

Abstract
The β-cell KATP channel is composed of two types of subunit – the inward rectifier K+ channel (Kir6.2) which forms the channel pore, and the sulphonylurea receptor (SUR1), which serves as a regulatory subunit. The N-terminus of Kir6.2 is involved in transduction of sulphonylurea binding into channel closure, and deletion of the N-terminus (Kir6.2ΔN14) results in functional uncoupling of the two subunits. In this study, we investigate the interaction of the hypoglycaemic agents repaglinide and glibenclamide with SUR1 and the effect of Kir6.2 on this interaction. We further explore how the binding properties of repaglinide and glibenclamide are affected by functional uncoupling of SUR1 and Kir6.2 in Kir6.2ΔN14/SUR1 channels. All binding experiments are performed on membranes in ATP-free buffer at 37°C.
Repaglinide was found to bind with low affinity (KD=59±16 nM) to SUR1 alone, but with high affinity (increased ∼150-fold) when SUR1 was co-expressed with Kir6.2 (KD=0.42±0.03 nM). Glibenclamide, tolbutamide and nateglinide all bound with marginally lower affinity to SUR1 than to Kir6.2/SUR1.
Repaglinide bound with low affinity (KD=51±23 nM) to SUR1 co-expressed with Kir6.2ΔN14. In contrast, the affinity for glibenclamide, tolbutamide and nateglinide was only mildly changed as compared to wild-type channels.
In whole-cell patch-clamp experiments inhibition of Kir6.2ΔN14/SUR1 currents by both repaglinide and nateglinde is abolished.
The results suggest that Kir6.2 causes a conformational change in SUR1 required for high-affinity repaglinide binding, or that the high-affinity repaglinide-binding site includes contributions from both SUR1 and Kir6.2. Glibenclamide, tolbutamide and nateglinide binding appear to involve only SUR1.
Keywords: Repaglinide, glibenclamide, SUR1, Kir6.2, binding, co-expression, coupling
Introduction
Repaglinide is a fast-acting prandial glucose regulator used in the treatment of type II diabetes (Ambavane et al., 2002). Repaglinide and sulphonylureas such as tolbutamide and glibenclamide share the common property that they are capable of closing ATP-sensitive potassium (KATP) channels. The KATP channel plays a key role in glucose-dependent insulin secretion from pancreatic beta cells (Ashcroft & Rorsman, 1990). KATP channels are open at low glucose concentrations, as metabolism is low, but close when glucose uptake and metabolism are stimulated by an increase in the plasma glucose concentration. This leads to membrane depolarization, activation of voltage-gated Ca2+ channels, Ca2+ influx and, in turn, insulin secretion. Inhibition of the KATP channel by repaglinide and sulphonylureas depolarizes the pancreatic β-cell and stimulates insulin secretion (Fuhlendorff et al., 1998). The hallmark of the KATP channel is that it is inhibited by intracellular ATP and activated by MgADP, thus enabling it to couple the metabolic state of the cell to the membrane potential and electrical activity (by sensing changes in intracellular ATP and ADP).
KATP channels consist of pore-forming Kir6.x subunits that associate with different kinds of regulatory sulphonylurea receptor subunits: SUR1, SUR2A or SUR2B (Seino & Miki, 2003). The β-cell KATP channel is composed of Kir6.2 and SUR1 (Inagaki et al., 1995; Sakura et al., 1995), the cardiac type of Kir6.2 and SUR2A (Inagaki et al., 1996), and the vascular smooth muscle type of Kir6.1 and SUR2B (Miki et al., 2002). SUR acts as a channel regulator, with the different SUR subtypes conferring differential sensitivity to the inhibitory effects of sulphonylurea drugs and the stimulatory actions of KATP channel openers and MgADP (Ashcroft & Gribble, 1998; Seino & Miki, 2003).
The precise regions of the protein involved in the functional coupling of SUR1 to Kir6.2 are not known. Co-expression of SUR1 with an N-terminally truncated Kir6.2 (Kir6.2ΔN14) results in channels insensitive to inhibition by tolbutamide and glibenclamide and with reduced ATP sensitivity secondary to an increased open probability (Babenko et al., 1999; Koster et al., 1999; Reimann et al., 1999). Interestingly, the Kir6.2ΔN14/SUR1 channels retain the ability to be stimulated by MgADP and diazoxide, suggesting that drug modulation of the channel can be induced by separate mechanisms (Reimann et al., 1999).
Studies on recombinant KATP channels suggest that drugs containing a sulphonylurea moiety (e.g. tolbutamide and glibenclamide) interact with residues in the cytoplasmic loop linking transmembrane segments 15 and 16 (TM15–16) of SUR1. Exchange of a single serine residue (S1237) in this loop abolishes the block by tolbutamide, renders the block by glibenclamide readily reversible and abolishes high-affinity [3H]glibenclamide binding to SUR1 (Ashfield et al., 1999). Photoaffinity labelling studies suggest that [125I]glibenclamide labels the N-terminal part of SUR1 (Aguilar-Bryan et al., 1995), and studies employing SUR1 expressed in insect cells suggest that the third cytoplasmic loop may also be involved in [3H]glibenclamide binding (Mikhailov et al., 2001).
Recent studies from our laboratory suggested that the binding site for repaglinide is not identical to that of sulphonylureas, since repaglinide binding and channel inhibition by this drug were unaffected by mutation S1237Y in SUR1, which abolishes KATP channel inhibition by tolbutamide (Hansen et al., 2002). In the present study, we examined the binding of repaglinide and glibenclamide when SUR1 was expressed alone or together with Kir6.2. Our results suggest that the Kir6.2-SUR1 protein interaction is required for high-affinity repaglinide, but not glibenclamide, binding.
Methods
Molecular biology
Human SUR1 cDNA (GenBank L78207) and human Kir6.2 (GenBank D50582) were cloned into pcDNA3.1(−) (Invitrogen). Mouse Kir6.2 (GenBank D50581) in which amino acids 2–14 were deleted (mKir6.2ΔN14) was transferred to pcDNA3.1(−) (Invitrogen) for mammalian expression (Reimann et al., 1999).
Cell culture and transfection
Human embryo kidney 293 (HEK293) cells were cultured in flasks at 37°C in a humidified atmosphere with 95% air and 5% CO2 in Dulbecco's modified Eagles medium with 4.5 g l−1 glucose (BioWhittaker) supplemented with 10% fetal calf serum, penicillin (100 U ml−1) and streptomycin (0.1 mg ml−1).
Transient transfections were performed using FuGene™ 6 Transfection Reagent (Roche) according to the manufacturer's instruction. Cells were seeded at 50% confluency and the following day transfected with mKir6.2ΔN14 plus hSUR1 at a molar ratio of 1 : 1. A plasmid containing GFP cDNA was added to the transient transfections for visual identification of transfected cells for the electrophysiological experiments. Experiments were performed 1–3 days after transfection. A cell line (HEK293) stably expressing human SUR1 and human Kir6.2 was used for the studies on the wild type (wt) hSUR1/hKir6.2 channels (Dabrowski et al., 2001).
In some transient transfection experiments, the molar ratio of Kir6.2 to SUR1 was varied. The amount of SUR1 was kept constant and the amount of Kir6.2 was varied in the following ratios (Kir6.2 : SUR1): 10 : 1, 1 : 1, 0.5 : 1 and 0.2 : 1.
Membrane preparation
The cells were harvested and centrifuged at 48,000 × g for 10 min at 4°C. The pellet was homogenized in ice-cold buffer (30 mM Tris–HCl, pH 7.4) using an Ultra Turrax for 20 s. Centrifugation and homogenization were repeated, the final pellet was resuspended in buffer and sucrose was added to a final concentration of 250 mM. Protein concentration was measured using the Bio-Rad Protein Assay. Membranes were stored at −80°C.
Binding experiments with [3H]repaglinide and [3H]glibenclamide
Binding experiments were performed in 96-well OptiPlates™ (Packard). Membranes were thawed on ice and incubated with [3H]repaglinide or [3H]glibenclamide in a total volume of 250 μl (200 μl membranes, 25 μl radioligand and 25 μl buffer/cold ligand – all diluted in 30 mM Hepes, pH 7.4), at 37°C for 60 min. The final concentration of membranes was 20–80 μg ml−1 of membrane protein. This results in a final buffer composition of 2.5–10 mM sucrose, 0.3–1.2 mM Tris and 29–30 mM Hepes. Bound [3H]repaglinide or [3H]glibenclamide was separated from free by rapid filtration on a Filtermate Harvester (Packard) through UniFilter®GF/B™ filterplates (Packard). Filterplates were washed five times with 450 μl water (room temperature) and dried. Scintillation cocktail (30 μl) (Microscint™, Packard) was added to each well and radioactivity was determined by counting the plates in a Microplate Scintillation and Luminescence Counter (Topcount-NTX™, Packard). Nonspecific binding was determined in the presence of 1 μM glibenclamide, and was <5% of the total binding. In competition experiments, 3 nM [3H]glibenclamide was used.
Electrophysiology
Whole-cell currents were recorded at 20–22°C using an EPC9 patch-clamp amplifier and Pulse+PulseFit v8.07 software. The extracellular bath solution contained (in mM): 140 NaCl, 5 KCl, 10 Hepes, 1.8 CaCl2 and 20 mannitol (pH 7.4 with NaOH). Cells were dialysed with intracellular solution containing (in mM): 120 KCl, 1 MgCl2, 5 EGTA, 2 CaCl2, 20 Hepes (pH 7.3 with KOH), 0.3 K2-ATP and 0.3 K2-ADP. Cells were clamped at −80 mV and currents were evoked by repetitive 200 ms, 10 mV depolarizing voltage steps. Signals were sampled at 20 kHz and filtered at 5 kHz.
Chemicals
Tolbutamide was purchased from Sigma (St Louis, U.S.A.) and glibenclamide from Research Biochemicals International (Natick, U.S.A.). Nateglinide was synthesized at Novo Nordisk A/S and repaglinide at Boehringer Ingelheim (Biberach/Riss, Germany). Concentrated stock solutions were prepared in DMSO for subsequent dilution in buffer. The concentration of DMSO in the experiments did not exceed 0.5% and had no effect on the binding or electrophysiological experiments. Radiolabelled glibenclamide was purchased from NEN Life Science Products (Boston, MA, U.S.A.).
Radiolabelled repaglinide was prepared in the Department of Isotope Chemistry, Novo Nordisk A/S, by catalytical tritiation of the repaglinide precursor S(+)-2-ethoxy-4-[2-[[3-methyl-1-[2-(piperidinyl)-phenyl]4-buten-yl]amino]2-oxoethyl]-benzoic acid kindly provided by Dr M. Mark, Boehringer Ingelheim. A specific activity of approximately 12 MBq μg−1 (144 Ci mmol−1) was estimated from mass spectroscopy of the final product.
Data analysis
Data are presented as mean±s.e.m. Competition-binding data were analysed in Prism™ (GraphPad) using the four-parameter logistic equation:
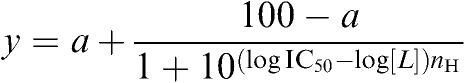 |
where y is specific binding, a is nonspecific binding, IC50 is the concentration that results in half-maximal inhibition, [L] is the concentration of drug, and nH is the Hill coefficient.
The experimental IC50 values for the competitive ligands were converted into Ki values using the Cheng–Prusoff equation:
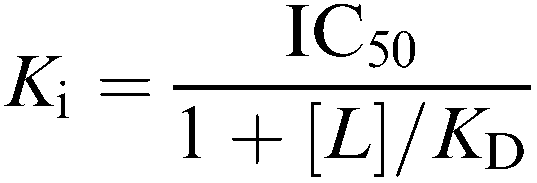 |
where [L] is the concentration of [3H]glibenclamide and the KD is the equilibrium dissociation constant for [3H]glibenclamide.
Ki and IC50 values were converted to log values (pKi=logKi) for statistical analysis (two-tailed unpaired Student's t-test).
Results
SUR1 and Kir6.2/SUR1 binding
To obtain information on the binding properties of repaglinide and glibenclamide, membranes were prepared from a stable HEK293 cell line co-expressing Kir6.2 and SUR1 or expressing SUR1 alone. In agreement with previous studies, saturation-binding experiments demonstrated high-affinity binding of both [3H]repaglinide and [3H]glibenclamide to Kir6.2/SUR1 (KD values of 0.42±0.03 nM (n=7) and 0.78±0.08 nM (n=7), respectively) (Figure 1). The density of receptor expression (Bmax) was 30±2 and 43±3 (n=5) pmol mg−1 of protein for repaglinide (n=6) and glibenclamide, respectively. When SUR1 was expressed alone, the affinity of [3H]repaglinide binding was about 150-fold lower than that of Kir6.2/SUR1 (KD=59±16 nM (n=6); Bmax=67±39 pmol mg−1 membrane protein) whereas the affinity of [3H]glibenclamide binding was only slightly changed (KD=3.27±0.3 nM (n=6); Bmax=45±15 pmol mg−1 membrane protein (Figure 1 and Table 2).
Figure 1.

Saturation analysis of [3H]glibenclamide (a) and [3H]repaglinide (b) binding to SUR1 expressed alone (open circles) or co-expressed with Kir6.2 (filled circles). Inset: Scatchard analysis of data. Single representative experiments.
Table 2.
KD values for [3H]repaglinide and [3H]glibenclamide binding
| SUR1 KD (nM) | Kir6.2/SUR1 KD (nM) | Kir6.2ΔN14/SUR1 KD (nM) | |
|---|---|---|---|
| [3H]repaglinide | 59±16 | 0.42±0.03 | 51±23 |
| [3H]glibenclamide | 3.27±0.30 | 0.78±0.08 | 1.91±0.18 |
Data are means±s.e.m. (n=3–7).
We examined the affinity of tolbutamide and nateglinide for SUR1 and Kir6.2/SUR1 by determining their ability to displace [3H]glibenclamide binding. For direct comparison, displacement studies with cold repaglinide and glibenclamide were also performed. With the exception of repaglinide, all drugs showed a monophasic displacement of [3H]glibenclamide binding with Hill coefficients close to unity (Figure 2). As shown in Table 1, the Ki values for tolbutamide, nateglinide and glibenclamide were in the same range for SUR1 and for Kir6.2/SUR1. Repaglinide showed a monophasic displacement of [3H]glibenclamide binding to SUR1, indicating that it interacted with a single binding site with a Ki (72 nM) similar to the KD (59 nM) obtained in saturation-binding experiments with SUR1 and [3H]repaglinide. In contrast, repaglinide displacement of [3H]glibenclamide binding to Kir6.2/SUR1 was biphasic, suggesting that it displaces glibenclamide from both a high- and a low-affinity binding site. The high-affinity binding site had a Ki (0.06 nM) similar to the KD found for [3H]repaglinide to Kir6.2/SUR1 (Figure 1). The low-affinity site had a Ki that was of the same order as that found for SUR1 alone.
Figure 2.

Inhibition of [3H]glibenclamide binding to SUR1 expressed alone (open circles) or co-expressed with Kir6.2 (filled circles) by repaglinide (a), glibenclamide (b), tolbutamide (c) and nateglinide (d).
Table 1.
Ki values for inhibition of [3H]glibenclamide binding to membranes expressing SUR1, Kir6.2/SUR1 and Kir6.2ΔN14/SUR1
| SUR1 Ki (nM) | Kir6.2/SUR1 Ki (nM) | Kir6.2ΔN14/SUR1 Ki (nM) | |
|---|---|---|---|
| Repaglinide | 72±9 | Site 1: 0.06±0.03 | 51±10 |
| Site 2: 28±9 | |||
| Glibenclamide | 3.8±1.9NS | 0.8±0.3 | 2.4±2.0NS |
| Tolbutamide | 79,000±6000* | 33,000±1000 | 48,000±1000* |
| Nateglinide | 2100±100* | 730±50 | 1200±30* |
Data are means±s.e.m. (n=3–5).
pKi values significantly different from Kir6.2/SUR1,
P<0.005 (t-test).
NS=not significantly different from Kir6.2/SUR1.
Co-expression with Kir6.2ΔN14
High-affinity inhibition of Kir6.2/SUR1 currents by tolbutamide and glibenclamide is abolished by N-terminal deletion of Kir6.2 (Reimann et al., 1999). We therefore examined the effect of repaglinide and nateglinide on Kir6.2ΔN14/SUR1 whole-cell currents in HEK293 cells (Figure 3). Concentrations of repaglinide (10 μM) and nateglinide (300 μM) producing maximal inhibition on wild type Kir6.2/SUR1 channels (mean inhibition of 98±1% (n=5) and 97±2 (n=4) for repaglinide and nateglinide, respectively) had only small effects on the N-terminally deleted channel. However, the Kir6.2ΔN14/SUR1 current could be blocked by BaCl2 (Figure 3), a nonspecific K+ channel blocker (Rudy, 1988).
Figure 3.

Whole-cell currents through Kir6.2ΔN14/SUR1 channels (upper panel) or Kir6.2/SUR1 channels (lower panel, from Hansen et al., 2002) expressed in HEK293 cells. The dotted line is the zero current level. Application of repaglinide, nateglinide and BaCl2 is indicated by solid horizontal lines.
We also determined the affinity of [3H]repaglinide and [3H]glibenclamide binding in HEK293 cells transiently expressing SUR1 and Kir6.2ΔN14 (KD=51±23 nM (n=3) and 1.91±0.18 nM (n=3), respectively). The results demonstrate that the binding affinities of Kir6.2ΔN14/SUR1 most closely resemble the affinities obtained for SUR1 expressed alone (Table 2). The Ki values obtained from displacement of [3H]glibenclamide bound to Kir6.2N14/SUR1 for nateglinide and tolbutamide were also similar to their affinities for SUR1 expressed alone (Table 1).
Kir6.2 to SUR1 ratio experiments
It can be seen in Figure 2 that repaglinide displaces [3H]glibenclamide from Kir6.2/SUR1 membranes in a biphasic manner. Experiments in which the ratio of Kir6.2 to SUR1 was varied in transient transfections are shown in Figure 4. These results show that the IC50 value and the Hill coefficient for repaglinide displacement of [3H]glibenclamide depend on the ratio of Kir6.2 to SUR1.
Figure 4.

Repaglinide displacement of [3H]glibenclamide from membranes expressing two different ratios of Kir6.2 to SUR1 (a). Data are from single representative experiments performed in triplicate. SUR1 data are taken from Figure 2. Log IC50 values (b) and Hill coefficients (c) for repaglinide displacement of [3H]glibenclamide (n=4–6).
Discussion
The results of this study indicate that the formation of the high-affinity binding site for repaglinide is dependent on the presence of both the SUR1 and Kir6.2 subunits of the KATP channel. Furthermore, by using a truncated form of Kir6.2, we showed that the N-terminus of Kir6.2 is involved in the formation of the high-affinity repaglinide-binding site. In contrast, the binding of KATP blockers such as nateglinide and the sulphonylureas, tolbutamide and glibenclamide, was only slightly affected by the presence of Kir6.2. Thus, the mode of action of repaglinide differs from that of these drugs.
Consistent with previous binding and electrophysiological studies, we find that repaglinide binds to Kir6.2/SUR1 channels with a KD in the low nanomolar range. It is likely that this high-affinity repaglinide-binding site mediates KATP channel closure because the functional effects of repaglinide on KATP currents and on insulin release occur at low nanomolar concentrations (Gromada et al., 1995; Leclercq-Meyer et al., 1997). A low-affinity binding site was also detected in displacement-binding studies (Figure 2). Fuhlendorff et al. (1998) also report biphasic displacement of [3H]glibenclamide from βTC3 cells, and Hu et al. (2000) report a low Hill coefficient (nH=0.58) for repaglinide displacement of [3H]glibenclamide bound to RIN-m5F cell membranes. It is possible that the low-affinity component of repaglinide binding arises from a fraction of SUR that is not complexed to Kir6.2, or that a fraction of the Kir6.2/SUR channels exist in an uncoupled state. In order to clarify this point, we performed experiments in which the ratio of Kir6.2 to SUR1 was varied. Although the molar ratio of Kir6.2 to SUR1 plasmids in the transfection might not reflect the actual protein ratio of Kir6.2 to SUR1, the results clearly show that the ratio of Kir6.2 to SUR1 influences the competition curve for repaglinide displacement of [3H]glibenclamide. We have also observed variations in the repaglinide competition curve for a stable cell-line expressing Kir6.2/SUR1 depending on the passage number, which is likely to reflect changes in expression of Kir6.2 and/or SUR1. It should be noted that the reason why the low-affinity repaglinide component is not seen in the saturation experiments is because the concentrations of radioligand used do not cover the low-affinity site.
In contrast to co-expression of Kir6.2 and SUR1, no high-affinity repaglinide binding was detected when SUR1 was expressed alone. Instead, a single binding site with a 100-fold reduced affinity was observed. One explanation for the observed low-affinity site could be that cell surface expression of monomeric SUR1 is downregulated by endoplasmic reticulum (ER) retention signals present in both Kir6.2 and SUR1 subunits, as has been reported in studies using Xenopus oocytes (Zerangue et al., 1999). The lipid environment is different in the plasma membrane from that in internal membranes, which could influence high-affinity repaglinide binding since the lipid composition of the membrane is known to regulate KATP channels (Ashcroft, 1998; Koster et al., 1999). Another factor potentially affecting repaglinide binding is whether SUR1 is present in monomeric or tetrameric form. However, because repaglinide also binds with low affinity to SUR1 co-expressed with Kir6.2ΔN14 (allowing functional SUR1-containing channels to be expressed in the plasma membrane), we favour the interpretation that repaglinide binds with high affinity only when octameric complexes of Kir6.2 and SUR1 are formed. Our results indicate that complex formation between Kir6.2 and SUR1 per se is not sufficient to produce high-affinity repaglinide binding. Instead, the N-terminus of Kir6.2 appears to be required. This provides further support for the idea that SUR1 may interact with the N-terminus of Kir6.2 (Babenko, 2002).
How does co-expression of Kir6.2 with SUR1 induce high-affinity repaglinide binding? There are at least two possibilities. First, the high-affinity binding site might involve residues from both SUR1 and from Kir6.2. If this is the case, then it appears that the N-terminus of Kir6.2 may contribute to the binding site, as high-affinity repaglinide binding is abolished when this is truncated. An alternative idea is that interaction of the N-terminus of Kir6.2 with SUR1 induces a conformational change in SUR1 that converts a low-affinity binding site to a high-affinity site.
In saturation studies using [3H]glibenclamide, we found only a four-fold decrease in KD (increase in affinity) upon co-expression of SUR1 with Kir6.2. Similar results were obtained in competition studies with unlabelled tolbutamide and nateglinide, indicating an almost identical Ki for inhibition of [3H]glibenclamide binding at SUR1 and Kir6.2/SUR1. Interestingly, glibenclamide binding to SUR2B has been reported to depend on the Kir6.x subtype co-expressed when measured in whole-cell assays but not in membranes prepared from the same cells (Hambrock et al., 2001).
At present, it is not clear if the unique binding properties of repaglinide reported in this study have any physiological or therapeutic relevance. However, depending on the extent to which SUR1 has a function in the absence of Kir6.2 complex formation, repaglinide crossreactivity is likely to be less. In this context, it is interesting to note that SUR1 has recently been reported to regulate nonselective cation channels in astrocytes (Simard & Chen, 2004).
In conclusion, these data support the idea that repaglinide has a functional high-affinity binding site, which differs from that of sulphonylureas and nateglinide. We hypothesize that formation of the high-affinity repaglinide-binding site only occurs when the Kir6.2 is functionally complexed with SUR1.
Acknowledgments
We gratefully acknowledge the expert technical assistance of Ms Tinna Fremming. F.M.A. is the Royal Society GlaxoSmithKline Research Professor.
Abbreviations
- HEK293 cell
human embryo kidney 293 cell
- KATP channel
ATP-sensitive potassium channel
- Kir
inwardly rectifying K+ channel
- SUR
sulphonylurea receptor
References
- AGUILAR-BRYAN L., NICHOLS C.G., WECHSLER S.W., CLEMENT J.P., BOYD A.E., GONZLEZ G., HERRERA-SOSA H., NGUY K., BRYAN J., NELSON D.A. Cloning of the beta cell high-affinity sulfonylurea receptor: a regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- AMBAVANE V., PATIL R., AINAPURE S.S. Repaglinide: a short acting insulin secretagogue for postprandial hyperglycaemia. J. Postgraduate Med. 2002;48:246–248. [PubMed] [Google Scholar]
- ASHCROFT F.M. Ion Channels: exciting times for PIP2. Science. 1998;282:1059–1060. doi: 10.1126/science.282.5391.1059. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., GRIBBLE F.M. Correlating structure and function in ATP-sensitive K+ channels. Trends Neurosci. 1998;21:288–294. doi: 10.1016/s0166-2236(98)01225-9. [DOI] [PubMed] [Google Scholar]
- ASHCROFT F.M., RORSMAN P. ATP-sensitive K+ channels: a link between B-cell metabolism and insulin secretion. Biochem. Soc. trans. 1990;18:109–111. doi: 10.1042/bst0180109. [DOI] [PubMed] [Google Scholar]
- ASHFIELD R., GRIBBLE F.M., ASHCROFT S.J., ASHCROFT F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the K(ATP) channel. Diabetes. 1999;48:1341–1347. doi: 10.2337/diabetes.48.6.1341. [DOI] [PubMed] [Google Scholar]
- BABENKO A.P. SUR-dependent modulation of KATP channels by an N-terminal KIR6.2 peptide. Defining intersubunit gating interactions. J. biol. chem. 2002;277:43997–44004. doi: 10.1074/jbc.M208085200. [DOI] [PubMed] [Google Scholar]
- BABENKO A.P., GONZALEZ G., BRYAN J. The tolbutamide site of SUR1 and a mechanism for its functional coupling to K(ATP) channel closure. FEBS Lett. 1999;459:367–376. doi: 10.1016/s0014-5793(99)01215-6. [DOI] [PubMed] [Google Scholar]
- DABROWSKI M., WAHL P., HOLMES W.E., ASHCROFT F.M. Effect of repaglinide on cloned beta cell, cardiac and smooth muscle types of ATP-sensitive potassium channels. Diabetologia. 2001;44:747–756. doi: 10.1007/s001250051684. [DOI] [PubMed] [Google Scholar]
- FUHLENDORFF J., RORSMAN P., KOFOD H., BRAND C.L., ROLIN B., MACKAY P., SHYMKO R., CARR R.D. Stimulation of insulin release by repaglinide and glibenclamide involves both common and distinct processes. Diabetes. 1998;47:345–351. doi: 10.2337/diabetes.47.3.345. [DOI] [PubMed] [Google Scholar]
- GROMADA J., DISSING S., KOFOD H., FROKJAER-JENSEN J. Effects of the hypoglycaemic drugs repaglinide and glibenclamide on ATP-sensitive potassium-channels and cytosolic calcium levels in beta TC3 cells and rat pancreatic beta cells. Diabetologia. 1995;38:1025–1032. doi: 10.1007/BF00402171. [DOI] [PubMed] [Google Scholar]
- HAMBROCK A., LOFFLER-WALZ C., RUSS U., LANGE U., QUAST U. Characterization of a mutant sulfonylurea receptor SUR2B with high affinity for sulfonylureas and openers: Differences in the coupling to Kir6.x subtypes. Mol. Pharmacol. 2001;60:190–199. doi: 10.1124/mol.60.1.190. [DOI] [PubMed] [Google Scholar]
- HANSEN A.M.K., CHRISTENSEN I.T., HANSEN J.B., CARR R.D., ASHCROFT F.M., WAHL P. Differential interactions of nateglinide and repaglinide on the human beta-cell sulphonylurea receptor 1. Diabetes. 2002;51:2789–2795. doi: 10.2337/diabetes.51.9.2789. [DOI] [PubMed] [Google Scholar]
- HU S.L., WANG S.Y., FANELLI B., BELL P.A., DUNNING B.E., GEISSE S., SCHMITZ R., BOETTCHER B.R. Pancreatic beta-cell K-ATP channel activity and membrane-binding studies with nateglinide: a comparison with sulfonylureas and repaglinide. J. Pharmacol. Exp. Therap. 2000;293:444–452. [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., NAMBA N., INAZAWA J., GONZALEZ G., AGUILAR-BRYAN L., SEINO S., BRYAN J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- INAGAKI N., GONOI T., CLEMENT J.P., WANG C.Z., AGUILAR-BRYAN L., BRYAN J., SEINO S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+ channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- KOSTER J.C., SHA Q., NICHOLS C.G. Sulfonylurea and K(+)-channel opener sensitivity of K(ATP) channels. Functional coupling of Kir6.2 and SUR1 subunits. J. Gen. Physiol. 1999;114:203–213. doi: 10.1085/jgp.114.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LECLERCQ-MEYER V., LADRIERE L., FUHLENDORFF J., MALAISSE W.J. Stimulation of insulin and somatostatin release by two meglitinide analogs. Endocrine. 1997;7:311–317. doi: 10.1007/BF02801324. [DOI] [PubMed] [Google Scholar]
- MIKHAILOV M.V., MIKHAILOVA E.A., ASHCROFT S.J.H. Molecular structure of the glibenclamide binding site of the beta-cell K-ATP channel. FEBS Lett. 2001;499:154–160. doi: 10.1016/s0014-5793(01)02538-8. [DOI] [PubMed] [Google Scholar]
- MIKI T., SUZUKI M., SHIBASAKI T., UEMURA H., SATO T., YAMAGUCHI K., KOSEKI H., IWANAGA T., NAKAYA H., SEINO S. Mouse model of Prinzmetal angina by disruption of the inward rectifier Kir6.1. Nat. Med. 2002;8:466–472. doi: 10.1038/nm0502-466. [DOI] [PubMed] [Google Scholar]
- REIMANN F., TUCKER S.J., PROKS P., ASHCROFT F.M. Involvement of the N-terminus of Kir6.2 in coupling to the sulphonylurea receptor. J. Physiol. 1999;518:325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUDY B. Diversity and ubiquity of K channels. Neuroscience. 1988;25:729–749. doi: 10.1016/0306-4522(88)90033-4. [DOI] [PubMed] [Google Scholar]
- SAKURA H., AMMALA C., SMITH P.A., GRIBBLE F.M., ASHCROFT F.M. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic beta-cells, brain, heart and skeletal muscle. FEBS Lett. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- SEINO S., MIKI T. Physiological and pathophysiological roles of ATP-sensitive K+ channels. Progr. Biophys. Mol. Biol. 2003;81:133–176. doi: 10.1016/s0079-6107(02)00053-6. [DOI] [PubMed] [Google Scholar]
- SIMARD J.M., CHEN M. Regulation by sulfanylurea receptor type 1 of a non-selective cation channel involved in cytotoxic edema of reactive astrocytes. J. Neurosurg. Anesthesiol. 2004;16:98–99. doi: 10.1097/00008506-200401000-00021. [DOI] [PubMed] [Google Scholar]
- ZERANGUE N., SCHWAPPACH B., JAN Y.N., JAN L.Y. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane KATP channels. Neuron. 1999;22:537–548. doi: 10.1016/s0896-6273(00)80708-4. [DOI] [PubMed] [Google Scholar]