

Abstract
This manuscript presents the preclinical profile of lumiracoxib, a novel cyclooxygenase-2 (COX-2) selective inhibitor.
Lumiracoxib inhibited purified COX-1 and COX-2 with Ki values of 3 and 0.06 μM, respectively. In cellular assays, lumiracoxib had an IC50 of 0.14 μM in COX-2-expressing dermal fibroblasts, but caused no inhibition of COX-1 at concentrations up to 30 μM (HEK 293 cells transfected with human COX-1).
In a human whole blood assay, IC50 values for lumiracoxib were 0.13 μM for COX-2 and 67 μM for COX-1 (COX-1/COX-2 selectivity ratio 515).
Lumiracoxib was rapidly absorbed following oral administration in rats with peak plasma levels being reached between 0.5 and 1 h.
Ex vivo, lumiracoxib inhibited COX-1-derived thromboxane B2 (TxB2) generation with an ID50 of 33 mg kg−1, whereas COX-2-derived production of prostaglandin E2 (PGE2) in the lipopolysaccharide-stimulated rat air pouch was inhibited with an ID50 value of 0.24 mg kg−1.
Efficacy of lumiracoxib in rat models of hyperalgesia, oedema, pyresis and arthritis was dose-dependent and similar to diclofenac. However, consistent with its low COX-1 inhibitory activity, lumiracoxib at a dose of 100 mg kg−1 orally caused no ulcers and was significantly less ulcerogenic than diclofenac (P<0.05).
Lumiracoxib is a highly selective COX-2 inhibitor with anti-inflammatory, analgesic and antipyretic activities comparable with diclofenac, the reference NSAID, but with much improved gastrointestinal safety.
Keywords: Lumiracoxib, COX-2, cyclooxygenase-2 selective inhibitor, preclinical
Introduction
Prostaglandins play key roles in modulating both physiological and inflammatory processes. They serve as autocrine factors regulating platelet aggregation, vascular tone and oedema, secretions from exocrine glands and the neuronal response to pain, to name but a few. Prostaglandin synthesis depends on the activity of cyclooxygenase (COX), which exists in two isoforms: COX-1 and COX-2. These two isoforms are highly homologous in sequence and differ only in one amino acid at their COX catalytic sites. While differences have been identified in the requirement for lipid peroxide activation (Chen et al., 1999) and in substrate preferences (Smith et al., 2000), both isoforms have identical catalytic efficiency and requirement (Km) for arachidonic acid. The most profound difference between the two isoforms relates to their expression (Smith & Langenbach, 2001). COX-1 is constitutively expressed in most tissues throughout the body (Pairet & Engelhardt, 1996). In contrast, COX-2 has a restricted pattern of distribution under normal physiological conditions, yet is highly induced at sites of inflammation and cell proliferation (Pairet & Engelhardt, 1996).
As a result of these different patterns of expression and pathophysiological roles, COX-2 selective inhibitors were developed as a means of achieving anti-inflammatory and analgesic activity, while potentially sparing the COX-1-dependent physiological functions of gastrointestinal (GI) cytoprotection and platelet aggregation. This hypothesis has been supported by clinical trials in which other COX-2 selective inhibitors have been reported to cause a lower incidence of GI side effects than nonselective COX inhibitors (Simon et al., 1998; Bombardier et al., 2000; Hunt et al., 2003). However, data from some studies have also suggested that the COX-2 selective inhibitor, rofecoxib, may be associated with an increased cardiovascular risk (Bombardier et al., 2000; Mamdani et al., 2004; Topol, 2004; Fitzgerald, 2004). However, this is not the case with lumiracoxib. In the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET) of lumiracoxib vs naproxen and ibuprofen (the largest GI outcomes study in patients with OA performed to date), lumiracoxib was associated with a 79% reduction in the incidence of upper GI ulcer complications with no increase in cardiovascular risk (Farkouh et al., 2004; Schnitzer et al., 2004). These recent findings seem to indicate that any possible increase in cardiovascular risk associated with some COX-2 selective inhibitors may be drug-specific rather than class-specific.
The studies presented here describe the preclinical pharmacology of lumiracoxib, a novel highly selective COX-2 inhibitor. Lumiracoxib has a chemical structure distinct from other COX-2 selective inhibitors, in that it lacks a sulphur-containing group but has a carboxylic acid moiety, a feature characteristic of many traditional nonsteroidal anti-inflammatory drugs (NSAIDs) (Figure 1). Lumiracoxib has been found to bind and interact with the COX-2 enzyme via mechanisms different from other COX-2 selective inhibitors and carboxylate-containing nonselective COX inhibitors. The carboxylate group of lumiracoxib forms hydrogen bonds with the catalytic Tyr385 and with Ser530 on COX-2, rather than with the larger hydrophobic side pocket (as utilised by other COX-2 selective inhibitors developed to date, such as celecoxib) or with Arg120 (as utilised by carboxylate-containing profen-class nonselective NSAIDs, such as flurbiprofen) (Clark et al., 2004). Lumiracoxib is shown to be highly selective for COX-2 over COX-1 using a variety of in vitro, in vivo and ex vivo biochemical measurements and to be effective in relieving pain and inflammation in a variety of rodent models. Importantly, lumiracoxib is associated with improved GI tolerability compared with the nonselective COX inhibitor, diclofenac.
Figure 1.

Chemical structure of compounds studied in this report.
Methods
In vitro, ex vivo and in vivo studies
The cyclooxygenase inhibitors described in this report were prepared at Novartis Pharmaceuticals Corporation, and were authenticated using mass and NMR spectroscopy. Unless otherwise specified, all chemicals and compounds were sourced from Sigma Chemical Co., St Louis, MO, U.S.A. For in vitro experiments, compounds were first dissolved in dimethyl sulphoxide (DMSO) and diluted into buffer such that the final concentration of DMSO was 0.1%. For ex vivo and in vivo analyses the compounds were suspended in fortified cornstarch (3% cornstarch (w v−1), 5% polyethylene glycol 400 (w v−1), 0.34% Tween 80 (w v−1)) or 1% tragacanth (w v−1)) and administered in a volume of either 2 or 5 ml kg−1. Ovine COX-1 was obtained from Cayman Chemical Company (Ann Arbor, MI, U.S.A.). Recombinant human COX-2 was prepared at Novartis according to the procedure described by Wennogle et al. (1995). Other preparations used included stably transfected human COX-1-expressing human embryonic kidney (HEK) 293 cells (ATCC, Rockville, MD, U.S.A.), human dermal fibroblasts (Clonetics, San Diego, CA, U.S.A.), calcium ionophore A23187 and lipopolysaccharide (LPS; Sigma Chemical Co., St Louis, MO, U.S.A.; Difco Laboratories, Detroit, MI, U.S.A.).
All in vivo procedures were approved by local Novartis Animal Care and Use Committees in accordance with guidelines in place in the country where the studies were conducted. Male Sprague–Dawley or Wistar rats and female Lewis rats (Harlan, Indianapolis, IN, U.S.A.; Charles River Breeding Laboratories, Wilmington, MA, U.S.A.) were housed in accredited facilities under standard conditions for rodents. Animals were acclimatised for at least 3 days prior to use.
Inhibition of purified COX-1 and COX-2
Inhibition of enzyme activity was determined by measuring oxygen (O2) consumption in a modification of the methods of Kulmacz & Lands (1985) and Callan et al. (1996). Purified enzyme preparations with control activities of 80 nmol of O2 consumption min−1 were pretreated with inhibitors at 37°C for various lengths of time (0–60 min). Arachidonic acid (20 μM) was then added, and O2 consumption measured using an oxygen electrode (Yellow Springs Instruments, Yellow Springs, OH, U.S.A.). The reaction of inhibitor with enzyme was assumed to be a simple second-order reaction during both the preincubation period and the time to highest observed O2 consumption velocity (Vopt). An exponential approach to equilibrium was used, with e–Ikt for the preincubation period, and e−Iktopt/‘1/‘1+Km/s'' for time to Vopt, where I is the inhibitor concentration, k is the second-order rate constant (kon), t is the preincubation time, topt is the time to optimal velocity after the addition of arachidonic acid, Km is the Michaelis–Menten constant for arachidonic acid and s is the arachidonic acid concentration. The values for the second-order rate constant k and equilibrium Ki were simultaneously solved from:
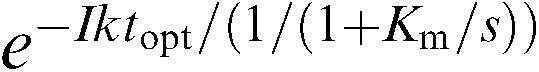 |
where Vobs is the observed velocity in the presence of inhibitor, V0 is the velocity in the absence of inhibitor, and F0 is the fraction of uninhibited enzyme at equilibrium:
The half-life (t1/2) for the enzyme-inhibitor complex was calculated using:
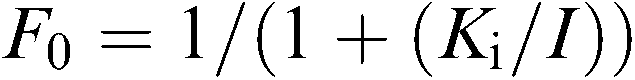 |
Constants were fitted for kon, Ki, a Michaelis–Menten constant for the maximal calculated velocity (Vmax), and the time to Vopt. Km was 8 μM for COX-1 and 5 μM for COX-2. For the data sets, the average s.d. of observed Vmax ranged from 6 to 9%.
Cellular assays for COX-1 and COX-2 inhibition
HEK 293 cells, and human dermal fibroblasts (stimulated with interleukin-1β (IL-1β; 10 ng ml−1) overnight to upregulate COX-2) were pretreated with carrier, diclofenac (0.0003–3 μM), naproxen, lumiracoxib or celecoxib (0.003–30 μM) for 15 min at 37°C. Arachidonic acid (40 μM) was then added, and after a further 15 min incubation, supernatants were harvested for measurement of prostaglandin E2 (PGE2) by radioimmunoassay (Perseptive Diagnostics, Framingham, MA, U.S.A.). Mean inhibition of PGE2 production compared to vehicle-treated control cells for each concentration was calculated and plotted vs the log of the compound concentration, and the IC50 value was calculated using a four-parameter logistic function using nonlinear regression analysis. Typical control PGE2 values both for HEK 293 cells and human dermal fibroblasts were 30 ng ml−1 following arachidonic acid addition and <0.1 ng ml−1 in the absence of arachidonic acid.
COX-1 and COX-2 inhibition in human whole blood assay
Thromboxane B2 (TxB2) production from platelets is a surrogate measure of COX-1 activity, while PGE2 production is a surrogate measure of COX-2 activity (Patrono et al., 1980; Patrignani et al., 1994). The production of TxB2 and PGE2 were measured in human blood from healthy volunteers. Blood (500 μl) was dispensed into Beckman deep well polypropylene titre plates and incubated in a 5% CO2 incubator at 37°C. After a 1-h equilibration time, test compound (range 0.1 nM–300 μM in half log increases) or DMSO carrier (5 μl) was added. The resulting mixture was then preincubated for a further 1 h, before being divided into two aliquots – one to be assayed to determine COX-1 activity and the other for COX-2 activity. To determine COX-1 activity, A23187 (50 μM) was added to one of the aliquots, and this mixture was then incubated for a further 1 h at 37°C with continuous shaking, after which TxB2 production was assessed. To determine COX-2 activity, LPS (10 μg ml−1) was added to the other aliquot, and the resulting mixture was incubated at 37°C overnight, after which PGE2 production was measured.
The samples were centrifuged at 250 × g for 10 min at 4°C to collect supernatant and PGE2 or TxB2 concentrations were determined in the supernatant using enzyme-linked chemiluminescence immunoassays (Assay Designs Inc., Ann Arbor, MI, U.S.A.). The mean±s.e.m. unstimulated TxB2 level was 5±1 ng ml−1 and the A23187-stimulated level was 210±40 ng ml−1. The mean±s.e.m. unstimulated PGE2 level was 0.20±0.06 ng ml−1 and the LPS-stimulated level was 17±5 ng ml−1. The level of prostaglandin in each sample was normalised to the percent inhibition associated with each test compound. The data from each donor were then pooled and fitted to a four-parameter logistic function using nonlinear regression analysis.
In vivo and ex vivo studies
Pharmacokinetics of lumiracoxib
Male Sprague–Dawley rats (225–250 g) were fasted overnight, orally dosed with 5 mg kg−1 of lumiracoxib or diclofenac (sodium salt) as comparator. Blood was collected at scheduled time points postdose. Plasma was separated by centrifugation (15 min at 250 × g). Samples (50 μl) containing internal standard were deproteinised by addition of acetonitrile and filtered through 0.45 μm filters before chromatographic separation using a C18 reverse-phase HPLC column (YMC ODS-AM 2 × 50 mm, 5 μm particle size). The effluent was introduced into the ion source of the mass spectrometer (LCQ Ion Trap, ThermoFinnigan, Piscataway, NJ, U.S.A.) and subjected to atmospheric pressure electrospray ionisation in the negative mode. Compounds were detected by mass spectrometric (MS) detection (LC/MS/MS (liquid chromatography/mass spectrometry/mass spectrometry)) after fragmentation of the parent ions. Calibration curves and quality control samples were prepared by spiking known amounts of analyte to internal standard (ordinate) vs the nominal plasma concentration (abscissa) and fitted by regression analysis to allow quantitation of the analyte. In all cases, the regression coefficient was >0.99. Pharmacokinetic parameters were derived from plasma concentration vs time curves fitted by noncompartmental modelling (WinNonlin Professional 3.0, Pharsight Corp., Cary, NC, U.S.A.).
TxB2 measurements
Serum TxB2 was measured in male Sprague–Dawley rats (195–225 g) 4 h after administration by gavage of lumiracoxib, diclofenac or vehicle. The rats were killed by carbon dioxide inhalation, before blood was collected by cardiac puncture, with one sample taken per animal. The sample was allowed to clot for 1 h at 37°C in a shaking water bath before serum was separated by centrifugation (15 min at 250 × g). TxB2 levels were measured by radioimmunoassay (NEN, Wilmington, DE, U.S.A.). The data were converted to percent inhibition and fitted to a three-parameter logistic function using nonlinear regression analysis. The control TxB2 value (clotting-induced) was 293 ng ml−1.
LPS-stimulated PGE2 production in the rat air pouch
PGE2 concentration was measured in LPS-stimulated dorsal air pouches in female Lewis rats (150–180 g). Pouches were produced by subcutaneous (s.c.) injection of 10 ml of air. At 24 h after inflation, lumiracoxib (0.2–2 mg kg−1) or diclofenac (0.06–0.6 mg kg−1) was administered by gavage. After 1 h, 8 μg of LPS suspended in sterile phosphate-buffered saline was injected directly into each pouch (negative control pouches were injected with sterile phosphate-buffered saline alone). After 3 h, pouch contents were harvested, and PGE2 content was measured using an enzyme immunoassay (Cayman Chemical Company, Ann Arbor, MI, U.S.A.). The data were converted to percent inhibition and fitted to a four-parameter logistic function using nonlinear regression analysis. Control PGE2 values were 0.5 ng per pouch (unstimulated) and 4–6 ng per pouch (stimulated).
Carrageenan-induced paw oedema
Male Sprague–Dawley rats (200–225 g) were fasted overnight and then orally dosed with test compound (0.1–3.0 mg kg−1). After 1 h, 0.1 ml of 1% carrageenan was injected into the subplantar region of the left hind paw. After a further 3 h, the rats were killed and the hind paws removed at the paw hairline and weighed. Oedema was determined by subtracting the weight of the noninflamed right paw from the weight of the carrageenan-injected left paw, and percent inhibition was determined for each animal relative to the mean of vehicle-treated control rats. ED30 values were estimated by back-transforming a linear regression model for percent inhibition as a function of log (dose). Control values were 1.6 g (normal paw) and 3–5 g (inflamed paw).
Complete Freund's adjuvant (CFA)-induced hyperalgesia
The nociceptive pressure threshold was determined on the left hind paw of fasted male Wistar rats (200–250 g), using paw-pressure apparatus (Analgesy-meter, Ugo Basile, Milan, Italy) and a wedge-shaped probe, just prior to intraplantar injection of 50 μl of CFA. The end point was taken as paw withdrawal or struggling. Predose nociceptive pressure threshold readings were taken 24 h after CFA administration, with further measurements taken 1 h after oral administration of lumiracoxib or diclofenac. Each compound was assayed in three separate dose–response studies, and the dose at which 30% inhibition of the predose hyperalgesia was achieved (D30) for each compound was calculated from the combined data. The mean control values were 111 g (naïve) and 37.1 g (predose).
LPS-induced pyresis
Male Sprague–Dawley rats (130–150 g) received LPS by s.c. injection (100 μg kg−1) and 2 h later had their temperature taken. The rats were then placed in temperature-matched groups. After a further 2 h, lumiracoxib, diclofenac or vehicle were administered by gavage, with final temperature measurements taken 2 h after treatment. The change in temperature was calculated for each animal and the percent inhibition compared to the mean of the vehicle-treated control group (typically 1.8–2.3°C).
Acute and chronic adjuvant-induced arthritis
Arthritis was induced in female Lewis rats (125–145 g) by intradermal injection of heat-killed Mycobacterium tuberculosis cells at the base of the tail. Control rats were injected with the mineral oil alone.
Animals were dosed once daily with lumiracoxib, diclofenac or vehicle beginning 14 days after adjuvant injection and continuing until day 17. Paw volumes were measured using a plethysmometer (Ugo Basile, Milan, Italy) prior to the initiation of dosing, and again on day 18. Percent inhibition of paw volumes in treated rats relative to control was determined, and ED50 values calculated as described below.
To model the effect of lumiracoxib treatment on inflammation associated with chronic arthritis, lumiracoxib was administered once daily by gavage (2 mg kg−1 day−1) beginning on day 12 and continuing until the day prior to study termination (day 28). The same dose of diclofenac was used as a comparator. Paw volumes were measured at scheduled time points (days 12, 15, 18, 21, 24 and 28). At study end, radiographs (lateral to medial view) were taken of each hind paw (Faxitron X-ray Corporation, Wheeling, IL, U.S.A.), before the hind paws were processed for histological evaluation. Radiographs were scored for soft tissue changes, bone erosion and periosteal new bone formation on a scale of 1 (normal) to 4 (severe changes). Stained sections were scored for periosteal new bone formation, bone and cartilage erosion, connective tissue changes and pannus formation. Average left–right values were used for calculation of group means and for statistical purposes. All assessments of drug effects were performed in a blinded manner. Paw volume control values were 1.25 ml for normal control rats (i.e. rats that received vehicle alone) and 2.5 ml for arthritis control rats (paw volume at day 14 for rats that received adjuvant plus vehicle).
Gastrointestinal tolerability
To measure gastric ulceration, fasted male Sprague–Dawley rats (195–225 g) were treated with lumiracoxib (1–100 mg kg−1) by gavage. After 4 h, the stomachs were removed and gross gastric lesions counted and measured to provide the total lesion length per rat. Each experiment included a comparison with diclofenac (0.3–100 mg kg−1), a known ulcerogen. All assessments of drug effects were performed in a blinded manner.
To measure the effect of the test compounds on intestinal permeability, rats were given test compound or vehicle, either in a single dose or once daily for 4 consecutive days. Immediately following the last dose, each rat was administered chromium-51-labelled EDTA (51Cr-EDTA; 5 μCi per rat). The rats were placed in individual metabolic cages and given food and water ad libitum. Urine was collected over a 24-h period. To quantify the effect of treatment on intestinal permeability, the level of 51Cr-EDTA in the urine of compound-treated rats was compared with that in rats that received vehicle. The percentage permeability for the vehicle-treated rats was 1.9±0.1 in the single-dose study and 1.59±0.06 in the four-dose group.
Statistics
Statistical comparisons between the vehicle-treated control and compound-treated groups were made by ANOVA followed by either Dunn's (intestinal permeability), Dunnett's (chronic adjuvant arthritis) or Tukey's (carrageenan oedema) post hoc tests. P-values less than 0.05 were considered statistically significant. Plots for percent inhibition vs dose were fitted by linear or nonlinear regression for calculation of IC50, ED30, ED50 or D30 values.
Results
Inhibition of purified COX-1 and COX-2
The mechanism and kinetics of inhibition of COX activity were studied using purified enzyme preparations of ovine COX-1 and human recombinant COX-2. Changes in O2 concentrations were monitored to follow enzymatic activity in real time. This approach allowed for kinetic characterisation of interactions between inhibitor and enzyme.
Lumiracoxib was rapidly and reversibly bound to COX-1 with micromolar affinity (Table 1 ). The COX-2 selective inhibitor celecoxib displayed similar kinetic behaviour and potency. In contrast, the interaction of diclofenac with COX-1 was different, showing time-dependent inhibition and tight binding with an affinity of 0.01 μM. For COX-2, all of the inhibitors studied showed time-dependent inhibition, with a defined initial rate of interaction followed by the formation of complexes with various degrees of stability. Lumiracoxib demonstrated potent inhibition of COX-2 (Ki=0.06 μM) forming a tight complex as indicated by its long dissociation t1/2. Diclofenac's Ki value was 0.01 μM, while celecoxib was less potent (Ki=0.2 μM) forming a weaker complex with COX-2 as evidenced by its relatively short dissociation t1/2.
Table 1.
Kinetic parameters for inhibition of COX-1 and COX-2 enzymatic activities
| COX-1 | COX-2 | |||||
|---|---|---|---|---|---|---|
| Ki (μM) | kon (s−1 μM–1) | t1/2 (min) | Ki (μM) | kon (s−1 μM−1) | t1/2 (min) | |
| Lumiracoxib | 3.2 (4.3, 2.0) | >105 (>105, >105) | ≪1 (≪1, ≪1) | 0.06 (0.05, 0.07) | 0.005 (0.004, 0.006) | 42 (48, 36) |
| Diclofenac | 0.01 (0.01, 0.01) | 0.1 (0.1, 0.1) | 6 (6, 6.1) | 0.01 (0.01, 0.01) | 0.006 (0.006, 0.006) | 152 (154, 150) |
| Celecoxib | 15 (16, 14) | >105 (>105, >105) | ≪1 (≪1, ≪1) | 0.20 (0.13, 0.26) | 0.01 (0.01, 0.01) | 5.1 (6.2, 4.0) |
Purified preparations of ovine COX-1 and human recombinant COX-2 were pretreated with compounds for varying time periods (0–60 min). Changes in the optimal velocity of O2 consumption were measured following the addition of the substrate arachidonic acid (20 μM). Kinetic parameters were calculated as described in Methods section.
The results are presented as the average of two experiments (the first value), and the results of each individual experiment (the values in parentheses).
Inhibition of COX-1 and COX-2 in cell-based assays
Lumiracoxib inhibited COX-2-dependent PGE2 production, assessed using IL-1β-stimulated dermal fibroblasts, with an IC50 value of 0.14 μM (Figure 2). This was similar to the IC50 of 0.31 μM observed for celecoxib. Lumiracoxib did not inhibit COX-1-dependent PGE2 production (assessed using COX-1 stably transfected HEK 293 cells) even at 30 μM, the highest concentration tested. The same concentration of celecoxib reduced COX-1-dependent PGE2 production by 50%.
Figure 2.

Inhibition of COX-1 and COX-2 in cell-based assays. Human HEK 293 cells stably expressing human COX-1, and human dermal fibroblasts, stimulated with IL-1β to induce COX-2, were pretreated with compounds for 30 min and then arachidonic acid was added to generate PGE2 production. Each concentration of compound was tested in quadruplicate and normalised to percent inhibition. The results were pooled from a series of experiments (lumiracoxib, n=7 experiments; diclofenac, n=7; celecoxib, n=2; naproxen, n=2). Results shown are mean percent inhibition (±s.e.m.) for COX-1 and COX-2.
Naproxen and diclofenac inhibited PGE2 production in both IL-1β-induced dermal fibroblasts and stably transfected HEK 293 cell cultures. Naproxen IC50 values in the COX-1 and COX-2 cellular assays were similar, being approximately 2 μM. Diclofenac inhibited PGE2 production in both cell types with IC50 values of 0.13 μM in the COX-1 cell-based assay and 0.01 μM in the COX-2 cell-based assay.
Inhibition of COX-1 and COX-2 in human whole blood assay
Assessing inhibition of COX activity in human whole blood has become a standard approach to gauge the selectivity of a compound (Flower, 2003). For this assay, heparinised human blood was divided into two aliquots: one for measuring TxB2 production and a second for measuring PGE2 production. All compounds tested produced concentration-dependent inhibition of prostaglandin synthesis where the span from 25 to 80% inhibition was achieved over a concentration range of approximately 1.5 log units (Figure 3). Lumiracoxib was a highly selective inhibitor of PGE2 (IC50=0.13 μM) achieving approximately 100% inhibition of PGE2 synthesis at concentrations of approximately 1 μM, while showing little inhibition of TxB2 production at concentrations up to 10 μM.
Figure 3.

In vitro inhibition of COX-1 and COX-2 in human blood assays. Results shown are mean percent inhibition (±s.e.m.) for COX-1 and COX-2. The IC50 values are presented in Table 2. TxB2 production was stimulated with the addition of A23187 (50 μM) and assessed after 1 h incubation; PGE2 production was induced with LPS (10 μg ml−1) and assessed after overnight incubation. Prostanoid production was normalised to percent inhibition and the results from a number of donors were pooled (lumiracoxib, n=52 donors; diclofenac, n=52; celecoxib, n=14; naproxen, n=15).
Table 2 compares the relative potencies of lumiracoxib and other clinically used COX inhibitors. The nonselective COX inhibitors studied include diclofenac, ibuprofen and naproxen. While absolute potencies differ from one drug to another, these compounds show little preference in their selectivity for either of the two COX isoforms. In contrast, the COX-2 selective inhibitors show similar potencies towards COX-2, but differ in their COX-1 inhibitory potency. As such, lumiracoxib is the least active agent against COX-1 but has the highest overall COX-2 selectivity (COX-2 : COX-1 ratio 515).
Table 2.
Summary of inhibitory potencies for thromboxane B2 (TxB2) and prostaglandin E2 (PGE2) production in the human whole blood assay
| Treatment (donors) | TxB2 (COX-1) | PGE2 (COX-2) | COX-2 selectivity |
|---|---|---|---|
| IC50 (μM) | IC50 (μM) | ||
| Nonselective COX inhibitors | |||
| Diclofenac (52) | 0.097 (±0.001) | 0.013 (±0.0001) | 7 |
| Ibuprofen (10) | 17 (±5) | 9 (±0.3) | 2 |
| Naproxen (15) | 10 (±0.3) | 21 (±0.7) | 0.5 |
| Selective COX-2 inhibitors | |||
| Celecoxib (14) | 7 (±0.3) | 0.19 (±0.1) | 37 |
| Etodolac (11) | 42 (±3) | 0.95 (±0.09) | 45 |
| Etoricoxib (14) | 69 (±5) | 0.26 (±0.02) | 265 |
| Lumiracoxib (52) | 67 (±2) | 0.13 (±0.002) | 515 |
| Meloxicam (14) | 3 (±0.1) | 0.11 (±0.004) | 26 |
| Nimesulide (7) | 35 (±3) | 0.69 (±0.05) | 51 |
| Rofecoxib (14) | 24 (±0.5) | 0.17 (±0.01) | 141 |
| Valdecoxib (16) | 27 (±2) | 0.1 (±0.005) | 270 |
| Metabolite of lumiracoxib | |||
| 4′-hydroxy lumiracoxib (11) | 86 (±9) | 0.5 (±0.01) | 172 |
Inhibition of TxB2 and PGE2 production was determined in separate aliquots of blood that had been pretreated with compounds for 1 h. TxB2 production was stimulated with the addition of A23187 (50 μM) and assessed after 1 h incubation. PGE2 production was induced with LPS (10 μg ml−1) and assessed after overnight incubation. Prostanoid production was normalised to percent inhibition and the results from the donors pooled. The IC50 indicated here is the mean value for the number of donors indicated in parentheses, along with the s.e.mean. The COX-2 selectivity represents the ratio of IC50 for COX-1 divided by COX-2.
Three major metabolites of lumiracoxib have been identified in rat and human. These metabolites are the 4′-hydroxy, 5-carboxy and 4′-hydroxy-5-carboxy metabolites (Mangold et al., 2004). Only the 4′-hydroxy metabolite demonstrated COX inhibitory activity in the human whole blood assay, although the COX-2 selectivity of this metabolite was only a third that of the parent compound (Table 2). The other two metabolites did not reach 30% inhibition for COX-1 or COX-2 activity, even at the highest concentrations tested (300 μM).
Pharmacokinetic assessment in rat
Diclofenac was selected as the reference nonselective COX-1 and COX-2 inhibitor to gauge the potency and efficacy of lumiracoxib in standard models of NSAID activity in the rodent. Following oral administration of a 5 mg kg−1 dose, both lumiracoxib (free acid) and diclofenac (sodium salt) were rapidly absorbed, reaching peak plasma levels between 1 and 0.5 h, respectively (Figure 4). Although peak plasma levels of lumiracoxib and diclofenac were comparable (4.4 and 4.3 μg ml−1, respectively), plasma levels of diclofenac declined relatively quickly, resulting in a greater overall exposure for lumiracoxib (area-under-curve of the concentration vs time curve (AUC) 18±2 μg ml−1 h−1) compared with diclofenac (AUC 5.1±0.5 μg ml−1 h−1).
Figure 4.

Concentrations of lumiracoxib and diclofenac in plasma of rats following a single oral administration of a 5 mg kg−1 dose. Results are expressed as means±s.e.m. (n=4). Samples were processed through a C18 reverse-phase HPLC column, and the plasma levels were determined by LC/MS/MS.
Ex vivo and in vivo inhibition of COX-1 and COX-2
Inhibition of COX-1-derived TxB2 generation ex vivo
Inhibition of COX-1 activity following oral dosing was measured ex vivo using platelet-derived TxB2 as a surrogate. Both diclofenac and lumiracoxib treatment resulted in a dose-dependent inhibition of serum TxB2. However, the respective ID50 values for diclofenac (5 mg kg−1) and lumiracoxib (33 mg kg−1) showed lumiracoxib to be a much less potent inhibitor of COX-1 than diclofenac (Figure 5). For example, no discernible inhibition of COX-1-dependent TxB2 production was seen with lumiracoxib 3 mg kg−1 (but complete inhibition of COX-2-dependent PGE2 production), while, in contrast, diclofenac at 5 mg kg−1 produced up to 50% inhibition of COX-1-dependent TxB2 production.
Figure 5.

Inhibition of ex vivo COX-1-dependent TxB2 and in vivo COX-2-dependent PGE2 in rat. Results shown are mean percent inhibition (±s.e.m.) for serum TxB2 (n=5–6 rats per dose) or air pouch PGE2 (n=9–12 rats per dose). TxB2 levels were determined ex vivo by radioimmunoassay of extracts from clotted blood sampled from rats 4 h after dosing. COX-2 activity was assessed by measuring PGE2 concentration in LPS-stimulated dorsal air pouches 4 h after dosing.
Inhibition of COX-2-derived PGE2 production in the LPS-stimulated rat air pouch
S.c. injection of air into rats causes the formation of an in vivo chamber, termed an air pouch, which over several days spontaneously develops a highly reactive lining tissue composed primarily of macrophage- and fibroblast-like cells (Sedgwick et al., 1983). When LPS is injected into this experimentally produced tissue compartment, it rapidly induces the production of PGE2, a process which is largely derived from COX-2 originating in the lining tissue (Masferrer et al., 1994; Appleton et al., 1995).
When this model was used for the in vivo assessment of COX-2 inhibition, lumiracoxib provided more than 90% inhibition at the highest dose tested (range 0.2–2 mg kg−1, ID50 0.24 mg kg−1; Figure 5), an efficacy similar to that seen for the highest tested dose of diclofenac (range 0.06–0.6 mg kg−1, ID50 0.12 mg kg−1).
In vivo measures of anti-inflammatory efficacy
Carrageenan-induced paw oedema
The carrageenan-induced rat paw oedema assay is widely used to evaluate the anti-inflammatory activity of NSAIDs and other anti-inflammatory agents in vivo (Mukherjee et al., 1996).
Lumiracoxib dose-dependently reduced carrageenan-induced paw oedema compared with vehicle-treated rats, showing an ED30 value of 0.35 mg kg−1 (Figure 6a). Diclofenac also reduced paw oedema (ED30 0.53 mg kg−1), with no statistically significant difference in the ED30 values for the two compounds. The maximum efficacy seen with lumiracoxib was identical to diclofenac.
Figure 6.

In vivo anti-inflammatory efficacy of lumiracoxib and diclofenac. Inflammation, hyperalgesia and pyresis were induced as described in the Methods section. Percent inhibition of (a) carrageenan-induced paw oedema measured 4 h after dosing (mean from two experiments, n=6 rats per treatment group in each experiment), (b) hyperalgesia 1 h after dosing (mean from three experiments, n=6 rats per treatment group in each experiment) and (c) pyresis 2 h after dosing (mean from four experiments, n=3 rats per treatment group in each experiment). The values shown are mean percent inhibition (±s.e.m.) for lumiracoxib and diclofenac.
CFA-induced hyperalgesia
Several groups have shown that an elevation in the expression of COX-2 gene products occurs in both the periphery and spinal cord following peripheral CFA-injection into the rat paw and that selective COX-2 inhibitors produced analgesic activity (Hay et al., 1997; Samad et al., 2001). The aim was to employ a model of persistent nociceptive or inflammatory pain in the absence of frank inflammation, which mimics the concept of central sensitisation. CFA produces short-lasting oedema (hours) and hyperalgesia lasting from a few hours up to 7–10 days. Hyperalgesia was measured 24 h following CFA injection, when oedema had subsided but there was establishment of peripheral and central sensitisation. Anti-nociception activity in inflamed tissue was evaluated by comparing the change in pressure threshold from baseline for the inflamed paw of test compound-treated rats with that in vehicle-treated rats.
Treatment with either lumiracoxib or diclofenac inhibited hyperalgesia compared with predose levels, with maximal reversal of 60–65%. For both compounds, inhibition was dose dependent (Figure 6b), with D30 values of 5.1 and 5.5 mg kg−1, for lumiracoxib and diclofenac, respectively.
LPS-induced pyresis
Nonselective COX inhibitors, such as diclofenac, are antipyretic in both human and animal models of pyresis (Bettini et al., 1986). Lumiracoxib (0.3–10 mg kg−1) and diclofenac (0.03–1 mg kg−1) produced similar levels of inhibition at the highest doses tested. The effect was dose dependent (Figure 6c), with ED50 values calculated as 1 and 0.13 mg kg−1 for lumiracoxib and diclofenac, respectively.
Acute and chronic adjuvant-induced arthritis
Intradermal injection of rats with heat-killed M. tuberculosis (adjuvant) reproducibly induces chronic, inflammatory arthritis accompanied by degradation of cartilage and bone that eventually leads to joint destruction (Whitehouse, 1988). The disease is driven largely by prostaglandin production, as evidenced by inhibition of disease severity by NSAIDs (Billingham, 1983) and COX-2 selective inhibitors (Anderson et al., 1996).
Administration of lumiracoxib or diclofenac beginning 14 days after adjuvant injection (i.e. during the period of rapidly increasing inflammation severity) limited disease progression. After a 4-day course of treatment, progression of inflammation was either slowed or reversed, depending on the dose of compound administered. ED50 values, measured after 4 days' treatment, were 5 mg kg−1 for lumiracoxib and 3 mg kg−1 for diclofenac (Figure 7a).
Figure 7.

Effects of lumiracoxib and diclofenac treatment on clinical severity of adjuvant-induced arthritis. (a) Rats were injected with M. tuberculosis and arranged into groups of equal severity on day 14. Rats were treated daily with lumiracoxib or diclofenac for 4 consecutive days. Values represent the mean percent inhibition (±s.e.m.) of paw volume 24 h after the final dose of compounds (mean from two experiments, n=4 rats per treatment group in each experiment). (b) Rats were injected with adjuvant on day 1 and treated with lumiracoxib or diclofenac (both at 2 mg kg−1) once daily from day 12 until termination of the experiment. Arthritis and normal control rats were given vehicle alone. Paw volumes were measured periodically during the course of disease. Values shown are mean volumes±s.e.m. (mean from four experiments, n=6 rats per treatment group in each experiment).
In chronic disease, both compounds significantly decreased clinical severity, as measured by paw volumes (Figure 7b). Mean histological scores of stained hind paw sections collected at study termination were reduced by 45 and 47% (both P<0.05) in rats treated with lumiracoxib and diclofenac, respectively. Radiograph scores were reduced by 54% (P<0.05) in both lumiracoxib- and diclofenac-treated rats.
Gastric ulceration
No ulcers were observed in rats treated orally with any of the lumiracoxib doses (1–100 mg kg−1). In contrast, oral administration of diclofenac at doses as low as 3 mg kg−1 caused a low incidence of ulcers (in three out of 18 rats) with a mean (±s.e.m.) ulcer length of 0.6±0.5 mm. Moreover, the incidence and size of ulcers in diclofenac-treated rats increased with the dose administered, resulting in severe gastric lesions (mean±s.e.m. ulcer length 28±3 mm) in 100% of rats treated with 100 mg kg−1 of diclofenac.
Intestinal permeability
The mean excretion of 51Cr-EDTA in urine of rats given a single dose of lumiracoxib (30 mg kg−1) was similar to that of vehicle-treated control rats (Table 3 ). A single dose of diclofenac (30 mg kg−1) significantly increased 51Cr-EDTA excretion compared with controls.
Table 3.
Changes in intestinal permeability following treatment with lumiracoxib and diclofenac
| Treatment | 51Cr-EDTA excretion in urine (%) | ||||
|---|---|---|---|---|---|
| Single dose | Four doses | ||||
| 30 mg kg−1 | 3 mg kg−1 | 10 mg kg−1 | 30 mg kg−1 | 100 mg kg−1 | |
| Lumiracoxib | 2.5±0.2* (n=18) | nt | 1.8±0.2* (n=12) | 4.3±0.4** (n=15) | 31.3±3.2** (n=9) |
| Diclofenac | 12.7±1.4** (n=24) | 5.0 ±1.1**> (n=12) | 12.8±1.5** (n=29) | nt | nt |
Rats were given test compound or vehicle – either in a single dose or once daily for 4 consecutive days. Immediately following the last dose, each rat was administered 5 μCi of 51Cr-EDTA. The level of 51Cr-EDTA was then measured in urine collected over a 24-h period.
Results shown are means±s.e.m. (n), where n=number of rats per dose. The % permeability for the vehicle-treated rats was 1.9±0.1 (24) in the single-dose study and 1.59±0.06 (51) in the four-dose group.
*P<0.05 vs diclofenac-treated rats; **P<0.05 vs vehicle-treated rats; nt=not tested.
In rats given daily doses over 4 days, excretion of 51Cr-EDTA in the 24-h period immediately following the last dose of lumiracoxib (10 mg kg−1) was also similar to excretion in vehicle-treated control rats, but diclofenac (3 or 10 mg kg−1) significantly increased 51Cr-EDTA excretion over the levels observed in control rats (Table 3). Higher doses of lumiracoxib (30 or 100 mg kg−1) caused significant increases in 51Cr-EDTA excretion relative to vehicle-treated controls. 51Cr-EDTA excretion could not be measured in rats treated with multiple doses of 30 or 100 mg kg−1 of diclofenac because of drug-induced toxicity.
Discussion
There has been considerable effort in the pharmaceutical industry to identify a COX-2 selective inhibitor with anti-inflammatory properties but without the adverse effects associated with traditional NSAIDs (Flower, 2003). Our studies describe the preclinical pharmacology of lumiracoxib, the first carboxyl, non-sulphur-containing COX-2 selective inhibitor with potent anti-inflammatory and analgesic activity (Table 4 ). Consistent with its weak activity on COX-1, the gastric tolerability of lumiracoxib is significantly better than that of diclofenac, a comparator that nonselectively inhibits both COX-1 and COX-2.
Table 4.
Summary of potencies for lumiracoxib and diclofenac in pharmacological models in the rat
| Model | Lumiracoxib (mg kg−1) | Diclofenac (mg kg−1) |
|---|---|---|
| Serum TxB2 (ID50)a | 33 (±1) | 5 (±1) |
| Air pouch PGE2 (ID50)b | 0.24 (±0.01) | 0.12 (±0.02) |
| Carrageenan oedema (ED30)c | 0.35 (±0.09) | 0.53 (±0.15) |
| CFA hyperalgesia (D30)d | 5.1 (±1.0) | 5.5 (±0.6) |
| LPS pyresis (ED50)e | 1 (±0.3) | 0.13 (±0.02) |
| Adjuvant arthritis (ED50)f | 5 (±1) | 3 (±1) |
Details of the methods used can be found in Methods section. Serum TxB2, air pouch PGE2 and carrageenan-induced paw oedema were all determined 4 h after administration of test compound. CFA-induced hyperalgesia was measured 1 h after oral administration of test compound. LPS-induced pyresis was assessed by determining the change in temperature of LPS-stimulated rats 2 h predose and 2 h postdose. Efficacy in adjuvant arthritis was determined by calculating the difference in paw volume on day 18 between vehicle- and test compound-treated rats.
Results are expressed as means (±s.e.mean).
Mean from three experiments, n=5 or 6 rats per treatment group in each experiment.
Mean from three experiments for lumiracoxib and two experiments for diclofenac, n=5 or 6 rats per treatment group in each experiment.
Mean from two experiments, n=6 rats per treatment group in each experiment.
Mean from three experiments, n=6 rats per treatment group in each experiment.
Mean from four experiments, n=3 rats per treatment group in each experiment.
Mean from two experiments, n=4 rats per treatment group in each experiment.
We established the biochemical potency and COX-2 selectivity of lumiracoxib in several assays using purified enzyme preparations, intact cells and in whole blood and tissues. Against purified enzymes, lumiracoxib proved to be a potent, time-dependent inhibitor of COX-2 and a weak, nontime-dependent, competitive inhibitor of COX-1. This kinetic profile has been observed for other COX-2 selective inhibitors (Gierse et al., 1999). In the cellular assay, lumiracoxib was shown to inhibit COX-2 activity but to have little effect on COX-1 activity.
The need for a standard assay to evaluate COX-2 selectivity led to the development and acceptance of the whole blood assay. In this assay, inhibition of platelet TxB2 (COX-1 dependent) and LPS-stimulated monocyte PGE2 (COX-2 dependent) production provide the necessary end points for determining potency and selectivity (Patrignani et al., 1994; Cryer & Feldman, 1998; Warner et al., 1999). To help gauge relative potency and COX-2 selectivity, lumiracoxib was compared with a variety of clinically used traditional NSAIDs and COX-2 selective inhibitors using this standard assay. Lumiracoxib demonstrated a 515-fold preference for inhibition of COX-2 as opposed to COX-1. Moreover, 100% inhibition of COX-2 was achieved at a lumiracoxib concentration that produced no COX-1 inhibition. This contrasts with celecoxib, diclofenac and naproxen where the drug concentration that maximally inhibits COX-2 also substantially inhibits COX-1 activity (Figure 3). It should be noted that the in vitro biochemical potency (IC50 0.13 μM) and COX-2 selectivity of lumiracoxib observed in the whole blood assay were comparable to those found ex vivo in human clinical trials, where the COX-2 IC50 was 0.18 μM and no inhibition of COX-1 was observed with supratherapeutic doses up to 800 mg (plasma Cmax 36 μM; Scott et al., 2002).
The degree of selectivity in the in vitro assays presented in this manuscript varied widely. Similar degrees of variability have been reported by others in sometimes closely related assay systems (Warner et al., 1999), and striking differences in measures of selectivity (approximately 1000-fold) have been reported between assays that utilised purified enzymes and whole blood assays (Gierse et al., 1999; Riendeau et al., 2001). The variation has been attributed to differences in substrate concentration, preincubation time, binding of drug by proteins and other factors in the various assay systems. Regardless of the explanation for the differences in selectivity, a similar rank ordering of compounds (with lumiracoxib as the most selective COX-2 inhibitor) is observed in the results from different laboratories, and the rank order as well as potency of comparator compounds in this report is consistent with published data (Chan et al., 1999; Warner et al., 1999; Patrono et al., 2001).
The biochemical selectivity for COX-2 inhibition by lumiracoxib was also demonstrated both ex vivo and in vivo. TxB2 generation by blood platelets is derived from COX-1 (Patrono et al., 1980; Patrignani et al., 1994), therefore, the ex vivo measurement of TxB2 inhibition in blood from animals given COX inhibitors can provide an index for COX-1 inhibition. In contrast, PGE2 production in the rat air pouch is derived from COX-2 (Masferrer et al., 1994), and inhibition of PGE2 production therefore serves as a surrogate for COX-2 inhibition. When tested 4 h after oral dosing, lumiracoxib reduced the COX-1-dependent TxB2 production in rat blood with an ID50 of 33 mg kg−1. This contrasts with the potency of 0.24 mg kg−1 for COX-2-dependent PGE2 production in the rat air pouch also measured at 4 h postdose. Moreover, the 2 mg kg−1 dose of lumiracoxib that achieved maximal inhibition of COX-2 activity caused no inhibition of COX-1 activity.
Lumiracoxib is rapidly absorbed and quickly reaches peak plasma levels. This can be attributed to its solubility at the site of absorption (water solubility at pH 3 <0.01 g l−1; water solubility at pH 6.8=0.17 g l−1; Novartis data on file), degree of ionisation (pKa 4.7; Mangold et al., 2004) and the relative lipid solubility of its ionised and nonionised forms (log P at pH 6.8=1.76; Novartis data on file). Differences in overall exposure between lumiracoxib and diclofenac suggests a relatively faster clearance of diclofenac possibly associated with known differences in the metabolic oxidative pathways of the two compounds (lumiracoxib is metabolised predominantly by cytochrome P450 [CYP] 2C9, whereas diclofenac is metabolised by CYP2C9 and CYP3A4) and in the metabolites formed (Degen et al., 1988; Boelsterli, 2003; Mangold et al., 2004; Scott et al., 2004b). After oral dosing in humans, the major metabolites of diclofenac are the 3′-hydroxy, 4′-hydroxy and 5-hydroxy metabolites (Stierlin et al., 1979). In comparison, the three major metabolites of lumiracoxib in rat and man are the 4′-hydroxy, 5-carboxy and 4′-hydroxy-5-carboxy metabolites (Mangold et al., 2004; Novartis data on file). Here, we report that only the 4′-hydroxy metabolite-inhibited COX activity in human blood with COX-2 potency and selectivity being three-fold less than that observed for lumiracoxib (Table 2). The exposure and half-life for the 4′-hydroxy metabolite was not studied in this report. In man, the plasma levels of 4′-hydroxy-lumiracoxib are only 10% of those of the parent molecule; thus, this metabolite is unlikely to contribute to pharmacological activity in man (Mangold et al., 2004).
The pharmacokinetics of lumiracoxib in healthy humans is similar to that observed in these preclinical studies, revealing a pharmacokinetic profile distinct from the other COX-2 selective inhibitors. In man, lumiracoxib has a much lower volume of distribution (approximately 9 l) than the other COX-2 selective inhibitors (celecoxib approximately 400 l, valdecoxib approximately 90 l, etoricoxib approximately 120 l and rofecoxib approximately 90 l) (Hartmann et al., 2003; Brune & Hinz, 2004). Whereas lumiracoxib demonstrates preferential distribution into inflamed synovial fluid (Scott et al., 2004a), rofecoxib, valdecoxib and etoricoxib distribute almost equally throughout the body, while celecoxib (due to its high lipophilicity) may be sequestered in body fat (Brune & Hinz, 2004). Lumiracoxib and etoricoxib are absorbed more rapidly than the other COX-2 selective inhibitors (Tmax 1–3 h), while lumiracoxib demonstrates the most rapid plasma elimination (t1/2 3–6 h) (Scott et al., 2002; Brune & Hinz, 2004). This rapid absorption and plasma elimination, combined with preferential distribution into inflamed tissue, means that in contrast to the other COX-2 selective inhibitors, there is reduced systemic COX-2 inhibition with lumiracoxib.
Lumiracoxib was found to be effective in a variety of standard rat models used to establish NSAID pharmacological activity, including hyperalgesia, oedema, pyresis and arthritis. The efficacy of lumiracoxib was identical to diclofenac, and the potency was generally comparable. The finding that a lower dose of lumiracoxib is required in the carrageenan oedema and air pouch models compared with that required in adjuvant-induced arthritis is thought to be a reflection of the differences in the amount of time during which the inhibitor needs to be present in the various models. In both the air pouch and the oedema models, efficacy is measured 4 h after compound administration. In contrast, to be effective in adjuvant-induced arthritis, a compound must be present for a much longer interval (approaching 24 h), since treatment must impact a continuous disease process lasting for days. When the half-life (4 h or five half-life intervals following the Tmax) is taken into account to adjust drug exposure, the difference between the acute models (4 h) vs adjuvant (daily for 4 days) all but disappears.
In the CFA-hyperalgesia model, the effective dose of lumiracoxib was much higher than seen in the other acute models. It is unlikely that the high dose reflects a non-COX-2 mechanism since many structurally dissimilar NSAIDs and COX-2 selective inhibitors require higher doses to show efficacy in this model (data not shown). It may, perhaps, be a consequence of a compartment that these drugs cannot readily access. Alternatively, the high dose may reflect a higher threshold of inhibition, which is needed to observe therapeutic activity in this model of pain. A comparable situation exists in man where higher doses of many NSAIDs and COX-2 selective inhibitors are required to treat acute pain compared to osteoarthritis pain (Abbott Laboratories Limited 2002; Merck, Sharp & Dohme Limited 2004a, 2004b, 2004c; Pharmacia Limited 2004). On the other hand, the high dose may reflect the difficulty in reducing COX-2 activity after the onset of inflammation.
GI safety of lumiracoxib was significantly superior to the nonselective NSAID diclofenac. Single doses of lumiracoxib did not cause apparent stomach ulcers at doses of 100 mg kg−1 (a dose 20 times that needed to achieve the ED50 for adjuvant arthritis), whereas ulcers were observed following doses of 3 mg kg−1 of diclofenac. Similarly, excretion of 51Cr-EDTA in urine – a measure of intestinal permeability – was lower in lumiracoxib-treated than in diclofenac-treated rats. Some have suggested that, instead of being solely due to inhibition of gastric protection afforded by COX-1, stomach ulceration caused by NSAIDs is partly attributable to topical toxicity as a consequence of their physical and chemical characteristics (Smale & Bjarnason, 2003). Namely, their hydrophobic and acidic characteristics disrupt the protective mucus layer and expose the underlying stomach epithelial cells to the acidic pH of stomach secretions (Lichtenberger et al., 1995). Additionally, acidic NSAIDs may accumulate due to ion trapping within intestinal enterocytes reaching concentrations that lead to uncoupling of mitochondrial oxidative phosphorylation (Krause et al., 2003). Neither of these hypothetical mechanisms seem to play a prominent role in the rodent model studied here since lumiracoxib and diclofenac have very similar hydrophobic (water/octanol partitioning) and acidic properties (pKa), yet have very different gastric liabilities.
Our findings agree with other published reports that show efficacy of COX-2 selective inhibitors in animal models (Futaki et al., 1993; Masferrer et al., 1994; Chan et al., 1999; Riendeau et al., 2001), but differs from some reports suggesting limited effectiveness of COX-2 selective inhibitors compared with nonselective COX inhibitors (Gilroy et al., 1998; Wallace et al., 1999; Pinheiro & Calixto, 2002). Lumiracoxib possesses a high COX-2 selectivity. In rodent models, which have served as standard tools in the development of traditional NSAIDs, lumiracoxib at doses well below those needed to inhibit COX-1 provides the same degree of efficacy as nonselective NSAIDs in reducing pain, fever and inflammation. In contrast to traditional NSAIDs, the high COX-2 selectivity of lumiracoxib translates into superior GI safety. In addition to differentiating lumiracoxib from nonselective NSAIDs, its different physical, chemical and pharmacokinetic characteristics may allow valuable distinctions to be made between lumiracoxib and currently available COX-2 selective inhibitors.
Acknowledgments
We acknowledge the assistance of James Wasvary and Kevin West for their invaluable contribution to the experimental work and preparation of this manuscript.
Abbreviations
- AUC
area-under-curve of the concentration vs time curve
- Cmax
maximum drug plasma concentration
- CFA
complete Freund's adjuvant
- 51Cr-EDTA
chromium-51 labelled EDTA
- COX
cyclooxygenase
- D30
dose at which 30% inhibition was achieved
- DMSO
dimethyl sulphoxide
- F0
fraction of uninhibited enzyme at equilibrium
- GI
gastrointestinal
- HEK
human embryonic kidney
- IL-1
interleukin-1
- Ki
inhibitor constant
- kon
second-order rate constant representing speed at which an inhibitor binds to an enzyme
- I
inhibitor concentration
- LC/MS/MS
liquid chromatography/mass spectrometry/mass spectrometry
- LPS
lipopolysaccharide
- NSAID
nonsteroidal anti-inflammatory drug
- O2
oxygen
- PGE2
prostaglandin E2
- s
arachidonic acid concentration
- t1/2
half-life
- topt
time to optimal velocity
- TxB2
thromboxane B2
- V0
velocity in the absence of inhibitor
- Vobs
observed velocity in the presence of inhibitor
- Vopt
highest observed O2 consumption velocity
- Vmax
Michaelis–Menten constant for the maximal calculated velocity
References
- ABBOTT LABORATORIES LIMITED 2002. Brufen 400 mg Tablets UK Summary of Product Characteristics, February 2002
- ANDERSON G.D., HAUSER S.D., MCGARITY K.L., BREMER M.E., ISAKSON P.C., GREGORY S.A. Selective inhibition of cyclooxygenase (COX)-2 reverses inflammation and expression of COX-2 and interleukin 6 in rat adjuvant arthritis. J. Clin. Invest. 1996;97:2672–2679. doi: 10.1172/JCI118717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- APPLETON I., TOMLINSON A., MITCHELL J.A., WILLOUGHBY D.A. Distribution of cyclooxygenase isoforms in murine chronic granulomatous inflammation. Implications for future anti-inflammatory therapy. J. Pathol. 1995;176:413–420. doi: 10.1002/path.1711760413. [DOI] [PubMed] [Google Scholar]
- BETTINI R., GROSSI E., RAPAZZINI P., GIARDINA G. Diclofenac sodium versus acetylsalicylic acid: a randomized study in febrile patients. J. Int. Med. Res. 1986;14:95–100. doi: 10.1177/030006058601400208. [DOI] [PubMed] [Google Scholar]
- BILLINGHAM M.E. Models of arthritis and the search for anti-arthritic drugs. Pharmacol. Ther. 1983;21:389–428. doi: 10.1016/0163-7258(83)90062-1. [DOI] [PubMed] [Google Scholar]
- BOELSTERLI U.A. Diclofenac-induced liver injury: a paradigm of idiosyncratic drug toxicity. Toxicol. Appl. Pharmacol. 2003;192:307–322. doi: 10.1016/s0041-008x(03)00368-5. [DOI] [PubMed] [Google Scholar]
- BOMBARDIER C., LAINE L., REICIN A., SHAPIRO D., BURGOS-VARGAS R., DAVIS B., DAY R., FERRAZ M.B., HAWKEY C.J., HOCHBERG M.C., KVIEN T.K., SCHNITZER T.J. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N. Engl. J. Med. 2000;343:1520–1528. doi: 10.1056/NEJM200011233432103. [DOI] [PubMed] [Google Scholar]
- BRUNE K., HINZ B. Selective cyclooxygenase-2 inhibitors: similarities and differences. Scand. J. Rheumatol. 2004;33:1–6. doi: 10.1080/03009740310004766. [DOI] [PubMed] [Google Scholar]
- CALLAN O.H., SO O.Y., SWINNEY D.C. The kinetic factors that determine the affinity and selectivity for slow binding inhibition of human prostaglandin H synthase 1 and 2 by indomethacin and flurbiprofen. J. Biol. Chem. 1996;271:3548–3554. doi: 10.1074/jbc.271.7.3548. [DOI] [PubMed] [Google Scholar]
- CHAN C.C., BOYCE S., BRIDEAU C., CHARLESON S., CROMLISH W., ETHIER D., EVANS J., FORD-HUTCHINSON A.W., FORREST M.J., GAUTHIER J.Y., GORDON R., GRESSER M., GUAY J., KARGMAN S., KENNEDY B., LEBLANC Y., LEGER S., MANCINI J., O'NEILL G.P., OUELLET M., PATRICK D., PERCIVAL M.D., PERRIER H., PRASIT P., RODGER I., TAGARI P., THERIEN M., VICKERS P., VISCO D., WANG Z., WEBB J., WONG E., XU L.-J., YOUNG R.N., ZAMBONI R., RIENDEAU D. Rofecoxib [Vioxx, MK-0966; 4-(4′-methylsulphonylphenyl)-3-phenyl-2-(5H)-furanone]: a potent and orally active cyclooxygenase-2 inhibitor. Pharmacological and biochemical profiles. J. Pharmacol. Exp. Ther. 1999;290:551–560. [PubMed] [Google Scholar]
- CHEN W., PAWELEK T.R., KULMACZ R.J. Hydroperoxide dependence and cooperative cyclooxygenase kinetics in prostaglandin H synthase-1 and -2. J. Biol. Chem. 1999;274:20301–20306. doi: 10.1074/jbc.274.29.20301. [DOI] [PubMed] [Google Scholar]
- CLARK K., KULATHILA R., KOEHN J., RIEFFEL S., STRAUSS A., HU S., KALFOGLOU M., SZETO D., LASALA D., SABIO M., WANG X., MARSHALL P. 2004. Crystal structure of the lumiracoxib : cyclooxygenase-2 complex. American Chemical Society (ACS) Book of Abstracts 2004; 22–26 August, Philadelphia, U.S.A. American Chemical Society: Philadelphia, U.S.A. (Abstract 178)
- CRYER B., FELDMAN M. Cyclooxygenase-1 and cyclooxygenase-2 selectivity of widely used nonsteroidal anti-inflammatory drugs. Am. J. Med. 1998;104:413–421. doi: 10.1016/s0002-9343(98)00091-6. [DOI] [PubMed] [Google Scholar]
- DEGEN P.H., DIETERLE W., SCHNEIDER W., THEOBALD W., SINTERHAUF U. Pharmacokinetics of diclofenac and five metabolites after single doses in healthy volunteers and after repeated doses in patients. Xenobiotica. 1988;18:1449–1455. doi: 10.3109/00498258809042267. [DOI] [PubMed] [Google Scholar]
- FARKOUH M.E., KIRSHNER H., HARRINGTON R.A., RULAND S., VERHEUGT F.W., SCHNITZER T.J., BURMESTER G.R., MYSLER E., HOCHBERG M.C., DOHERTY M., EHRSAM E., GITTON X., KRAMMER G., MELLEIN B., GIMONA A., MATCHABA P., HAWKEY C.J., CHESEBRO J.H. , on behalf of the TARGET Study Group Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), cardiovascular outcomes: randomised controlled trial. Lancet. 2004;364:675–684. doi: 10.1016/S0140-6736(04)16894-3. [DOI] [PubMed] [Google Scholar]
- FITZGERALD G.A. Coxibs and cardiovascular disease. N. Engl. J. Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- FLOWER R.J. The development of COX2 inhibitors. Nat. Rev. Drug. Discov. 2003;2:179–191. doi: 10.1038/nrd1034. [DOI] [PubMed] [Google Scholar]
- FUTAKI N., YOSHIKAWA K., HAMASAKA Y., ARAI I., HIGUCHI S., IIZUKA H., OTOMO S. NS-398, a novel non-steroidal anti-inflammatory drug with potent analgesic and antipyretic effects, which causes minimal stomach lesions. Gen. Pharmacol. 1993;24:105–110. doi: 10.1016/0306-3623(93)90018-s. [DOI] [PubMed] [Google Scholar]
- GIERSE J.K., KOBOLDT C.M., WALKER M.C., SEIBERT K., ISAKSON P.C. Kinetic basis for selective inhibition of cyclo-oxygenases. Biochem. J. 1999;339:607–614. [PMC free article] [PubMed] [Google Scholar]
- GILROY D.W., TOMLINSON A., WILLOUGHBY D.A. Differential effects of inhibitors of cyclooxygenase (cyclooxygenase 1 and cyclooxygenase 2) in acute inflammation. Eur. J. Pharmacol. 1998;355:211–217. doi: 10.1016/s0014-2999(98)00508-1. [DOI] [PubMed] [Google Scholar]
- HARTMANN S., SCOTT G., RORDORF C., CAMPESTRINI J., BRANSON J., KELLER U.Lumiracoxib demonstrates high absolute bioavailability in healthy subjects European Collaboration: Towards Drug Development and Rational Drug Therapy. Proceedings of the Sixth Congress of the European Association for Clinical Pharmacology and Therapeutics 2003Berlin: Springer-Verlag; 124ed. Tulunay, F.C. & Orme, M. p(Abstract P-199) [Google Scholar]
- HAY C.H., TREVETHICK M.A., WHEELDON A., BOWERS J.S., DE BELLEROCHE J.S. The potential role of spinal cord cyclooxygenase-2 in the development of Freund's complete adjuvant-induced changes in hyperalgesia and allodynia. Neuroscience. 1997;78:843–850. doi: 10.1016/s0306-4522(96)00598-2. [DOI] [PubMed] [Google Scholar]
- HUNT R.H., HARPER S., WATSON D.J., YU C., QUAN H., LEE M., EVANS J.K., OXENIUS B. The gastrointestinal safety of the COX-2 selective inhibitor etoricoxib assessed by both endoscopy and analysis of upper gastrointestinal events. Am. J. Gastroenterol. 2003;98:1725–1733. doi: 10.1111/j.1572-0241.2003.07598.x. [DOI] [PubMed] [Google Scholar]
- KRAUSE M.M., BRAND M.D., KRAUSS S., MEISEL C., VERGIN H., BURMESTER G.R., BUTTGEREIT F. Nonsteroidal antiinflammatory drugs and a selective cyclooxygenase 2 inhibitor uncouple mitochondria in intact cells. Arthritis Rheum. 2003;48:1438–1444. doi: 10.1002/art.10969. [DOI] [PubMed] [Google Scholar]
- KULMACZ R.J., LANDS W.E. Stoichiometry and kinetics of the interaction of prostaglandin H synthase with anti-inflammatory agents. J. Biol. Chem. 1985;260:12572–12578. [PubMed] [Google Scholar]
- LICHTENBERGER L.M., DIAL E.J., ROMERO J.J., LECHAGO J., JARBOE L.A., WOLFE M.M. Role of luminal ammonia in the development of gastropathy and hypergastrinemia in the rat. Gastroenterology. 1995;108:320–329. doi: 10.1016/0016-5085(95)90056-x. [DOI] [PubMed] [Google Scholar]
- MAMDANI M., JUURLINK D.N., LEE D.S., ROCHON P.A., KOPP A., NAGLIE G., AUSTIN P.C., LAUPACIS A., STUKEL T.A. Cyclo-oxygenase-2 inhibitors versus non-selective non-steroidal anti-inflammatory drugs and congestive heart failure outcomes in elderly patients: a population-based cohort study. Lancet. 2004;363:1751–1756. doi: 10.1016/S0140-6736(04)16299-5. [DOI] [PubMed] [Google Scholar]
- MANGOLD J.B., GU H., RODRIGUEZ L.C., BONNER J., DICKSON J., RORDORF C. Pharmacokinetics and metabolism of lumiracoxib in healthy male subjects. Drug Metab. Dispos. 2004;32:566–571. doi: 10.1124/dmd.32.5.566. [DOI] [PubMed] [Google Scholar]
- MASFERRER J.L., ZWEIFEL B.S., MANNING P.T., HAUSER S.D., LEAHY K.M., SMITH W.G., ISAKSON P.C., SEIBERT K. Selective inhibition of inducible cyclooxygenase 2 in vivo is antiinflammatory and nonulcerogenic. Proc. Natl. Acad. Sci. U.S.A. 1994;91:3228–3232. doi: 10.1073/pnas.91.8.3228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MERCK, SHARP & DOHME LIMITED 2004a. VIOXXACUTE 25 and 50 mg Tablets UK Summary of Product Characteristics, July 2004
- MERCK, SHARP & DOHME LIMITED 2004b. Vioxx Tablets and Oral Suspension UK Summary of Product Characteristics, July 2004
- MERCK, SHARP & DOHME LIMITED 2004c. Arcoxia 60, 90 & 120 mg Tablets UK Summary of Product Characteristics, July 2004
- MUKHERJEE A., HALE V.G., BORGA O., STEIN R. Predictability of the clinical potency of NSAIDs from the preclinical pharmacodynamics in rats. Inflamm. Res. 1996;45:531–540. doi: 10.1007/BF02342223. [DOI] [PubMed] [Google Scholar]
- PAIRET M., ENGELHARDT G. Distinct isoforms (COX-1 and COX-2) of cyclooxygenase: possible physiological and therapeutic implications. Fundam. Clin. Pharmacol. 1996;10:1–17. doi: 10.1111/j.1472-8206.1996.tb00144.x. [DOI] [PubMed] [Google Scholar]
- PATRIGNANI P., PANARA M.R., GRECO A., FUSCO O., NATOLI C., IACOBELLI S., CIPOLLONE F., GANCI A., CREMINON C., MACLOUF J. Biochemical and pharmacological characterization of the cyclooxygenase activity of human blood prostaglandin endoperoxide synthases. J. Pharmacol. Exp. Ther. 1994;271:1705–1712. [PubMed] [Google Scholar]
- PATRONO C., CIABATTONI G., PINCA E., PUGLIESE F., CASTRUCCI G., DE SALVO A., SATTA M.A., PESKAR B.A. Low dose aspirin and inhibition of thromboxane B2 production in healthy subjects. Thromb. Res. 1980;17:317–327. doi: 10.1016/0049-3848(80)90066-3. [DOI] [PubMed] [Google Scholar]
- PATRONO C., PATRIGNANI P., GARCIA RODRIGUEZ L.A. Cyclooxygenase-selective inhibition of prostanoid formation: transducing biochemical selectivity into clinical read-outs. J. Clin. Invest. 2001;108:7–13. doi: 10.1172/JCI13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHARMACIA LIMITED 2004. Bextra 10, 20 & 40 mg Tablets UK Summary of Product Characteristics, April 2004
- PINHEIRO R.M., CALIXTO J.B. Effect of the selective COX-2 inhibitors, celecoxib and rofecoxib in rat acute models of inflammation. Inflamm. Res. 2002;51:603–610. doi: 10.1007/pl00012435. [DOI] [PubMed] [Google Scholar]
- RIENDEAU D., PERCIVAL M.D., BRIDEAU C., CHARLESON S., DUBE D., ETHIER D., FALGUEYRET J.P., FRIESEN R.W., GORDON R., GREIG G., GUAY J., MANCINI J., OUELLET M., WONG E., XU L., BOYCE S., VISCO D., GIRARD Y., PRASIT P., ZAMBONI R., RODGER I.W., GRESSER M., FORD-HUTCHINSON A.W., YOUNG R.N., CHAN C.C. Etoricoxib (MK-0663): preclinical profile and comparison with other agents that selectively inhibit cyclooxygenase-2. J. Pharmacol. Exp. Ther. 2001;296:558–566. [PubMed] [Google Scholar]
- SAMAD T.A., MOORE K.A., SAPIRSTEIN A., BILLET S., ALLCHORNE A., POOLE S., BONVENTRE J.V., WOOLF C.J. Interleukin-1beta-mediated induction of COX-2 in the CNS contributes to inflammatory pain hypersensitivity. Nature. 2001;410:471–475. doi: 10.1038/35068566. [DOI] [PubMed] [Google Scholar]
- SCHNITZER T.J., BURMESTER G.R., MYSLER E., HOCHBERG M.C., DOHERTY M., EHRSAM E., GITTON X., KRAMMER G., MELLEIN B., MATCHABA P., GIMONA A., HAWKEY C.J. , on behalf of the TARGET Study Group Comparison of lumiracoxib with naproxen and ibuprofen in the Therapeutic Arthritis Research and Gastrointestinal Event Trial (TARGET), reduction in ulcer complications: randomised controlled trial. Lancet. 2004;364:665–674. doi: 10.1016/S0140-6736(04)16893-1. [DOI] [PubMed] [Google Scholar]
- SCOTT G., RORDORF C., BLOOD P., BRANSON J., MILOSAVLJEV S., GREIG G.Dose escalation study to assess the safety, tolerability, pharmacokinetics and pharmacodynamics of COX189 in healthy subjects Ann. Rheum. Dis. 200261Suppl. 1242(Abstract FRI0300)11830430 [Google Scholar]
- SCOTT G., RORDORF C., REYNOLDS C., KALBAG J., LOOBY M., MILOSAVLJEV S., WEAVER M., HUFF J.P., RUFF D.A. Pharmacokinetics of lumiracoxib in plasma and synovial fluid. Clin. Pharmacokinet. 2004a;43:467–478. doi: 10.2165/00003088-200443070-00003. [DOI] [PubMed] [Google Scholar]
- SCOTT G., YIH L., YEH C.M., MILOSAVLJEV S., LAURENT A., RORDORF C. Lumiracoxib: pharmacokinetic and pharmacodynamic profile when coadministered with fluconazole in healthy subjects. J. Clin. Pharmacol. 2004b;44:193–199. doi: 10.1177/0091270003262110. [DOI] [PubMed] [Google Scholar]
- SEDGWICK A.D., SIN Y.M., EDWARDS J.C., WILLOUGHBY D.A. Increased inflammatory reactivity in newly formed lining tissue. J. Pathol. 1983;141:483–495. doi: 10.1002/path.1711410406. [DOI] [PubMed] [Google Scholar]
- SIMON L.S., LANZA F.L., LIPSKY P.E., HUBBARD R.C., TALWALKER S., SCHWARTZ B.D., ISAKSON P.C., GEIS G.S. Preliminary study of the safety and efficacy of SC-58635, a novel cyclooxygenase 2 inhibitor: efficacy and safety in two placebo-controlled trials in osteoarthritis and rheumatoid arthritis, and studies of gastrointestinal and platelet effects. Arthritis Rheum. 1998;41:1591–1602. doi: 10.1002/1529-0131(199809)41:9<1591::AID-ART9>3.0.CO;2-J. [DOI] [PubMed] [Google Scholar]
- SMALE S., BJARNASON I. Determining small bowel integrity following drug treatment. Br. J. Clin. Pharmacol. 2003;56:284–291. doi: 10.1046/j.1365-2125.2003.01942.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SMITH W.L., DEWITT D.L., GARAVITO R.M. Cyclooxygenases: structural, cellular, and molecular biology. Annu. Rev. Biochem. 2000;69:145–182. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- SMITH W.L., LANGENBACH R. Why there are two cyclooxygenase isozymes. J. Clin. Invest. 2001;107:1491–1495. doi: 10.1172/JCI13271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STIERLIN H., FAIGLE J.W., SALLMANN A., KUNG W., RICHTER W.J., KRIEMLER H.P., ALT K.O., WINKLER T. Biotransformation of diclofenac sodium (Voltaren) in animals and in man. I. Isolation and identification of principal metabolites. Xenobiotica. 1979;9:601–610. doi: 10.3109/00498257909042327. [DOI] [PubMed] [Google Scholar]
- TOPOL E.J. Failing the Public Health – Rofecoxib, Merck, and the FDA. N. Engl. J. Med. 2004;351:1707–1709. doi: 10.1056/NEJMp048286. [DOI] [PubMed] [Google Scholar]
- WALLACE J.L., CHAPMAN K., MCKNIGHT W. Limited anti-inflammatory efficacy of cyclo-oxygenase-2 inhibition in carrageenan-airpouch inflammation. Br. J. Pharmacol. 1999;126:1200–1204. doi: 10.1038/sj.bjp.0702420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WARNER T.D., GIULIANO F., VOJNOVIC I., BUKASA A., MITCHELL J.A., VANE J.R. Nonsteroid drug selectivities for cyclo-oxygenase-1 rather than cyclo-oxygenase-2 are associated with human gastrointestinal toxicity: a full in vitro analysis. Proc. Natl. Acad. Sci. U.S.A. 1999;96:7563–7568. doi: 10.1073/pnas.96.13.7563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WENNOGLE L.P., LIANG H., QUINTAVALLA J.C., BOWEN B.R., WASVARY J., MILLER D.B., ALLENTOFF A., BOYER W., KELLY M., MARSHALL P. Comparison of recombinant cyclooxygenase-2 to native isoforms: aspirin labeling of the active site. FEBS Lett. 1995;371:315–320. doi: 10.1016/0014-5793(95)00930-8. [DOI] [PubMed] [Google Scholar]
- WHITEHOUSE M.W.Adjuvant-induced polyarthritis in rats Handbook of Animal Models for the Rheumatic Diseases 1988Raton: CRC Press; ed. Greenwald, R.A. & Diamond H.S., Vol. 1. Boca [Google Scholar]