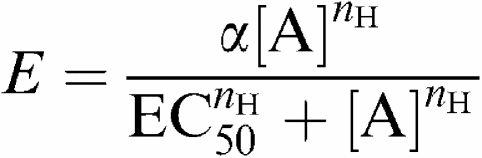 |
was then fitted to the E/[A] curve data in order to estimate maximum effect (α), curve midpoint (EC50) and Hill slope parameter (nH); other terms in the equation are effect (E) and agonist concentration ([A]). Individual estimates of curve parameters were obtained from each curve and then averaged to provide mean data. Quoted values are therefore the mean±standard deviation (s.d.) of n separate experiments, each derived from a separate animal. As errors around slope estimates are log-normally distributed, slope data are expressed as the geometric mean with 95% confidence intervals. Where curve parameters could not be estimated, the mean effect at the maximum concentration tested is quoted.
Agonist relative potencies (RP) were calculated as the ratio of agonist EC50 : PGE2 EC50. Therefore, the RP of PGE2 =1.0; agonists more potent than PGE2 have RP<1.0, while compounds less potent than PGE2 have RP >1.0. Correlation of agonist potencies was performed by calculation of the product-moment correlation coefficient (r2).
Antagonist competition data were analysed using a modified version of the Schild equation (Lew & Angus, 1995) in order to generate an antagonist affinity (pKb) according to the equation
where pEC50 is the agonist E/[A] curve midpoint in control and treated tissues, [B] is the antagonist concentration, n is the ‘Schild slope' and −log c is the difference between the antagonist pKb and the agonist control curve pEC50.
Single agonist E/[A] curves were generated in each tissue; therefore comparisons were made between data generated in different tissues from the same animal. Quoted values are therefore the mean±s.d. of n separate experiments, each derived from a separate animal.
Statistical significance was assessed using Student's t-test, with P<0.05 taken as indicating significance.
Drugs used
Pargyline, indomethacin, PE, Nω-nitro-L-arginine, methyl ester (L-NAME), 5-hydroxytryptamine, hydrochloride salt (5-HT), PGE2, PGI2, PGD2 and PGF2α were purchased from Sigma, Poole, Dorset, U.K. Potassium chloride (KCl; AnalaR grade) was obtained from BDH, Lutterworth, Leics., U.K. PGE1, BW245C ((4S)-(3-[(3R,S)-3-cyclohexyl-3-hydropropyl]-2,5-dioxo)- 4-imidazolidineheptanoic acid), sulprostone (N-(methylsulphonyl)-9-oxo-11α,15R-dihydroxy-16-phenoxy-17,18,19,20-tetranor-prosta-5Z,13E-dien-1-amide), misoprostol (9-oxo-11α,16-dihydroxy-16-methyl-prost-13E-en-1-oic acid, methyl ester), butaprost (9-oxo-11α,16R-dihydroxy-17-cyclobutyl-prost-13E-en-1-oic acid, methyl ester), 17-phenyl trinor PGE2, 11-deoxy PGE1, 16,16-dimethyl PGE2, 19-R-hydroxy PGE2, fluprostenol ((±)-9α,11α,15R-trihydroxy-16-(3-(trifluoromethyl)phenoxy)-17,18,19,20-tetranor-prosta-5Z,13E-dien-1-oic acid) and cloprostenol (9α,11α,15R-trihydroxy-16-(3-(chlorophenoxy))-17,18,19, 20-tetranor-prosta-5Z,13E-dien-1-oic acid, sodium salt) were purchased from Cayman Chemical Company, Ann Arbor, Michigan, U.S.A. Iloprost (6,9α-methylene-11α,15S-dihydroxy-16-methyl-prosta-5E,13E-dien-18-yn-1-oic acid, trometamol salt) was purchased from Amersham, Bucks., U.K. Cicaprost (5-[(E)-(1S, 5S, 6S, 7R)-7-hydroxy-6-[(3S, 4S)-3-hydroxy-4-methylnona-1,6-diinyl]-bicyclo[3.3.0]octan-3-yliden]-3-oxapentanoic acid, ZK96480) was the kind gift of Schering A.G., Berlin, Germany. Butaprost free acid, GR32191B ([1R-[1α(Z),2β,3β,5α]]-(+)-7-[5-([1,1′-biphenyl]-4-ylmethoxy)-3-hydroxy-2-(1-piperidinyl)-cyclo pentyl]-4-heptenoic acid hydrochloride salt), GR63799X ([1R-[1α(Z),2β(R*),3α]-(−)-4-benzoylamino)phenyl-7-[3-hydroxy-3-phenoxy-propoxy)-5-oxocyclopentyl]-4-heptenoate), BWA868C90 (3-benzyl-5-(6-carboxyhexyl)-1-(2-cyclohexyl-2-hydroxyethylamino)-hydantoin) and GW627368X (N-{2-[4-(4,9-diethoxy-1-oxo-1,3-dihydro-2H-benzo[f]isoindol-2-yl)phenyl]-acetyl}benzenesulphonamide) were prepared in the Department of Medicinal Chemistry, GlaxoSmithkline Research and Development, Stevenage, U.K.
Indomethacin, GW627368X and GR32191B were dissolved at 10 mM in dimethyl sulphoxide (DMSO). Iloprost was dissolved at 1 mM in Tris buffer pH 8.3. All other prostanoids were dissolved at 10 mM in 100% ethanol and stored at −20°C. For tissue bath studies, dilutions of drugs and PE were made freshly on each day of study in Krebs solution containing indomethacin and GR32191B as described above. Diluted compounds were stored in the dark at 4°C for the duration of an experiment. Pargyline was dissolved at 0.5 M in dH2O and stored at −20°C. Potassium chloride was dissolved at 4 M in Krebs solution and stored at room temperature.
Results
Effect of spasmogens and acetylcholine (ACh) on PGE2 responses
PGE2 fully relaxed PE-contracted tissues in a well-defined, stable, concentration-related manner (Figure 2; pEC50=8.6±0.2, nH=1.9 (1.5–2.4); α=98.1±3.2%). When 5-hydroxytryptamine was the spasmogen (5-HT; 3 μM), at a concentration which produced tone equivalent in magnitude to that generated by 1 μM PE, similar PGE2 responses were obtained although the potency of PGE2 was significantly reduced (pEC50=7.9±0.6, nH=1.3 (1.0–1.9), α=89.6± 31.3%; P<0.05).
In tissues contracted with PE, ACh (1 μM) elicited transient relaxations (31±24% of PE tone) followed by rapid desensitisation. Ultimately, a small increase in tension of 9±17% relative to the starting PE tone was established. Exposure to ACh significantly reduced the potency of PGE2 (pEC50= 7.7±0.7, nH=1.35 (0.6–4.2), α=100.9±2.0%; P<0.05). Mechanical removal of the endothelium abolished ACh-induced relaxation but revealed small, long-lasting contractile responses to this agent (7.0±6.0% of PE tone). Endothelium removal did not significantly affect PGE2 E/[A] curve potency in ACh-treated tissues (pEC50=7.1±0.8, nH=1.5 (0.6–3.8), α=96.9±6.3%).
EP4 receptor antagonist vs PGE2
The EP4 receptor antagonist, GW627368X (10 μM), did not produce any significant change in basal or PE-elevated tone. However, occasional ‘spikes' of contraction were observed in 36% of antagonist-treated tissues. GW627368X (30–300 nM) produced parallel rightward displacement of PGE2 E/[A] curves. Higher concentrations of antagonist (1–10 μM) did not produce any further displacement (Figure 3, Panel a). Nonlinear regression analysis of the antagonist-sensitive phase yielded a pKb of 9.2±0.2 and a slope of 0.8±0.3. A plot of log(CR-1) vs the negative log of the antagonist concentration (Figure 3, Panel b) reveals two linear phases, clearly demonstrating the presence of EP4 receptor antagonist-resistant responses to PGE2.
Figure 3.

Antagonism of PGE2 concentration–effect (E/[A]) curves in PSV by GW627368X. Panel a: Mean PGE2 E/[A] curves in the presence of vehicle and GW627368X 1, 3, 10, 30, 100, 300 nM, 1, 3, and 10 μM (n=3). Tissues were incubated with antagonist for 60 min prior to exposure to PGE2 as described in Methods. Panel b: Antagonist competition analysis using nonlinear regression as described in Methods. An additional line (shown in dots and dashes) illustrates the antagonist resistant phase of the data (100 nM–10 μM GW627368X).
Prostanoid receptor agonists in the absence and presence of the EP4 receptor antagonist
A panel of 18 prostanoid receptor agonists also relaxed PSV in a concentration-related manner, the results of which are shown in Table 2 and Figure 4. All agonists except fluprostenol produced essentially full relaxation of the tissue. Generally, high potency responses were generated. For example, PGE1 and PGE2 relaxed PSV maximally with EC50 values of 2 and 3 nM, respectively. Without exception, curve slopes were steep (in the range 1.3–3.6) and not significantly different from that for PGE2. Fluprostenol elicited small relaxations (1–35%) at high concentrations of agonist (30 μM). The rank order of potency for agonists in the absence of GW627368X was PGE2=PGE1>11-deoxy PGE1>16,16-dimethyl PGE2= BW245C >misoprostol=17-phenyl-ω-trinor PGE2=19-(R)-hydroxy PGE2>iloprost=cicaprost=PGD2>GR63799X⩾ PGF2α=sulprostone=butaprost ME⩾butaprost FA.
Table 2.
Agonist E/[A] curve data in PSV in the presence and absence of the EP4 receptor antagonist GW627368X (10 μM)
| i.p.o. 10 μM GW627368X | ||||
|---|---|---|---|---|
| Agonist | pEC50 | pEC50 | α | Fold shift |
| PGE1 | 8.7±0.5 | 6.7±0.3 | 104±4 | 100 |
| PGE2 | 8.6±0.2 | 6.9±0.1 | 98±3 | 50 |
| PGD2 | 6.0±0.5 | 5.3±0.3 | 85±7 | 5 |
| PGF2α | 5.7±0.2 | 12±4% relaxation at 30 μM | >10 | |
| PGI2 | 5.6±0.4 | 5.4 (n=2) | 81±15 | 2 |
| Iloprost | 6.0±0.5 | 6.7±0.3 | 12±6 | |
| Cicaprost | 5.9±0.5 | 5.7±0.2 | 75±20 | 2 |
| Sulprostone | 5.8±1.0 | 3±5% relaxation at 30 μM | >10 | |
| Misoprostol | 6.3±0.4 | 5.8±0.1 | 95±12 | 3 |
| GR63799X | 5.8±0.1 | 5.2±0.1 | 32±19 | 4 |
| Fluprostenol | 16±14% relaxation at 30 μM | 5±5% relaxation at 30 μM | ||
| Cloprostenol | 5.4±0.3 | 15±36% relaxation at 30 μM | >3 | |
| Butaprost ME | 5.8±0.2 | 5.4±0.1 | 77±19 | 3 |
| Butaprost FA | 5.6±0.3 | 5.8±0.1 | 82±33 | |
| BW245C | 6.8±0.1 | Biphasic – see text | ||
| 17-phenyl-ω-trinor PGE2 | 6.2±0.4 | 24±11% relaxation at 30 μM | >30 | |
| 11-deoxy-PGE1 | 7.6±0.9 | 5.7±0.2 | 99±4 | 80 |
| 16,16-dimethyl-PGE2 | 7.2±0.6 | 6.2±0.6 | 97±3 | 10 |
| 19-(R)-hydroxy-PGE2 | 6.3±0.1 | 6.0±0.2 | 90±8 | 2 |
Data are mean±s.d., n=3 or 4. No significant differences in agonist maximum effects (α) or Hill slope parameter (nH) were observed in tissues not treated with antagonist.
Figure 4.

Illustrative data showing agonist concentration effect curves in PSV in the absence (closed symbols) and presence (open symbols) of 10 μM GW627368X. Panel a: PGE1, 17-phenyl-ω-trinor PGE2, 11-deoxy PGE1. Panel b: Butaprost methyl ester, butaprost free acid, PGD2. Panel c: Fluprostenol, cloprostenol, PGF2α. Panel d: Iloprost, cicaprost, PGI2.
In order to determine which agonists were eliciting these responses through the EP4 receptor, the same 18 agonists were tested in the presence of 10 μM GW627368X. This resulted in a markedly different rank order of potencies and maximum effects (Table 2 and Figure 4). Many of the agonists were sensitive to GW627368X, but the degree of rightward shift was extremely variable (Table 2). As PGE2, PGE1, 11-deoxy PGE1, 16,16-dimethyl PGE2, BW245C, 17-phenyl trinor PGE2, iloprost and PGF2α E/[A] curve pEC50's were markedly shifted by GW627368X (>3-fold or >50% decrease in maximum response), responses to these agonists in the absence of the antagonist can be assumed to be mediated predominantly by EP4 receptors. Relative potencies for these agonists were correlated with those previously generated at hEP4 receptors expressed in HEK cells (Table 3; Wilson et al., 2004), yielding a correlation coefficient (r2) of 0.74.
Table 3.
Agonist relative potencies in PSV in the absence of the EP4 receptor antagonist GW627368X (10 μM) together with comparative data generated at human recombinant EP4 receptors expressed in HEK 293 (T) cells (Wilson et al., 2004)
| Agonist | PSV | HEK hEP4 |
|---|---|---|
| PGE2 | 1 | 1 |
| PGE1 | 1 | 2 |
| 11-deoxy-PGE1 | 13 | 3 |
| 16,16-dimethyl-PGE2 | 30 | 40 |
| 17-phenyl-ω-trinor PGE2 | 320 | 120 |
| BW245C | 80 | 200 |
| PGF2α | 1000 | 5000 |
| Iloprost | 500 | 10,000 |
| GR63799X | 790 | 200 |
| Misoprostol | 250 | 400 |
| Cicaprost | 630 | 790 |
| PGI2 | 1260 | 2500 |
| 19-(R)-hydroxy-PGE2 | 250 | 3200 |
| PGD2 | 500 | 6300 |
| Sulprostone | 790 | 32,000 |
| Cloprostenol | 2000 | >63,000 |
| Fluprostenol | >63,000 | |
| Butaprost FA | 1260 | >200,000 |
| Butaprost ME | 790 | >2,000,000 |
A subset of compounds (indicated in italics) were shifted by >3-fold by 10 μM GW627368X and are therefore assumed to be acting predominantly through EP4 receptors.
Agonist profiling was also carried out in the presence of 10 μM GW627368X. This concentration of antagonist is 25,000-fold greater than its pKb at EP4 receptors in this tissue and it is therefore expected to effectively block responses mediated by these receptors. Under these conditions, PGE2 produced a maximal relaxation of the tissue and was the most potent prostanoid agonist tested (pEC50=6.9±0.1; Table 4). The rank order of potency of prostanoid agonists generated in the presence of GW627368X (PGE2>PGE1=iloprost>16,16-dimethyl PGE2>19-(R)-hydroxy PGE2>misoprostol=11-deoxy PGE1>butaprost>PGD2>GR63799X>17-phenyl-ω-trinor PGE2=PGF2α=sulprostone) agrees well with the rank order of agonist relative potencies (c.f. PGE2=1) generated at hEP2 receptors expressed in CHO cells (r2=0.72) (Wilson et al., 2004).
Table 4.
Agonist relative potencies in PSV in the presence of the EP4 receptor antagonist GW627368X (10 μM) together with comparative data generated at human recombinant EP2 receptors expressed in CHO K1 cells (Wilson et al., 2004)
| Agonist | PSV i.p.o. 10 μM GW627368X | CHO hEP2 |
|---|---|---|
| PGE2 | 1 | 1 |
| PGE1 | 2 | 1 |
| Butaprost FA | 13 | 2 |
| 16,16-dimethyl-PGE2 | 5 | 3 |
| 19-(R)-hydroxy-PGE2 | 8 | 3 |
| 11-deoxy-PGE1 | 16 | 8 |
| GR63799X | 50 | 10 |
| Misoprostol | 13 | 13 |
| BW245C | 16 | |
| PGF2α | 20 | |
| PGD2 | 40 | 25 |
| Butaprost ME | 32 | >30 |
| 17-phenyl-ω-trinor PGE2 | 63 | |
| Iloprost | 2 | >100 |
| Sulprostone | >100 | |
| PGI2 | 32 | ∼320 |
| Cloprostenol | ∼320 | |
| Cicaprost | 16 | >1000 |
| Fluprostenol | >1000 |
Studies with iloprost
Iloprost behaved as a full agonist, pEC50=6.0±0.5, nH=1.3 (0.3–5.8), α=99±6%) (Figure 5). However, in the presence of 10 μM GW627368X, the maximum response was only 12.3±5.9% relaxation (pEC50=6.7±0.3, nH=3.7 (2.4–5.7). Pre-incubation with both GW627368X (10 μM) and the DP receptor antagonist BWA868C (10 μM) did not alter iloprost responses (GW627368X vs iloprost: pEC50=6.7±0.3, nH=3.7 (2.4–5.7), α=12.3±5.9%; GW627368X+BWA868C vs iloprost: pEC50=6.7±0.1, nH=2.2 (1.4–3.7), α=28.3±40.1%; Figure 5, Panel a).
Figure 5.

Panel a: Iloprost concentration–effect curves in rings of PE (1 μM) precontracted PSV treated with vehicle, GW627368X (10 μM) or GW627368+BWA868C84 (10 and 1 μM, respectively). Under each set of conditions, a maximal response to PGE2 (10 μM) was generated at the end of each iloprost curve. Panel b: PGE2 concentration effect curves in GW627368X (10 μM) treated rings of piglet saphenous vein in the presence or absence of iloprost (10 μM). All data are mean±s.d.; n=3.
In GW627368X (10 μM)-treated tissues, a single 10 μM concentration of iloprost elicited a rapid relaxation of PE induced tone. Ultimately, tissue tone stabilised to 14.2±16.2% relaxation. Under these conditions, no additional rightward shift of PGE2 E/[A] curves was observed (GW627368X vs PGE2: pEC50=6.9±0.1, nH=1.7 (1.6–2.1), α=97.8±3.0%; GW627368X+iloprost vs PGE2: pEC50=7.0±0.8, nH=1.9 (1.6–2.2), α=105.5±15.8%; Figure 5, Panel b).
Studies with BW245C
In control tissues, BW245C produced maximal relaxations and monophasic E/[A] curves (pEC50=6.8±0.1, nH=1.8± (1.4–2.4), α=102.1±3.8%; Figure 6). However, in the presence of 10 μM GW627368X, BW245C produced biphasic response curves: phase one pEC50=6.6; α=24%; phase two pEC50=5.1; α=112%. Addition of the selective DP receptor antagonist BWA868C (1 μM) resulted in a 50-fold rightward displacement of the most potent portion of the biphasic curve (Figure 6).
Figure 6.

BW245C90 concentration–effect curves in rings of PE (1 μM) precontracted PSV treated with vehicle, GW627368X (10 μM) or GW627368+BWA868C84 (10 and 1 μM, respectively). All data are mean±s.d.; n=3 or 4.
In order to detect the presence of non-TP contractile prostanoid receptors, sulprostone, misoprostol, GR63799X and PGE2 were tested for their effects at 1 μM on basal tone. In all cases, no significant effect was observed (Figure 7).
Figure 7.

Changes in basal tissue tension in rings of PSV induced by the indicated prostanoid receptor agonists (10 μM). Data are mean±s.d. and are expressed as % of 80 mM KCl-induced tone.
Discussion
PSV contains EP prostanoid receptors coupled to smooth muscle relaxation (EP2 and/or EP4). Although PGE2 relaxed control tissues with high potency, our data indicate generally lower prostanoid agonist potencies compared with data previously published by Coleman et al. (1994a) and Jones et al. (2000). This probably reflects our use of a higher concentration of PE (approximate EC80). Indeed, substitution of 5-HT for PE which produced larger and more sustained contractions, and pretreatment of both endothelium intact and denuded tissues with ACh which elicited small additional contractions, reduced PGE2 potency. It would then follow that prostanoid-induced relaxations are highly sensitive to functional antagonism in this tissue. However, irrespective of the degree of functional antagonism, agonist rank orders of potency and antagonist affinity values will be constant and so these conditions are suitable for receptor classification purposes (Kenakin, 1993).
Functional antagonism of agonist responses may also arise from activation of contractile prostanoid receptors by agonists. This could confound agonist potency ratios and therefore we sought to exlude this possibility. Inclusion of 1 μM GR32191B (150–600-fold its TP receptor affinity; Lumley et al., 1989) in the bathing medium prevented activation of TP receptors. Others have identified contractile EP3 receptors in certain vascular preparations (Jones et al., 1998), while Walch et al. (2001) have suggested a role for EP1 receptors in mediating contractions of human pulmonary veins, and Li et al. (2004) have identified EP1 receptor mRNA in vascular smooth muscle cells. The lack of contractile effects of misoprostol (EP2/EP3/EP4 agonist), sulprostone (EP1/EP3 agonist) and PGE1 on basal tone suggests that EP1 and EP3 receptors are either absent or represent a minor component of the overall receptor complement in this tissue. Under the right conditions, a synergistic interaction with another agonist might unmask EP1 or EP3 receptor-mediated contractile responses. Typically, synergism of this type in vascular tissues takes place between Gi and Gq coupled contractile receptors such as TP and 5-HT1D receptors (for a review, see Maclennan et al., 1993). In this study, an EC80 concentration of PE, presumably activating Gq coupled α1-adrenoceptors, was used. Under these conditions, Gi-coupled EP3 responses could have been amplified and become measurable. No such synergism was observed. Interestingly, rapidly reversing relaxations were observed with the EP1/IP agonist iloprost. It is tempting to speculate that these represent the effect of an EP1-mediated contractile response. However, no such rapid reversal was seen with 17-phenyl PGE1 or sulprostone (agonists with some selectivity for EP1 receptors) or with other nonselective agonists, such as PGE2 itself. Therefore, the rapid reversal of relaxatory responses to iloprost cannot be easily attributed to an EP1-mediated action. Indeed agonist E/[A] curve slopes for all compounds were generally steep and not indicative of an action at a functionally antagonising receptor. The mechanism leading to the occasional 'spikes' of contractile activity seen in some tissues treated with the EP4 receptor antagonist GW627368X is not clear and may have arisen from a non-prostanoid mechanism, from spontaneous neurotransmitter release, or from some action of a prostanoid on a contractile receptor. The inclusion of 3 μM indomethacin in the bathing medium makes inhibition of endogenously synthesised prostanoid action an unlikely explanation. However, such contractions may arise if GW627368X possessed inverse agonist properties and inhibited a constitutive pro-relaxatory EP4 receptor tone. Our data do not provide any insight into this possibility. Taken together, then, while we have not used antagonists to specifically exclude the presence of EP1 or EP3 receptors in PSV, the weight of evidence suggests that no significant population of them exists. Therefore, characterisation of the relaxant receptors is not likely to have been confounded by opposing contractile activity of the agonists studied.
GW627368X antagonised responses to PGE2, confirming the presence of EP4 receptors in this tissue. However, the profile of PGE2 E/[A] curve displacement by GW627368X clearly indicated the presence of at least two relaxant prostanoid receptors, with the second receptor being apparently insensitive to concentrations of the antagonist as high as 3 μM. Our attention immediately focused on the possibility that the second receptor was of the EP2 type since it was coupled to tissue relaxation and responded to PGE2 with moderate potency (pEC50=6.9±0.1). In order to confirm our hypothesis, we examined the relative potencies of a range of selective and nonselective prostanoid receptor agonists in the absence and presence of 10 μM GW627368X.
Generally, high potency responses were generated by prostanoid agonists in PSV not treated with GW627368X (control tissues). Without exception, curve slopes were steep (in the range 1.3–3.6), suggesting activation of a single receptor type. The rank order of potency for agonists in the absence of GW627368X was PGE2=PGE1>11-deoxy PGE1>16,16-dimethyl PGE2=BW245C>misoprostol=17-phenyl-ω-trinor PGE2=19-(R)-hydroxy PGE2>iloprost=cicaprost=PGD2>GR63799X⩾PGF2α=sulprostone=butaprost methyl ester ⩾butaprost free acid. Responses to most compounds were antagonised by GW627368X, indicating that they were EP4 receptor agonists. Responses to PGE2, PGE1, 11-deoxy PGE1, 16,16-dimethyl PGE2, BW245C, 17-phenyl trinor PGE2, iloprost and PGF2α were markedly diminished or right-shifted by exposure to GW627368X. Therefore, in antagonist-naïve tissues, activation of non-EP4 prostanoid receptors contributes little to the overall responses to these agonists and E/[A] curves can be assumed to be predominantly EP4 receptor mediated. Although the degree of correlation observed between agonist relative potencies generated in PSV with those generated in HEK-EP4 cells is modest (r2=0.54; Wilson et al., 2004), when the set of agonists was restricted to those most sensitive to GW627368X, the degree of correlation increased (r2=0.74).
Agonist profiling in the presence of 10 μM GW627368X confirmed our hypothesis that EP2 prostanoid receptors were present, but revealed further complexity in the pharmacology of PSV. The concentration of antagonist used is 25,000-fold greater than its pKb in this tissue and it is therefore expected to effectively block responses mediated by EP4 receptors. The persistence of relaxatory agonist responses in the presence of EP4 receptor blockade suggests that at least one other non-EP4 relaxatory prostanoid receptor is present in PSV. Above an antagonist concentration of 0.1 μM, no further rightward shift of PGE2 curves could be generated, showing that the receptor(s) responsible are insensitive to GW627368X. In the presence of a saturating concentration of antagonist, PGE2 produced a maximal relaxation of the tissue and was the most potent prostanoid agonist tested (pEC50=6.9±0.1), suggesting that the likely identity of the receptor mediating this response is EP2. The rank order of potency of prostanoid agonists generated in the presence of GW627368X (PGE2>PGE1=iloprost>16,16-dimethyl PGE2>19-(R)-hydroxy PGE2>misoprostol=11-deoxy PGE1>butaprost>PGD2>GR63799X>17-phenyl-ω-trinor PGE2=PGF2α=sulprostone) agrees well with the rank order of potency generated at hEP2 receptors expressed in CHO cells (Wilson et al., 2004). The greater degree of correlation (r2=0.72) observed for these predominantly EP2 receptor-mediated responses reflects the removal of contaminating EP4 receptor-mediated responses by the antagonist.
Before discussing data that point to further complexity in the prostanoid receptor complement of PSV, we wish to highlight aspects of the pharmacology of some of the agonists used. Misoprostol and PGD2 produced maximal relaxation of both control and antagonist-treated tissues and were approximately 0.6 log units more potent in control tissues. Similar separation of potencies was achieved with GR63799X, but this compound was only able to elicit a maximum response of 32±19% in the presence of GW627368X. A combination of EP2 and EP4 receptor agonism could therefore generate relaxation in response to these compounds. However, because their E/[A] curves were steep, it is reasonable to suggest that responses in control tissues were predominantly the result of EP4 receptor activation. Artefactual curve-steepening under the influence of pro-contractile prostanoid receptors is not suspected because agonists for pro-contractile prostanoid receptors were without effect. GR63799X has not previously been noted as an efficacy-driven (partial) agonist at the EP2 receptor. This agonist produced very small agonist responses at recombinant hEP2 receptors expressed in CHO cells (Wilson et al., 2004). Consistent with this observation, it has been noted that it is 1000-fold less potent than PGE2 at the EP2 receptor in cat trachea (Bunce et al., 1991) but only 50-fold less potent than PGE2 in PSV. Potency differences between full and partial agonists can be magnified in well-coupled assay systems. Therefore, leaving species differences aside, these data are consistent with the notion that EP2 receptors in PSV are poorly coupled.
Butaprost methyl ester, butaprost free acid and 19-(R)-hydroxy PGE2 were insensitive to the EP4 receptor antagonist. Therefore, either these compounds are devoid of activity at porcine EP4 receptors or they activate porcine EP2 receptors with greater or equal potency. Butaprost free acid and methyl ester have been shown to be virtually devoid of agonist activity at hEP4 receptors expressed in HEK293(T) cells (Wilson et al., 2004), while 19-(R)-hydroxy PGE2 produced low-potency agonism in this system (relative potency c.f. PGE2=3200). Indeed, butaprost free acid is highly selective for mouse (Kiriyama et al., 1997) and human (Abramowitz et al., 2000; Wilson et al., 2004) EP2 receptors over other prostanoid receptors. The low potency of butaprost methyl ester in rings of PSV has been noted previously (Milne et al., 1995) and was thought to indicate the absence of EP2 receptors from this preparation. Taken together, the present findings suggest that an alternative explanation is the presence of a population of EP2 receptors with poor coupling to smooth muscle relaxation and that these agonists are acting solely via EP2 receptors in PSV.
The IP receptor agonists iloprost, cicaprost and PGI2 (prostacyclin) have all been found to be agonists in HEK-hEP4 cells (Wilson et al., 2004). All three compounds were found to be sensitive to EP4 receptor antagonism, though to varying degrees. The small decreases in maximum response observed for cicaprost and PGI2 suggest that EP4 agonism contributes little to the overall responses to these agents. However, the dramatic alteration of iloprost responses in the presence of GW627368X suggests that this agonist acts mainly through EP4 receptors in PSV. The slopes of iloprost E/[A] curves in the absence of GW627368X were the shallowest of all the compounds tested, suggesting the involvement of more than one receptor in responses to this compound. The small relaxations that persisted in antagonist-treated tissues confirm this view.
The identity of the receptor(s) mediating responses to the IP receptor agonists in the presence of GW627368X is not clear. If IP receptors mediated these responses, then one would expect iloprost to be a more efficacious agonist than we observed. At recombinant hEP2 receptors, these compounds were essentially devoid of agonist activity (Wilson et al., 2004) and, as we have already discussed, EP2 receptors in PSV appear to be poorly coupled, making this receptor an unlikely candidate. However, we reasoned that if EP2 receptors were responsible then iloprost would be acting as a partial agonist at them. If this were the case, then it should be possible to observe further antagonist shifts of PGE2 curves in the presence of GW627368X+iloprost. The failure of iloprost to produce any such shift in PGE2 responses further eliminates the possibility of EP2 receptor involvement. Involvement of DP receptors may also be eliminated by the failure of BWA868C to antagonise iloprost responses. In the light of these findings, care should be exercised in the interpretation of potent responses to IP agonists in vascular preparations. For example, those observed by Jones et al. (2000) in PSV may be interpreted in terms of EP4 receptor activation. It is therefore possible to speculate that the clinical utility of prostacyclin (Epoprostenol® in primary pulmonary hypertension may be at least partly due to its EP4 agonist properties.
Iloprost was unique among the compounds that we tested in producing rapidly reversing relaxations: it may either be a substrate for an endogenous metabolic enzyme or may be simultaneously activating another receptor that is coupled to smooth muscle contraction. Although iloprost is known to possess affinity for EP1 and EP3E receptors (Abramowitz et al., 2000) and is a known EP1 receptor agonist (Sheldrick et al., 1988), our data show that these receptors are not present. It is possible to speculate that the different response stability observed may reflect fundamental differences in the transduction of efficacy induced by iloprost in the manner recently described by Kenakin (2002). Thus, agonists acting at a given receptor may elicit different cellular responses by possessing a unique spectrum of efficacies for the many biochemical processes stimulated. Therefore, we propose that PSV does contain IP prostanoid receptors and that iloprost elicits rapidly desensitising activation of them.
We have previously shown that the DP receptor agonist BW245C (Whittle et al., 1983) possesses EP2 and EP4 receptor agonism (Wilson et al., 2004). These findings have been confirmed here by the sensitivity of this compound to EP4 receptor antagonism in PSV and are similar to the data obtained by Lydford et al. (1996a, 1996b) in rabbit saphenous vein. The generation of biphasic BW245C E/[A] curves in the presence of GW627368X indicates the presence of a third relaxatory receptor type in PSV. The ability of BWA868C to antagonise the lower potency phase suggests that BW245C activates DP receptors in PSV.
In conclusion, we have demonstrated that PSV contains predominantly EP4 receptors but also contains EP2, DP and IP receptors coupled to smooth muscle relaxation with low efficacy. Human and porcine EP2 and EP4 receptors possess a considerable degree of pharmacological equivalence, underlining the utility of this preparation in the development of coronary bypass models. We have also demonstrated that BW245C is a mixed EP2/4 and DP receptor agonist at porcine receptors and that IP receptor agonists are also agonists at porcine EP4 receptors. These studies therefore contribute to our understanding of both the PSV and of the prostanoid receptor agonists currently available.
Acknowledgments
We are grateful to Dr M. Hayes and Dr M. Todd (Biotransformations & Natural Products Chemistry, GlaxoWellcome) for assistance with the preparation of butaprost free acid, and to Dr F. McDonald (Schering, AG) for the gift of cicaprost.
Abbreviations
- cAMP
cyclic adenosine monophosphate
- CHO
Chinese hamster ovary
- DMSO
dimethyl sulphoxide
- E/[A] curve
concentration–effect curve
- HEK
human embryonic kidney
- 5-HT, 5-hydroxytryptamine
serotonin
- L-NAME, Nω-nitro-L-arginine
methyl ester
- PE
phenylephrine
- PSV
piglet saphenous vein
- r2
correlation coefficient
References
- ABRAMOWITZ M., ADAM M., BOIE Y., CARRIERE D.D., GODBOUT C., LAMONTAGNE S., ROCHETTE C., SAWYER N., TREMBLAY N.M., BELLEY M., GALLANT M., DUFRESNE C., GAREAU Y., RUEL R., JUTEAU H., LABELLE M., OUIMET N., METTERS K. The utilization of recombinant prostanoid receptors to determine the affinities and selectivities of prostaglandins and related analogs. Biochim. Biophys. Acta. 2000;1483:285–293. doi: 10.1016/s1388-1981(99)00164-x. [DOI] [PubMed] [Google Scholar]
- ANGELINI G.D., BRYAN A.J., WILLIAMS H.M., MORGAN R., NEWBY A.C. Distention promotes platelet and leukocyte adhesion and reduces short-term patency in pig arteriovenous bypass grafts. J. Thor. Cardiovasc. Surg. 1990;99:433–439. [PubMed] [Google Scholar]
- BUNCE K.T., CLAYTON N.M., COLEMAN R.A., COLLINGTON E.W., FINCH H., HUMPHRAY J.M., HUMPHREY P.P., REEVES J.J., SHELDRICK R.L., STABLES R. GR63799X – a novel prostanoid with selectivity for EP3 receptors. Adv. Prost. Thromb. Leuk. Res. 1991;21:379–382. [PubMed] [Google Scholar]
- COLEMAN R.A., GRIX S.P., HEAD S.A., LOUTIT J.B., MALLETT A., SHELDRICK R.L.G. A novel inhibitory prostanoid receptor in piglet saphenous vein. Prostaglandins. 1994a;47:151–168. doi: 10.1016/0090-6980(94)90084-1. [DOI] [PubMed] [Google Scholar]
- COLEMAN R.A., SMITH W.L., NARUMIYA S. International Union of Pharmacology VIII. Classification of prostanoid receptors: properties, distribution and structure of the receptors and their subtypes. Pharmacol. Rev. 1994b;46:205–229. [PubMed] [Google Scholar]
- GIBLIN G.M.P., JONES H.T., MASON A.M., MILLER N.D., ROOMANS S., SHANAHAN S.E., WALKER A.L. New naphthalene derivatives are EP4 antagonists – useful for the treatment of e.g. rheumatoid arthritis, osteoarthritis, gouty arthritis, myositis or rheumatic fever (WO200250032-A1) 2002a.
- GIBLIN G.M.P., WILSON R.J., FOORD S.M., SWARBRICK M., WALKER A.L., BAMFORD M., ROOMANS S., MASON A.M., MILLER N.D., JONES H.T., SHANAHAN S.E., RASMUSSEN S., SMITH L., SPALDING D., RANSHAW L., FENWICK R., ANCLIFF R., SAEZ V., FRYE S., STRATTON S., LEWELL X., CARTWRIGHT K.-A., RHODES S., ROBERTS N., GREEN R.A novel, selective, non-prostanoid EP4 receptor antagonist 2002bPoster at the 224th ACS National Meeting, August 18–22, 2002, Boston, Abstract MEDI-306
- GILES H., LEFF P. The biology and pharmacology of PGD2. Prostaglandins. 1988;35:277–300. doi: 10.1016/0090-6980(88)90093-7. [DOI] [PubMed] [Google Scholar]
- GILES H., LEFF P., BOLOFO M.L., KELLY M.G., ROBERTSON A.D. The classification of prostaglandin DP-receptors in platelets and vasculature using BW A868C, a novel, selective and potent competitive antagonist. Br. J. Pharmacol. 1989;96:291–300. doi: 10.1111/j.1476-5381.1989.tb11816.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HIRAI H., TANAKA K., YOSHIE O., OGAWA K., KENMOTSU K., TAKAMORI Y., ICHIMASA M., SUGAMURA K., NAKAMURA M., TAKANO S., NAGATA K. Prostaglandin D2 selectively induces chemotaxis in T-helper type 2 cells, eosinophils, and basophils via seven-transmembrane receptor CRTH2. J. Exp. Med. 2001;193:225–261. doi: 10.1084/jem.193.2.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HUMBLES A.A., NIALS A.T., COLEMAN R.A. Pharmacological characterisation of the prostanoid receptors in the rabbit isolated ear artery. Br. J. Pharmacol. 1991;104:418. [Google Scholar]
- JONES R.L., CHAN K.-M., RUDD J.A. Investigation of prostanoid EP4 and IP1 systems in blood vessels of piglets. Br. J. Pharmacol. 2000;131:63. [Google Scholar]
- JONES R.L., QIAN Y.M., CHAN K.M., YIM A.P.C. Characterization of a prostanoid EP3-receptor in guinea-pig aorta – partial agonist action of the non-prostanoid ONO-AP-324. Br. J. Pharmacol. 1998;125:1288–1296. doi: 10.1038/sj.bjp.0702189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JONES R.L., QIAN Y.-M., WONG H.N.C., CHAN H.W., YIM A.P.C. Prostanoid action on the human pulmonary vascular system. Clin. Exp. Pharmacol. Phys. 1997;24:969–972. doi: 10.1111/j.1440-1681.1997.tb02730.x. [DOI] [PubMed] [Google Scholar]
- KENAKIN T. Pharmacologic Analysis of Drug–Receptor Interaction. New York: Raven; 1993. Methods of drug and receptor classification; pp. 344–384. [Google Scholar]
- KENAKIN T. Drug efficacy at G protein coupled receptors. Ann. Rev. Pharmacol. Toxicol. 2002;42:349–379. doi: 10.1146/annurev.pharmtox.42.091401.113012. [DOI] [PubMed] [Google Scholar]
- KIRIYAMA M., USHIKUBI F., KOBAYASHI T., HIRATA M., SUGIMOTO Y., NARUMIYA S. Ligand binding specificities of the eight types and subtypes of the mouse prostanoid receptors expressed in Chinese hamster ovary cells. Br. J. Pharmacol. 1997;122:217–224. doi: 10.1038/sj.bjp.0701367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEW M.J., ANGUS J.A. Analysis of competitive agonist–antagonist interactions by non-linear regression. Trends Pharmacol. Sci. 1995;16:328–337. doi: 10.1016/s0165-6147(00)89066-5. [DOI] [PubMed] [Google Scholar]
- LI R.-C., CINDROVA-DAVIES T., SKEPPER J.N., SELLERS L.A. Prostacyclin induces apoptosis of vascular smooth muscle cells by a cAMP-mediated inhibition of extracellular signal-regulated kinase activity and can counteract the mitogenic activity of endothelin-1 or basic fibroblast growth factor. Circ. Res. 2004;94:759–767. doi: 10.1161/01.RES.0000121568.40692.97. [DOI] [PubMed] [Google Scholar]
- LIU Y.J., JACKSON D.M., BLACKHAM A. Effects of BW A868C, a selective prostaglandin DP receptor antagonist in dog isolated vascular preparations. Eur. J. Pharmacol. 1996;303:187–192. doi: 10.1016/0014-2999(96)00037-4. [DOI] [PubMed] [Google Scholar]
- LUMLEY P., WHITE B.P., HUMPHREY P.P. GR32191, a highly potent and specific thromboxane A2 receptor blocking drug on platelets and vascular and airways smooth muscle in vitro. Br. J. Pharmacol. 1989;97:783–794. doi: 10.1111/j.1476-5381.1989.tb12017.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYDFORD S.J., MCKECHNIE K.C.W., DOUGALL I.G. Pharmacological studies on prostanoid receptors in the rabbit isolated saphenous vein: a comparison with the rabbit isolated ear artery. Br. J. Pharmacol. 1996a;117:13–20. doi: 10.1111/j.1476-5381.1996.tb15148.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LYDFORD S.J., MCKECHNIE K.C.W., LEFF P. Interaction of BW A868C, a prostanoid DP-receptor antagonist, with two receptor subtypes in the rabbit isoltaed saphenous vein. Prostaglandins. 1996b;52:125–139. doi: 10.1016/0090-6980(96)00058-5. [DOI] [PubMed] [Google Scholar]
- MACLENNAN S.J., BOLOFO M.L., MARTIN G.R. Amplifying interactions between spasmogens in vascular smooth muscle. Biochem. Soc. Trans. 1993;21:1145–1150. doi: 10.1042/bst0211145. [DOI] [PubMed] [Google Scholar]
- MILNE S.A., ARMSTRONG R.A., WOODWARD D.F. Comparison of the EP receptor subtypes mediating relaxation of the rabbit jugular and pig saphenous veins. Prostaglandins. 1995;49:225–237. doi: 10.1016/0090-6980(95)00018-6. [DOI] [PubMed] [Google Scholar]
- NARUMIYA S., SUGIMOTO Y., USHIKUBI F. Prostanoid receptors: structures, properties and functions. Phys. Rev. 1999;79:1193–1226. doi: 10.1152/physrev.1999.79.4.1193. [DOI] [PubMed] [Google Scholar]
- QIAN Y.-M., JONES R.L., CHAN K.-M., STOCK A.I., HO J.K.S. Potent contractile actions of prostanoid EP3-receptor agonists on human isolated pulmonary artery. Br. J. Pharmacol. 1994;113:369–374. doi: 10.1111/j.1476-5381.1994.tb16997.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- OLIVA D., NICOSIA S. PGI2-receptors and molecular mechanisms in platelets and vasculature: state of the art. Pharm. Res. Commun. 1987;19:735–765. doi: 10.1016/0031-6989(87)90010-5. [DOI] [PubMed] [Google Scholar]
- ROUAUD C., DELAFORGE M., ANGER-LEROY M., LE FILLIATRE G., FINET M., HANF R. The cyclo-oxygenase-dependent regulation of rabbit vein contraction: evidence for a prostaglandin E2-mediated relaxation. Br. J. Pharmacol. 1999;126:35–44. doi: 10.1038/sj.bjp.0702265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHELDRICK R.L.G., COLEMAN R.A., LUMLEY P. Iloprost – a potent EP1- and IP-receptor agonist. Br. J. Pharmacol. 1988;94:334. [Google Scholar]
- SMITH J.B., ARAKI H., LEFER A.M. Thromboxane A2, prostacyclin and aspirin: effects on vascular tone and platelet aggregation. Circulation. 1980;62:V19–V25. [PubMed] [Google Scholar]
- WALCH L., MONTPREVILLE V.D., BRINK C., NOREL X. Prostanoid EP1- and TP-receptors involved in the contraction of human pulmonary veins. Br. J. Pharmacol. 2001;134:1671–1678. doi: 10.1038/sj.bjp.0704423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WHITTLE B.J.R., MONCADA S., MULLANE K., VANE J.R. Platelet and cardiovascular activity of the hydantoin BW 245C, a potent prostaglandin analogue. Prostaglandins. 1983;25:205–223. doi: 10.1016/0090-6980(83)90105-3. [DOI] [PubMed] [Google Scholar]
- WILSON R.J., GIBLIN G., FOORD S., SWARBRICK M., WALKER A., BAMFORD M., ROOMANS S., MASON A., MILLER N., JONES H., SHANAHAN S., RASMUSSEN S., SMITH L., SPALDING D., RANSHAW L., FENWICK R., ANCLIFF R., SAEZ V., FRYE S., STRATTON S., LEWELL X., CARTWRIGHT K.-A., RHODES S., ROBERTS N., GREEN R. GW627368X: a novel, potent and selective EP4 prostanoid receptor antagonist. Br. J. Pharmacol. 2003;138:84. [Google Scholar]
- WILSON R.J., RHODES S.A., WOOD R.L., SHIELD V.J., NOEL L.S., GRAY D.W., GILES H. Functional pharmacology of human prostanoid EP2 & EP4 receptors. Eur. J. Pharmacol. 2004;501:49–58. doi: 10.1016/j.ejphar.2004.08.025. [DOI] [PubMed] [Google Scholar]