

Abstract
4-(4-Fluorophenoxy)benzaldehyde semicarbazone (V102862) was initially described as an orally active anticonvulsant with robust activity in a variety of rodent models of epilepsy. The mechanism of action was not known. We used whole-cell patch-clamp techniques to study the effects of V102862 on native and recombinant mammalian voltage-gated Na+ channels.
V102862 blocked Na+ currents (INa) in acutely dissociated cultured rat hippocampal neurons. Potency increased with membrane depolarization, suggesting a state-dependent mechanism of inhibition. There was no significant effect on the voltage dependence of activation of INa.
The dissociation constant for the inactivated state (KI) was ∼0.6 μM, whereas the dissociation constant for the resting state (KR) was >15 μM.
The binding to inactivated channels was slow, requiring a few seconds to reach steady state at −80 mV.
The mechanism of inhibition was characterized in more detail using human embryonic kidney-293 cells stably expressing rat brain type IIA Na+ (rNav1.2) channels, a major Na+ channel α subunit in rat hippocampal neurons. Similar to hippocampal neurons, V102862 was a potent state-dependent blocker of rNav1.2 channels with a KI of ∼0.4 μM and KR ∼30 μM. V102862 binding to inactivated channels was relatively slow (k+≃1.7 μM−1 s−1). V102862 shifted the steady-state availability curve in the hyperpolarizing direction and significantly retarded recovery of Na+ channels from inactivation.
These results suggest that inhibition of voltage-gated Na+ channels is a major mechanism underlying the anticonvulsant properties of V102862. Moreover, understanding the biophysics of the interaction may prove to be useful in designing a new generation of potent Na+ channel blocker therapeutics.
Keywords: Anticonvulsant V102862, hippocampal neurons, HEK-293 cells, Na+ channel, state-dependent inhibition, inactivation, binding affinity, binding kinetics
Introduction
Voltage-gated Na+ channels play key roles in determining neuronal excitability (Catterall, 1992; 2000). The molecular biology of Na+ channels is complex. Native channels are comprised of α and β subunits, and probably other interacting proteins. In mammals, molecular studies have identified nine genes encoding distinct voltage-gated Na+ channel α subunits and three genes encoding β subunits (reviewed in Goldin et al., 2000; Goldin, 2002). The α subunits are large (∼260 kDa) membrane proteins comprised of four domains each containing six transmembrane regions, denoted S1–6. Expressed alone in mammalian cells, recombinant α subunits form functional voltage-gated channels indicating they contain the ion pore, voltage sensor and inactivation gate (e.g. Scheuer et al., 1990; West et al., 1992). The β subunits appear to play roles in modulating channel gating and regulating stability of expression (e.g. Isom et al., 1992; Patton et al., 1994). Site-directed mutagenesis studies indicate that the α subunit S4 region contains the voltage sensor and that the S5 and S6 regions line the channel pore (for review, see Catterall, 2000). Low-resolution, three-dimensional images of sodium channels generated by cryoelectron microscopy suggest a complex molecular structure, with multiple interconnecting pores (Sato et al., 2001). The different α subunits and β subunits have distinct spatial and temporal patterns of expression in central nervous system and peripheral nervous system and in non-neuronal tissues, implying that the different subtypes of channel have properties designed to serve distinct and specific functions (Catterall, 2000; Goldin, 2002).
Voltage- and use-dependent block of voltage-gated Na+ channels appears to be important in the mechanism of action of many anticonvulsants (e.g. phenytoin (PHT), carbamazepine (CBZ) and lamotrigine (LTG)), local anesthetics (e.g. lidocaine and bupivacaine) and Class I antiarrhythmics (Bean et al., 1983; Hondeghem & Katzung, 1984; Butterworth & Strichartz, 1990; Kuo & Bean, 1994; Xie et al., 1995; Kuo et al., 1997; 2000; Yang & Kuo, 2002). Drugs exploiting this mechanism of inhibition show enhanced blocking activity with increases in the level and/or frequency of membrane depolarization. The voltage dependence of inhibition can be understood by the modulated receptor model (Hille, 1977; Hondeghem & Katzung, 1984; Butterworth & Strichartz, 1990), which proposes that drugs bind to open or inactivated channels with higher affinity than to channels in resting states. Thus, dependence on membrane voltage is simply due to there being a higher proportion of channels in inactivated states vs resting states at depolarized voltages. Use-dependent block, seen during repetitive electrical discharges, is due to the accumulation of drug binding to open and inactivated channels during each depolarizing pulse. For Na+ channel blockers, use- and voltage dependence are considered critical for the ‘safety' of a drug at a molecular level, that is, the ability to block excessive/pathological firing patterns while at the same time minimizing effects on normal firing activity.
In 1993 and 1996, Dimmock et al. (1993; 1996) reported on (aryloxy)aryl semicarbazones as compounds with potent anticonvulsant activity in maximum electroshock- and pentylenetetrazole-induced seizure models in rodents. These compounds did not have obvious structural similarities with classical anticonvulsants, and their mechanism of action was not known. Initial binding experiments suggested interactions with γ-aminobutyric acid (GABA)-gated Cl− channels (Dimmock et al., 1993). However, these effects were only seen at high concentrations, and subsequent experiments revealed no positive modulation of native or recombinant GABAA channels expressed in Xenopus oocytes (E.R. Whittemore & R.M. Woodward, unpublished results). Among these compounds, 4-(4-fluorophenoxy)benzaldehyde semicarbazone, V102862 (formerly Co 102862), had a particularly attractive profile as an anticonvulsant, demonstrating potent activity in rodent models of tonic/clonic and partial-complex seizures with an encouraging side-effects profile vs ataxia (Carter et al., 1997).
In the current study, we investigated the mechanism of action of V102862 by assaying for interactions with voltage-gated INa in acutely dissociated rat hippocampal neurons. To understand the mechanism of inhibition in more detail, we also characterized the effects of V102862 on recombinant, rat brain type IIa Na+ channels (rNav1.2; Goldin et al., 2000), the most abundant Na+ channel α subunit in the rat hippocampal pyramidal layer (e.g. Catterall, 1992). Our experiments show that V102862 is a potent, broad-spectrum, state-dependent Na+ channel blocker.
Some of these findings have been presented in preliminary form at Meetings of the Society for Neuroscience (Ilyin et al., 1997; Ilyin & Woodward, 1998; Ilyin & Hodges, 1999).
Methods
Molecular biology
A conventional protocol was used to construct the cell line overexpressing rNav1.2 channels. Briefly, the plasmid pCMV-RIIA, which encodes the rat type IIa Na+ channel under control of the CMV promoter, was obtained from Dr Alan Goldin, University of California, Irvine, CA, U.S.A. The construct also contained the neomycinR gene for selection of stable cell lines with the neomycin analog G418. Bacterial growth, DNA preparations and restriction enzyme characterizations were performed using standard procedures (Sambrook et al., 1989). Human embryonic kidney (HEK)-293 cells were plated at a density of 3 × 105 cells well−1 in a six-well plate and transfected the following day with pCMV-RIIA DNA using Transfast reagent (Promega Inc., Madison, WI, U.S.A.). After 3 weeks of selection in 400 μg ml−1 G418, resistant colonies were isolated, expanded and plated in 35 mm dishes coated with poly-D-lysine for electrophysiological analysis. Several clones expressing tetrodotoxin (TTX)-sensitive inward currents were identified. Clone B2 (NaIIA-B2), which expressed the largest amplitude currents, was expanded and further electrophysiological analyses performed.
Cell preparation
Acute cultures of rat hippocampal neurons were prepared daily using a modification of procedures described previously (Kuo & Bean, 1994). Briefly, hippocampi were isolated from 3- to 11-day-old rat pup brains (Sprague–Dawley; Charles River Laboratories, Wilmington, MA, U.S.A.) and were sectioned, by hand, into 0.5–1 mm thick transverse slices (Whittemore & Koerner, 1991). Slices were incubated for at least 30 min at room temperature (20–24°C) in an oxygenated medium: (in mM) 124 NaCl, 3.3 KCl, 2.4 MgSO4, 2.5 CaCl2, 1.2 KH2PO4, 26 NaHCO3 (pH 7.4), continuously bubbled with 5% CO2/95% O2. Prior to recording, 4–5 slices were transferred to an oxygenated dissociation medium (in mM, 82 NaSO4, 30 K2SO4, 3 MgCl2, 2 HEPES, 26 NaHCO3, 0.001% phenol red, pH 7.4) containing 3 mg ml−1 protease XXIII (Sigma, St Louis, MO, U.S.A.) and incubated for 10–15 min at 37°C while continuously bubbling with 5% CO2/95% O2. Enzymatic digestion was terminated by transferring the slices to dissociation medium without bicarbonate, supplemented with 1 mg ml−1 bovine serum albumin and 1 mg ml−1 trypsin inhibitor (Sigma, St Louis, MO, U.S.A.). Slices were then transferred to a 35 mm culture dish containing dissociation medium without bicarbonate and triturated with a fire-polished glass Pasteur pipette to release single cells. Cells were allowed to settle in this dish for ∼30 min and were then used for electrical recordings.
HEK-293 cells were cultured using standard techniques, as described previously (e.g. Verdoorn et al., 1990). For electrophysiology, cells were plated onto 35 mm Petri dishes (precoated with poly-D-lysine) at a density of ∼2–4 × 104 cells dish−1 on the day of reseeding from confluent cultures. HEK-293 cells were suitable for recordings for 3–4 days after plating.
Whole-cell patch-clamp recordings
Whole-cell voltage-clamp recordings were made using conventional patch-clamp techniques (Hamill et al., 1981) at room temperature (22–24°C). Currents were recorded using an Axopatch 200A amplifier (Axon Instruments, Foster City, CA, U.S.A.) and were leak-subtracted (P/4 or by the built-in analog circuitry), low-pass filtered (3 kHz, 8-pole Bessel), digitized (20–50 μs intervals) and stored using Digidata 1200 B interface and Pclamp8/Clampex software (Axon Instruments, Union City, CA, U.S.A.).
In the recording chamber, hippocampal neurons were continuously superfused at a speed of about 1 ml min−1 with Tyrode's solution (in mM): 148 NaCl, 3.5 KCl, 2 CaCl2, 5 NaHCO3, 10 HEPES, 10 D-glucose, pH 7.4. Thin-walled pipettes were pulled from 100-μl Clay Adams Accu-Fill 90 Micropet disposable pipettes (Becton, Dickenson and Company, Parsipanny, NJ, U.S.A.), fire-polished and sylgarded (Dow-Corning, Midland, MI, U.S.A.). The pipettes were filled with an internal solution containing (in mM) 130 CsF, 20 NaCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES, with pH adjusted to 7.4 with CsOH.
The external solution for NaIIA-B2 (HEK-293) cells contained (in mM) 150 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, 10 D-glucose, 5 HEPES, with pH adjusted to 7.4 with NaOH. The patch-clamp pipettes were pulled from WPI, thick-walled borosilicate glass (WPI, Sarasota, FL, U.S.A.). The internal solution contained (in mM) 130 CsF, 20 NaCl, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 HEPES, with pH adjusted to 7.4 with CsOH; osmolality was set at ∼10 mmol kg−1 lower than that for the external solution. The pipette resistances ranged from 1.5 to 3 MΩ. Residual series access resistance was in the range of 0.5–1.2 MΩ after partial (75–80%) cancellation using built-in amplifier circuitry. The junction potential calculated using JPCalcW software (Cell MicroControl, Virginia Beach, VA, U.S.A.) was small (<7 mV); so, no correction of the holding voltage was made.
Drug solutions and intervening intervals of wash were applied through a linear array of flow pipes (Drummond Microcaps, 2-μl, 64-mm length) producing full solution exchange within a few hundred milliseconds. V102862 was dissolved in dimethylsulfoxide (DMSO) and then diluted in DMSO to make 1–30 mM stock solutions. The stock solutions were then diluted into working external saline solutions on each day of experimentation. PHT (sodium salt) and carbamazepine were from RBI (Natick, MA, U.S.A.) and LTG was synthesized in-house. Stock solutions of 20–200 mM were made up in DMSO. For all drugs, the concentration of DMSO in most experiments did not exceed 0.1% by volume, which had no measurable effect on INa. For isolated experiments, in particular when testing 6 or 8 μM V102862 or 300 μM PHT, DMSO was used up to 1%. In these experiments, data were corrected for a minor inhibition caused by the vehicle.
Statistics
Data are described as mean±standard error of the mean (n=number of cells) with statistical significance assessed (where needed) with paired t-test. P<0.05 indicates significance.
Results
V102862 inhibits INa in rat hippocampal neurons in a voltage-dependent manner
Depolarization of hippocampal neurons from −80 to −10 mV evoked transient inward currents that showed profound inactivation within a few milliseconds (Figure 1a). The currents were abolished by 400 nM TTX (data not shown) and reversed around the Na+ equilibrium potential (Figure 1c), indicating that they were due to activation of voltage-gated Na+ channels. At a concentration of 1 μM, V102862 had little effect on INa activated from a holding potential of −100 mV. However, as the holding potential was depolarized, V102862 produces increasing levels of inhibition. In the experiment illustrated in Figure 1a and b, 1 μM V102862 produced 17% inhibition of INa activated from a holding voltage of −80 mV, and 54% inhibition of currents activated from a holding potential of −60 mV. Note that over this range of holding potentials, the size of the peak INa declines two- to three-fold due to voltage-dependent inactivation of the Na+ channels (Figure 2b).
Figure 1.

Block of INa in hippocampal neurons by V102862. The currents shown in this and other figures are measured after ⩾1 min in control or drug-containing solutions, that is, under steady-state drug binding conditions. INa values were elicited by pulsing to −10 mV (maximal INa, panel c), either from holding potentials of −80 mV (a) or −60 mV (b): first under control conditions and then in the presence of 1 μM V102862 (lower and upper traces in each pair, respectively). (c) Current–voltage relationships taken in another hippocampal neuron in control, in the presence of 3 μM V102862 and upon washout. It illustrates that V102862 inhibits currents caused by different test voltages by equal amounts and does not affect appreciably the shape of current–voltage curve. Recordings were taken from a holding voltage of −70 mV by a series of test voltage pulses (25 ms long) incremented by 5 mV every other second. (d) Concentration-dependent inhibition of INa by 0.3–3 μM V102862 at −60 mV (data from a distinct neuron). Inset: The chemical structure of V102862.
Figure 2.

(a) Dose–response curves for inhibition of INa in hippocampal neurons by V102862 (0.1–3 μM) at different holding membrane voltages from −60 to −100 mV. The data are means±s.e.m. with n=3–5 for each concentration point. The lines are sigmoidal curve fits, 1/(1+([V102862]/KD)), to experimental mean values, where [V102862] is the concentration of V102862 and KD is the apparent dissociation constant. The KD values are 0.85 μM (−60 mV), 1.48 μM (−70 mV), 3.3 μM (−80 mV), 7.8 μM (−90 mV) and 15.4 μM (−100 mV), correspondingly. (b) Mean steady-state inactivation (availability) curve for Na channels in hippocampal neurons. The individual curves were taken by a series of 100 ms long depolarizing conditioning pulses followed up by a 15 ms test pulse to −10 mV. The points are an average of individual points taken from four distinct neurons (the arrow bars are omitted for the sake of clarity). The line is the fit of Boltzmann function, 1/[1+exp((V+67.5)/7.66)], where V is the conditioning voltage. (c) The apparent affinity of V102862, expressed as 1/KD, plotted against the holding potential. The KD values were taken from sigmoidal curve fits in (a). The line is a Boltzmann function, 1/KD=1.59/[1+exp(−(V+67.5)/7.66)], which, as predicted by the model (Kuo & Bean, 1994), is approximately a mirror image of the inactivation curve in (b).
At a fixed depolarized holding voltage, V102862 blocked INa elicited by pulsing to different test potentials to similar degrees. Thus, although the level of inhibition was strongly dependent on holding potential, it appeared to be independent of the activation potential (Figure 1c). The inhibition was reversible on drug washout (e.g. Figure 1c), but recovery required longer intervals of wash at depolarized voltages.
At any specific holding potential, levels of inhibition increased progressively with the concentration of V102862 (Figures 1d and 2a). Even at the highest concentration tested, the compound did not seem to affect the time course of the INa at any holding voltage (data not shown). The block by various concentrations of V102862 could be adequately fit by a simple one-to-one binding curve for each membrane voltage for the mean data presented in Figure 2a. The highest levels of inhibition were seen at a membrane voltage of −60 mV, where V102862 blocked the current with an apparent KD of ∼0.85 μM. At this potential, INa was reduced by ∼73% due to steady-state inactivation of the channels (Figure 2b). The weakest block was seen at the holding potential of −100 mV, where 3 μM V102862 inhibited current by only ∼15%. The apparent KD at this holding potential was >15 μM. Thus, the affinity of the drug increased on average by >18-fold in the range of membrane voltages between −100 and −60 mV. Actually, in some individual neurons, the affinity shift was more pronounced and exceeded 30-fold (data not shown).
These experiments demonstrated a clear correlation between the inhibitory potency of V102862 and the degree of steady-state inactivation of the Na+ channels: that is, the higher the degree of inactivation, the higher the apparent potency of V102862. This finding is consistent with the modulated-receptor hypothesis (Hille, 1977; Hondeghem & Katzung, 1984), that is, with the idea that V102862, like other anticonvulsants (e.g. Rogawski & Porter, 1990), binds to inactivated channels with significantly higher affinity than to channels in the resting state. The validity of the hypothesis can be assessed by reference to a simplified state diagram (Bean et al., 1983; Bean, 1984; Kuo & Bean, 1994), which is a particular version of the modulated-receptor model appropriate for interpreting most of our experiments:
Scheme 1
Here V is the voltage necessary to affect transition between states, R and I are the resting and inactivated states of the channel, B is the blocking agent and RB and IB are drug-bound (and inhibited) resting and inactivated states. At the two extremes (i.e. at very negative holding voltages when all channels are in state R and very depolarized voltages when all channels are in state I), block by V102862 will simply follow 1 : 1 binding to receptor with dissociation constants KR and KI, respectively. For intermediate voltages, where only some channels are in state I, block of the current is more complicated. Binding to inactivated channels is electrically silent, but due to the equilibrium between resting and inactivated channels, decrease in the fraction of channels in state I will cause a proportional decrease in the fraction in state R and thus a smaller peak current elicited by depolarizing test pulses. This scheme is an oversimplification because it ignores interactions between V102862 and open states. However, these seem negligible using our experimental protocols (discussed below). In addition, the simple model does not take account of the existence of multiple inactivated states. Nevertheless, this scheme is useful to interpret our data, since the main assumptions on 1 : 1 binding and tighter binding to inactivated state as compared to resting state hold for most of our experimental protocols.
Applying the simple model, the apparent dissociation constant KD at a given holding voltage is determined by combination of the actual binding reactions to resting and inactivated states:
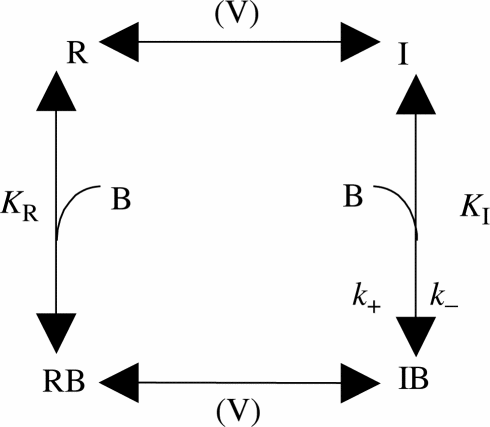 |
where h is the fraction of channels in state R in the absence of drug and KR and KI are the dissociation constants for the resting and inactivated states, respectively (Bean, 1984). If KR≫KI, KD∼KI/(1−h), or KD is equal to true dissociation constant KI divided by the degree of steady-state inactivation at the same voltage. The reciprocal of KD (the apparent affinity) plotted against holding voltage should have the same voltage dependence as the degree of steady-state inactivation, that is, it should be the mirror image of the conventional inactivation curve (Kuo & Bean, 1994). Figure 2c shows that this prediction is consistent with the data for V102862. Hence at strongly depolarized voltages, where inactivation approaches 100%, the apparent dissociation constant will approach the true KI of the blocker for the inactivated channel. From Figure 2c, the KI for V102862 is estimated as 1/1.59 or ∼0.63 μM. This figure is somewhat lower than 0.85 μM derived from the Hill equation fit to the data at −60 mV where >70% channels were in inactivated state.
The dissociation constant (KR) for resting states of the Na+ channel, made from the fit of the concentration–inhibition curve at a holding potential of −100 mV, was ∼15 μM. However, this value is only a rough estimate, since inhibition of the sodium current by V102862 was modest (<20%) at the highest concentration tested (8 μM – the limit of solubility), and inactivation of Na+ channels may not be completely removed at −100 mV. In general, the assumption on 1 : 1 binding seems to agree well with the data (Figure 2a).
Inhibition of INa by V102862 in hippocampal neurons is slow
At a holding voltage of −80 mV, V102862 blocked INa relatively slowly. As shown in Figure 3a, V108262 blocks currents at a rate that gradually increases with the drug concentration. However, even at the highest concentration, the steady-state inhibition is reached in a time scale of seconds. The plot in Figure 3b was used to estimate the rate constant of inhibition at −80 mV, which is equal to the slope of the linear fit to the data points, that is, ∼0.06 μM−1 s−1. The off-time constant did not appear to be dependent on the concentration of the drug. Note that at −80 mV, only about 20% of channels are in inactivated states (Figure 2b). If the development of block reflects mainly binding to the inactivated state, then the rate of block should increase with membrane depolarization. We did see acceleration of inhibition at more depolarized voltages (data not shown). At the voltage when all the channels enter inactivated state, the rate of block should saturate and lose its dependence on membrane voltage (see the section on the measurement of k+ in HEK-293 cells).
Figure 3.

(a) Onset and recovery from inhibition by V102862 at −80 mV. The cell was voltage-clamped at −80 mV and 200 pulses (each 5 ms in duration) were generated to −10 mV at 2 Hz. After approximately 25 s (base line), V102862 was applied via a fast solution exchange shooter for about 30–35 s. This was followed up by 30–35 s long wash interval. The protocol was repeated with increasing concentrations of V102862 every 2–4 min. The peak currents were normalized to mean current in control (before fast application of the drug) and plotted vs time. The superimposed time courses of inhibition are presented for four different concentrations of V102862. Mono-exponential fits (not presented) to both onset and recovery from inhibition returned the following time constants, correspondingly: 4.6 and 5.8 s for 0.3 μM (squares); 3.3 and 6.1 s for 1 μM (up-triangles); 2.5 and 7.8 s for 3 μM (down-triangles); and 1.5 and 7.5 s for 8 μM (diamonds). (b) The reciprocal of blocking time constant is plotted against V102862 concentration. The solid line is the best least-square fit with a slope of ∼0.06 μM−1 s−1.
V102862 is more potent than phenytoin
In eight separate hippocampal neurons, we tested V102862 side by side with PHT, an anticonvulsant widely used for treatment of generalized tonic–clonic seizures and partial seizures (Rogawski & Porter, 1990). Like V102862, inhibition of INa by PHT became stronger at depolarized membrane voltages, indicating a state-dependent mechanism of action (Figure 4). But at each voltage, ⩾10-fold higher concentrations of PHT were needed to cause inhibition comparable to the level seen with V102862.
Figure 4.

Partial dose–response curves for inhibition of INa by 0.3–3 μM V102862 (open symbols) and 100 and 300 μM PHT (solid symbols) taken in the same hippocampal neuron at different holding membrane voltages of −60, −70, −80 and −90 mV. Partial, two point inhibition–concentration curves for PHT are consistently shifted to the right for all holding voltages tested (from −60 to −90 mV), indicating significantly (10- to 30-fold) lower potency of the drug as compared to V102862. Similar shifts in potencies were measured in seven more neurons.
Characterization of HEK-293 cells expressing rNav1.2 sodium channels
Type I (Nav1.1) and Nav1.2 account for >85% of the sodium channels in adult rat brain (Gordon et al., 1987). Expression of rNav1.2 channels in mammalian cells therefore represents a useful in vitro model for characterizing the molecular pharmacology of anticonvulsants and other Na+ channel blockers (Ragsdale et al., 1991).
Depolarization of NaIIA-B2 cells from −90 to +40 mV triggered transient inward currents that inactivated within a few milliseconds (Figure 5). The currents were half-maximally activated by depolarization to −18.2±0.3 mV (n=13) with a steepness of 5.7±0.3 mV and reached maximum at ∼0 mV with a mean current density of 252±33 pA pF−1 (n=15). Using 100 ms depolarizing conditioning prepulses, half-maximal inactivation occurred at Vhalf=−53.9±1.7 mV with k=6.4±0.1 mV per e-fold change in membrane potential (n=17). The currents were completely blocked by 400 nM TTX and reversed at 65±2 mV (n=15), that is, at a voltage close to the Na+ equilibrium potential under these experimental conditions. The data indicate that the currents were due to activation of voltage-gated Na+ channels. Untransfected cells displayed only small (peak ⩽200 pA) inward currents that were partially blocked by 1 μM TTX. This indicated that Na+ channels in NaIIA-B2 cell line are encoded by the transfected cDNA and are not of native origin. The currents displayed fast activation and inactivation properties characteristic of Na+ channels reported in Chinese hamster ovary cells expressing the rat brain IIA channel (e.g. Ragsdale et al., 1991; West et al., 1992).
Figure 5.

Effects of membrane holding voltage on block of INa by 1 μM V102862 in NaIIA-B2 cells. INa was elicited by pulsing to 0 mV for 5 ms, either from holding potentials of −120 mV (a) or −63 mV (b) first under control conditions and then in the presence of the drug. (a) At −120 mV, all Na channels are in resting state (see the availability curve in (c)) and V102862 blocks the peak current only by 4%. (b) At −63 mV, about 50% of Na channels are in inactivated state (panel (c)). Current traces were scaled up (scaling factor equals 1.926; note also the difference in the current calibration bars for (a) and (b)) to match the size of the control current at −120 mV. At this voltage, 1 μM V102862 inhibits the peak current by 61%. (c) Steady-state inactivation curve taken in the same cell with a series of 100 ms long conditioning depolarizing prepulses and a 5 ms test pulse to 0 mV. The solid line is the fit of Boltzmann function, 1/{1+exp((V+63.6)/4.81)], where V is the conditioning voltage. (d) Same traces as in (b), but the current trace in the presence of the drug (dotted trace) was scaled up to match the peak of the current in control. The overlap demonstrates that V102862 does not significantly affect either activation or inactivation kinetics of Na+ current.
Voltage-dependent inhibition of rNav1.2 channels by V102862
Similar to results from hippocampal neurons, inhibition of INa by V102862 in NaIIA-B2 cells showed a clear dependence between apparent potency and membrane depolarization (Figure 5a and b). In the illustrated example, 1 μM V102862 blocked 4% of the current at −120 mV and 61% of the current at −63 mV. In this cell, −63 mV approximately matched Vhalf (Figure 5c); thus, at −63 mV, the maximal INa was reduced by ∼50% of its control value at −120 mV, due to inactivation. Clearly, 1 μM V102862 did not affect either the activation or inactivation kinetics of INa (Figure 5d). At each voltage, V102862 blocked INa in a concentration-dependent manner (Figure 6a). It is noteworthy that the assumption about 1 : 1 binding agrees well with the data in the steady state. Partial concentration–inhibition curves were shifted to the left with depolarizing voltages, indicating enhancement in the apparent potency of the block: IC50 values were 15.9 μM at −95 mV and 0.28 μM at −65 mV, that is, ∼60-fold increase in potency for a 30 mV positive shift in holding potential. The profound effect of membrane voltage is consistent with the hypothesis that V102862 has higher affinity for inactivated states of rNav1.2 channels than for resting states. This is further supported by the observation that the drug caused a progressive leftward shift in Vhalf of the steady-state inactivation curve for conditioning depolarizing pulses of relatively long duration (Figure 6b). For example, at 1 and 6 μM V102862, the shift was −5.9 and −13.8 mV, respectively, when a 5 s long prepulse was used. This translates in the reduction of channel availability at −70 mV from 82% in control to 59 and 22% at 1 and 6 μM V102862, respectively. There was no significant shift in Vhalf using a 100 ms long conditioning prepulse (data not shown). The requirement of prolonged depolarizing prepulses to produce a significant Vhalf shift suggested slow kinetics of binding to inactivated channels. V102862, like other anticonvulsants, works as an ‘inactivation state stabilizer', that is, it shifts the equilibrium in the state diagram (Scheme 1) toward inactivated states so that fewer channels are available for excitation in the presence of drug at physiologically relevant voltages.
Figure 6.

(a) Effects of membrane holding voltage on block of Na+ currents by 0.1–6 μM V102862. Na+ currents were elicited by pulsing to 0 mV, from holding potentials of −95, −80 or −65 mV. The lines were drawn according to the Hill equation with KD values of 15.9, 1.64 and 0.28 μM, respectively, and the slope fixed at 1.0. (b) Normalized inactivation curves measured with 5 s long prepulse. The cell was held at −120 mV and given an inactivating conditioning prepulse from −120 to −30 mV, immediately followed by 5 ms testing pulse to 0 mV every 15 s. The lines are fits to a Boltzmann function, 1/[1+exp((V−Vhalf)/k)]. The Vhalf and k values were −62.7 and 4.8, −68.6 and 4.8 and −76.5 mV and 5.3 mV in control, 1 μM V102862 and 6 μM V102862, respectively.
Block of resting channels
To assess the block of resting channels, we measured the partial concentration–inhibition curve at a highly negative membrane voltage, where residual inactivation is completely removed. According to Figures 5c and 6b, the holding voltage should be more negative than −100 mV. At these negative potentials, inhibition of INa, even at 3 μM V102862, is weak (<10%). Thus KR cannot be directly measured due to solubility limit of V102862 (∼8 μM at 1% DMSO). The best estimate for KR is obtained by measuring the fractional response (FR) for peak current at a very negative holding voltage (–120 mV) at the highest concentration (6–8 μM) of V102862. Then, assuming one-to-one binding of V102862 to resting channels, a simplified version of the Hill equation relating KR and FR is KR≃(FR/(1−FR))[V102862], where [V102862] is a single-point concentration of the blocker. The closer the single-point concentration is to the IC50, the better the estimate of KR. Using this approach, we estimated that KR for Nav1.2 channels is 29±3 μM (n=5).
Retardation of recovery of rNav1.2 channels from inhibition by V102862
Anticonvulsants and local anesthetics are known to affect the recovery (repriming) of Na+ channels from inactivation retarding it (e.g. Bean et al., 1983; Ragsdale et al., 1991; Kuo & Bean, 1994). This feature is thought to play a role in use-dependent block and filtering of pathological bursts of action potentials in episodes of hyperexcitability (Ragsdale et al., 1991; see for review Butterworth & Strichartz, 1990).
The rate of recovery from inactivation was assessed using a double-pulse protocol where the depolarizing prepulse was followed by a step back to the initial negative holding voltage for varying durations (recovery gap) and then by a test pulse to examine the extent of channel repriming (Figure 7). The holding (and gap) voltage was set at −120 mV to remove residual steady-state inactivation, the conditioning prepulse was set to −20 mV to drive all channels into inactivated states and the test pulse voltage was 0 mV. Duration of the conditioning prepulse was chosen to be sufficiently long (⩾1 s) to permit a steady-state level of binding of the drug to inactivated channels.
Figure 7.

Retardation of recovery from inactivation by V102862. In control and in the presence of 3 μM V102862, the cell was held at −120 mV and stepped to −20 mV for 1 s followed by recovery gap potential at −120 mV for variable length. Then, the cell was stepped to 0 mV for 10 ms to assess the available current and the cycle was repeated every 5 s. (a) Recovery gap was varied in Δt=0.2 or 10 ms increments. A total of 43 data points were taken with Δt=0.2 ms to acquire the fast component (within first 10 ms) of the recovery from inactivation; then, the recovery was remeasured with Δt=10 ms increments. The data sets were superimposed for both control and drug application. Mono-exponential fits (solid lines) were performed separately for each data set. In control, the fast component has τ1=2.3 ms and amplitude ∼0.85; the slow component has τ2∼130 ms. In the presence of 3 μM V102862, the amplitude of the fast component (τ1=2.3 ms) drops down to ∼0.13 and recovery proceeds at a slower pace with τ2∼560 ms. Obviously, within the first 10 ms, the majority (>85%) of the control currents have recovered while most currents in V102862 have not. (b) Normalized mean time course of recovery from inactivation with hyperpolarizing gap varied in Δt=50 ms increments. Mono-exponential fit to the data in the presence of 3 μM V102862 returns the mean retardation constant, τrepr, equal to 670 ms (10 cells; each point a mean of 10 measurements). Recovery in control also follows single exponential time course with the mean time constant of 200 ms (eight cells; each point a mean of ⩾3 measurements); it reflects the slow, low-magnitude component of biphasic recovery shown in (a).
Under control conditions, recovery of rNav1.2 channels from inactivation was biphasic. About 80–85% of channels recovered with the time constant of 2–5 ms. The remaining 15–20% of channels recovered more slowly, with the time constant of ⩾200 ms (Figure 7a). Presumably, the slow component of recovery reflects a slower inactivation process (e.g. Figure 4b in Kuo & Bean, 1994). In the presence of 3 μM V102862, the recovery process remained biphasic (Figure 7a), but while the time constant of the fast component appeared to be unchanged, its magnitude dropped from 85% in control to about 13% in the presence of the drug. Within the first 10 ms following the conditioning prepulse currents recovered to 86±2% in control experiments and only to 16±1% in the presence of 3 μM V102862 (n=5). The rapid phase of recovery was presumably due to rapid repriming of channels that did not bind V102862 during the conditioning prepulse. In contrast, the predominant fraction of channels reprimed with much slower kinetics with a time constant (τrepr) of about 560 ms. This slow component appears to reflect the relatively slow recovery of drug-bound channels. In the presence of the drug, the current never recovered to its initial control value. For example, in Figure 7a, it approached an asymptote of 85% of the recovery level in control. This residual block, which averaged 9±3% (n=5), was primarily due to low-affinity block of resting channels by V102862. This explanation is supported by calculating the KD value for the residual block using the modified logistic equation KD≅(FR/(1−FR))[V102862]=(0.91/0.09) × 3 μM=30 μM, which matches well with the KR of 29 μM measured earlier.
Given the amplitude and time scale of the retardation of recovery, in subsequent experiments we did not measure the fast component, but just the predominant decelerated process of repriming in the presence of V102862. For this purpose, the recovery gap duration was varied in 50 or 100 ms steps (Figure 7b). The time course of recovery in the presence of V102862 was well fitted with a mono-exponential giving a mean τrepr value of 680±43 ms (n=16; for the experiments including both 50 and 100 ms gap increments). The retardation of recovery was not obviously dependent on V102862 concentration, but was accelerated by membrane hyperpolarization (compare Bean et al., 1983; Ragsdale et al., 1991; Kuo et al., 1997).
Kinetics of binding of V102862 to inactivated rNav1.2 channels
Significant retardation of repriming from inactivation by V102862 provided us with another way to measure binding of the drug to inactivated channels by using a modified double-pulse protocol. From the comparison of the degree of recovery within the first 10 ms (>80% in control, ∼16% in the presence of the drug), the duration of the hyperpolarizing gap in the stimulation protocol was fixed at 5 ms. Such a gap permitted only negligible contribution of the recovery of nonliganded channels to the size of the current elicited by test pulse. Thus, the amplitude of this current was a reliable measure of the binding (and inhibition) of V102862 to inactivated rNav1.2 channels. Instead of gap interval, the duration of conditioning prepulse was increased in equal increments in subsequent cycles of stimulation applied every 10–15 s. The size of the increments was 80 ms for concentrations of V102862 ⩽1 μM and was decreased to 40 ms for higher drug concentrations to improve temporal resolution of measurements. The idea was to let the drug see inactivated states of the channel for increasing blocks of time. Thus, in subsequent stimulation episodes, the compound could exert more inhibition asymptotically approaching steady state. Test pulses following a short recovery gap delivered a satisfactory measure of drug-bound (and inhibited) inactivated channels. Figure 8a and b shows examples of actual current traces obtained from this type of experiment. Under control conditions, the size of currents showed only a slight trend to drop with the duration of the depolarizing prepulse, presumably reflecting concomitant accumulation of fast inactivating channels, which had not recovered during the 5 ms gap, and/or some development of slow inactivation (Figure 8a). In the presence of 1 μM V102862, the size of currents dropped with increasing duration of conditioning depolarization due to progressive binding (and inhibition) of the drug to inactivated channels. Binding reached an apparent steady state at the prepulse duration of 960 ms (the 12th stimulation cycle). Contamination with nonrecovered fast inactivating and slow inactivating channels could be corrected by calculating the difference between Na+ currents in control and in the presence of V102862. The corrected time course of inhibition could be well fit by a mono-exponential. Classical binding theory formulated by Langmuir assumes that there exists one homogenous kind of independent binding sites and that the concentration of ligand at the receptor is constant. In light of the known oversimplifications of the model (Scheme 1), our experimental conditions seem to meet these assumptions since at highly depolarized voltages all channels are in the same inactivated state and the concentration of V102862 is constant in steady-state conditions. Consistent with theory, the speed of inhibition should increase with higher drug concentrations and the rate of binding to inactivated channels can be approximated by the slope of the plot of 1/τ against drug concentration. Figure 9a (and Figure 8c) shows that the microscopic binding rates do rise linearly with drug concentration in support of the presumption in Figure 6a that V102862 binds to Na+ channels in a simple 1 : 1 mode. From the linear fit, the microscopic rate constant k+ (pseudo-first-order rate constant) for V102862 binding to inactivated channels was ∼1.7 μM−1 s−1.
Figure 8.

The binding rate of V102862 to inactivated rNav1.2 channels. (a) Control: the cell was pulsed for variable times (in increments of 80 ms) to −20 mV from the holding voltage of −120 mV every 15 s. Gap voltage (−120 mV, 5 ms) was applied after depolarizing prepulse to recover most inactivated and unliganded channels (see Figure 7a). The 10 ms testing pulse to 0 mV gave a measure of channels available for activation. A total of 12 successive traces (episodes) were superimposed; downward deflections represent inward Na currents evoked by test pulses in successive episodes. In control, only a slight tendency of currents to drop in size with increasing duration of conditioning prepulse is apparent. (b) Same protocol in the presence of 1 μM V102862. Progressive drop in the size of current over time reflects increase in the proportion of Na+ channels bound (and inhibited) to V102862. The mono-exponential fit to the peak current amplitudes (corrected for decay in control) returns the time constant, τ, and the steady-state level of inhibition. (c) Corrected time course of binding for 0.3, 1, 3 and 6 μM V102862 (different cell). The differences between the currents measured in the presence of the drug and the currents in control were normalized and plotted against the duration of conditioning prepulse. The lines are the mono-exponential fits. The time constants and the fractional responses in steady state are as follows: 511 ms and 0.57 (0.3 μM), 263 ms and 0.32 (1 μM), 163 ms and 0.22 (3 μM) and 65 ms and 0.11 (6 μM).
Figure 9.

(a) The macroscopic binding rates for V102862 (the inverses of the time constants as measured in Figure 8c) taken from four cells and plotted against concentration of the drug. The line is linear regression fit to mean values. The slope is ∼1.7 μM−1 s−1 and numerically equal to the pseudo-first-order constant k+ of V102862 binding to inactivated Na+ channels. (b) A set of semilogarithmic concentration–inhibition curves for inhibition of Na channels by V102862 taken for conditioning (inactivating) prepulses to −20 mV in four different cells. Fractional responses were measured in steady state in the experiments like the one shown in Figure 8a and b. The curves are the best sigmoidal curve fits to the data points: 1/{1+([V102862]/0.38)1.06}, 1/{1+([V102862]/0.39)0.94}, 1/{1+([V102862]/0.36)0.98} and 1/{1+([V102862]/0.36)0.95}.
Affinity of V102862 for inactivated rNav1.2 channels
Fractional inhibition measured at steady state in experiments like that shown in Figure 8a and b is valuable for estimating binding affinity of V102862 to inactivated rNav1.2 channels. Figure 9b presents a set of partial concentration–inhibition curves collected from four separate cells. Fitting the Hill equation to the data points gives individual dissociation constants for binding of the drug to inactivated Na+ channels; the mean value of KI is equal to 0.37+0.01 μM (n=4). If compared with resting block (KR ∼29 μM), V102862 is about 80-fold more potent in inhibiting inactivated than resting Na+ channels.
Use-dependent block with V102862
Neurons in seizure are typically more depolarized than normal cells and fire high-frequency trains of action potentials. In addition to voltage-dependent inhibition observed with low excitation frequencies (<0.5 Hz), state-dependent blockers can also manifest the phenomenon of use dependence, which enhances drug action during sustained higher frequency electrical activity mimicking the repetitive discharge in hyperexcitable neurons (e.g. Catterall, 1987; Ragsdale et al., 1991; Xie et al., 1995). We assessed the ability of V102862 to ‘filter' excessive electrical activity by measuring use-dependent block of trains of depolarizing voltage pulses of varying frequency. Figure 10 shows the amplitudes of currents evoked by a train of 20 pulses from −70 to 0 mV in control conditions and in the presence of increasing concentrations of V102862. The stimulation frequency was 2, 5 and 10 Hz and duration of each pulse was 5 ms. In control, there was a small steady decline in the current amplitude, which enhanced with increasing frequency. In the presence of V102862, there was an additional decline in the current due to accumulation of use-dependent block. The use-dependent block became more pronounced with increasing concentration of the drug and frequency of the pulse train. A stimulus frequency of 5 Hz was necessary to clearly detect use-dependent block with 3 μM V102862, whereas with 0.3 μM V102862 the block only became evident using the 10 Hz train.
Figure 10.

Frequency (use-) dependence of inhibition of trains of maximal rNav1.2 currents measured from the holding voltage of −70 mV with 5 ms depolarizing pulses to 0 mV. The peak currents in each train were normalized to the size of the first current in a series and plotted against the pulse number (20 pulses in total). The curves are the mono-exponential fits. The size of the current to the 20th depolarizing pulse is: 2 Hz – 0.95, 0.95, 0.91, 0.87; 5 Hz – 0.93, 0.9, 0.82, 0.74; 10 Hz – 0.91, 0.83, 0.7, 0.57 in control and 0.3, 1 and 3 μM V102862, respectively.
Inhibition of rNav1.2 channels by phenytoin, carbamazepine and lamotrigine
For comparison purposes, we characterized inhibition of rNav1.2 channels by the therapeutically important anticonvulsants PHT, CBZ and LTG. As anticipated, all the compounds blocked the channels by state-dependent mechanisms (see also Ragsdale et al., 1991; Kuo & Bean, 1994; Xie et al., 1995; Kuo et al., 1997). For these compounds, we have measured the key set of biophysical parameters determined for V102862, that is, KR, τrepr, KI and k+. The kinetic profile inherent with these drugs required some adjustments of stimulation protocols. Specifically, in the double-pulse protocol to measure retardation of recovery from inactivation (τrepr), the gap duration was varied in steps of 10 ms instead of 50 and 100 ms for V102862, since retardation of the recovery from inactivation by the anticonvulsants was not as prominent as with V102862. Correspondingly, the recovery gap duration in double-pulse protocol to measure kinetics of binding to inactivated channels was fixed at 5 ms. Bigger increments in conditioning prepulse duration were required since the kinetics of binding to inactivated states, in contrast to retardation of repriming, was slower for the classical anticonvulsants. Although overall solubility of the anticonvulsants was greater than that of V102862, it still did not permit precise measurement of KR, because the IC50 concentration for the resting block again exceeded solubility limits. As with V102862, KR was estimated using a single, high concentration of anticonvulsant (usually, 100–200 μM) at a holding voltage of −120 mV. Values are presented in Table 1. V102862 clearly has higher potency and a higher rate of binding to inactivated rNav1.2 channels than classical anticonvulsants (P<0.001; two-tailed t-test). In addition, V102862 causes a more pronounced retardation of channel recovery from inactivation.
Table 1.
Summary of effects of V102862, PHT, CBZ and LTG on rNav1.2 channels in HEK-293 cells
| KR (μM) | KI (μM) | k+ (μM−1 s−1) | τrepr (ms) | |
|---|---|---|---|---|
| V102862 | 29±3 (5) | 0.37±0.01 (4) | ∼1.7 (4) | 680±43 (16) |
| PHT | 1400±100 (3) | 20±2 (3) | ∼0.018 (3) | 109+13 (3) |
| CBZ | 1300±100 (5) | 46±5 (5) | ∼0.045 (4) | 20±1 (3) |
| LTG | 1800±250 (5) | 29±3 (3) | ∼0.024 (4) | 64±3 (3) |
Discussion and conclusions
The major finding of this study is that the novel anticonvulsant V102862 is a potent, state-dependent blocker of mammalian voltage-gated Na+ channels. Specifically, V102862 blocks native Na+ channels in rat hippocampal neurons and recombinant rNav1.2 channel α subunits expressed in a heterologous mammalian cell line (HEK-293 cells). In both cell types, the degree of inhibition is strongly dependent on holding voltage. In addition, V102862 revealed a distinct use-dependent block of rNav1.2 channels. These observations are consistent with the idea that the drug binds with high affinity to inactivated channels and low affinity to resting channels. Binding to open channels, as a potential component of voltage- and use-dependent inhibition (e.g. Butterworth & Strichartz, 1990), is unlikely for V102862. If this were happening, we would have seen an acceleration in time-dependent inactivation of INa in the presence of drug, and this was not apparent (compare Figure 7 in Yang & Kuo, 2002 and Figure 5d from this article).
In hippocampal neurons, mean inhibition of resting channels by V102862 was relatively weak with an apparent KR of >15 μM. V102862 inhibited the inactivated state of hippocampal neuronal Na+ channels ∼25-fold more potently with a KI value of ∼0.6 μM. V102862 was significantly more potent than PHT, although both compounds have similar features in their mechanism of inhibition (e.g. Kuo & Bean, 1994; Kuo et al., 1997). We found that PHT is ∼10-fold less potent in binding to inactivated channels; thus, its projected KI is ∼6 μM. This matches well the KD of ∼7 μM measured by Kuo & Bean (1994).
Expression of rNav1.2 channels in mammalian cells represents a convenient and logical model for in vitro profiling of anticonvulsants and other Na+ channel blockers (Gordon et al., 1987; Westenbroek et al., 1989). In HEK-293 cells, V102862 blocked inactivated channels with a KI of 0.37 μM, which is close to the value of 0.6 μM measured in rat hippocampal neurons. The drug was about 80-fold less potent in inhibiting Na+ channels in resting states (KR∼29 μM). That is, 80-fold higher concentrations of V102862 are required to cause significant reductions in the availability of resting Na+ channels for excitation. The levels of state dependence, or molecular ‘safety margin' (KR/KI), for the three anticonvulsants tested on rNav1.2 channels were also appreciable (∼70-fold for PHT, ∼30-fold for CBZ and ∼60-fold for LTG; Table 1), and this is consistent with their acceptable therapeutic index in humans. As with V102862, estimates of KR for the clinical anticonvulsants were not as reliable as the estimates for KI values, due to concerns about solubility; however, they are in good agreement with values of ∼600 μM for PHT (Kuo & Bean, 1994), ∼900 μM for CBZ (Kuo et al., 1997) and ∼1500 μM for LTG (Kuo & Lu, 1997) measured in rat hippocampal neurons.
The binding affinity of the three anticonvulsants to inactivated states of rNav1.2 channels measured here is somewhat lower than values reported previously (Kuo & Bean, 1994; Xie et al., 1995; Kuo & Lu, 1997; Kuo et al., 1997). Cell type-related discrepancies may underlie the ∼2-fold differences. V102862 binds rNav1.2 channels in HEK-293 cells with 54-, 124- and 78-fold higher affinity than PHT, CBZ and LTG, respectively. Although V102862 is distinctly more potent than the anticonvulsants referenced in this study, the physiological implication of this remains uncertain. Characteristic features of epileptic episodes, and other states of pathological hyperexcitability, are sustained depolarizations with superimposed high-frequency bursts of action potentials (for review, see Dichter & Ayala, 1987). These events lead to a progressive accumulation of Na+ channels in inactivated states, reducing availability of the channels for repetitive excitation. An additional reduction of channel availability by a use-dependent block is thought to lead to selective cessation of spike bursts in the depolarized neurons. At the high end of the dose range, therapeutic concentrations in the cerebrospinal fluid (CSF) of humans are ∼8 μM for PHT, ∼13 μM for CBZ and ∼20 μM for LTG (Johannessen & Strandjord, 1973; Sherwin et al., 1973). At these concentrations, when steady state of drug action is reached after prolonged depolarization, the fraction of available Na+ channels would be decreased by ∼29, ∼22 and ∼41% by PHT, CBZ and LTG, respectively, based on our estimates for KI (assuming 1 : 1 binding). The lack of knowledge of therapeutically relevant CSF concentrations for V102862 makes it difficult to speculate whether or not V102862 will look distinctly different from other anticonvulsants in reducing the availability of Na+ channels. We can state with certainty, however, that at 50- to 120-fold lower concentrations, V102862 will produce a comparable level of steady-state inhibition, which may result in an improved off-target side-effect profile. Consistent with this notion, V102862 has been demonstrated to be more potent than LTG and CBZ at blocking electrically induced convulsions in mice and rats with a correspondingly reduced propensity for motor impairment (Carter et al., 1997).
V102862 binds to inactivated rNav1.2 channels with k+ of ∼1.7 μM−1 s−1 (Table 1). This is 10–100 times slower than if drug binding were diffusion limited and probably precludes significant binding of V102862 to transiently inactivated channels during normal patterns of neuronal activity. Our measurements of k+ for the three anticonvulsants match well with the literature data obtained in rat hippocampal neurons (Kuo & Bean, 1994; Kuo et al., 1997; Kuo & Lu, 1997). Thus, at equimolar concentrations, V102862 binds rNav1.2 channels about 90-fold faster than PHT, 40-fold faster than CBZ and 75-fold faster than LTG. Given the effective levels of drugs in CSF (see above), macroscopic binding rates at room temperature would be 0.14 s−1 for PHT, 0.49 s−1 for CBZ and 0.48 s−1 for LTG. For V102862 to have a macroscopic binding rate comparable to CBZ and LTG, and appreciably faster than PHT, V102862 would need a CSF concentration of approximately 0.29 μM, that is, 0.29 × 1.6 μM−1 s−1≃0.5 s−1. Doses of V102862 producing higher concentrations would potentially be more effective than the referenced anticonvulsants against bursts of spikes superimposed on relatively short depolarizations in epileptogenic brain. At body temperature, binding will be faster than it is at room temperature for all the anticonvulsants (Kuo et al., 1997). However, we believe that a parallel shift in the on-rates to shorter time spans should take place and the rank of order will hold.
Retardation of the recovery from inactivation is also an important feature of the biophysics of the interaction of anticonvulsants with voltage-gated Na+ channels (e.g. Catterall, 1987; Ragsdale et al., 1991). In our experiments, the predominant fraction of rNav1.2 channels (∼80%) recovered from inactivation, with τrepr of approximately 680 ms in the presence of V102862, 109 ms in the presence of PHT, 20 ms in the presence of CBZ and 64 ms in the presence of LTG (Table 1); in control experiments, the time constant was 2–5 ms. The mechanism of retardation is not well understood (e.g. Ragsdale et al., 1991). Slow repriming could reflect either slow drug dissociation from inactivated or resting channels or slow transition of drug-bound channels from the inactivated to resting state. It is interesting, however, that the effect of V102862 on repriming is more pronounced than for the other anticonvulsants. The impact of this parameter on therapeutic utility and side effects warrants further study, as the pharmacological tools become available.
Use-dependent block of rNav1.2 channels by V102862 was apparent at concentrations ⩾1 μM and at frequencies of depolarizing pulse trains ⩾2 Hz. At 0.3 μM, V102862 produced clear use-dependent inhibition at 10 Hz. It is hard to compare the ‘filtering capacity' of V102862 with the literature data for other anticonvulsants due to differences in experimental protocols and stimulation frequency. In the future, it would be interesting to assess use-dependent block of action potentials in current-clamp mode in hippocampal neurons by therapeutically relevant concentrations of V102862.
Separate studies indicate that V102862 has similar biophysical parameters of state-dependent inhibition over a panel of TTX-sensitive Na+ channel isoforms. For example, KI values are in the range of 0.2–0.5 μM and k+ values vary between 1.1 and 1.8 μM−1 s−1 for human skeletal muscle 1 (Nav1.4), human peripheral nerve 1 (Nav1.7) and human brain type III (Nav1.3) Na+ channels (Ilyin & Woodward, 1998; V.I. Ilyin, D.D. Hodges & A. Kammesheidt, unpublished data). The affinity for the resting state is consistently between 70- and 100-fold lower than affinity to inactivated state(s) for these isoforms. Clearly, like other anticonvulsant state-dependent Na+ channel blockers, V102862 does not have a high level of subtype selectivity across the different Na+ channel isoforms tested. Interestingly, there is more variation in τrepr, that is, the range is from about ∼100 ms in hSkM1 to ∼1500 ms in hPN1. Moreover, V102862 blocks TTX-sensitive and TTX-resistant native Na+ currents in rat sensory dorsal root ganglion neurons as well as rat recombinant TTX-resistant SNS/PN3 (Nav1.8) channels expressed in HEK-293 cells with a KI of approximately 0.3 μM (V.I. Ilyin, D.D. Hodges & S. Malik, unpublished data). This implies that, in addition to its attractive anticonvulsant properties, V102862 may also have utility in treatment of a variety of pain conditions, including neuropathic pain (Carter et al., 1999).
In conclusion, V102862 is a potent, broad-spectrum state-dependent blocker of mammalian voltage-gated Na+ channels. A key feature of the mechanism of inhibition is that V102862 has up to 80-fold higher affinity for inactivated Na+ channels as compared to channels in resting states. The efficacy and safety of the drug in animals appear to stem from its high level of state dependence and a unique set of kinetic parameters. Understanding the molecular mechanism of V102862 should be useful in the design of new generations of Na+ channel blockers targeted at multiple therapeutic indications, in particular epilepsy and neuropathic pain.
Acknowledgments
We acknowledge Dr Anja Kammesheidt and Shiazha Malik for their expert help with recombinant sodium channel cell lines. We also thank Gregg Crumley for recloning rNav1.2 channel cell line and Daniela Leumer for her help with culturing NaIIA-B2 HEK-293 cells.
Abbreviations
- CBZ
carbamazepine
- CSF
cerebrospinal fluid
- h
fraction of channels in the resting state
- HEK
human embryonic kidney
- INa
sodium current
- KI
dissociation constant for the inactivated channel state
- KR
dissociation constant for the resting state
- k
Boltzmann parameter, slope factor of the availability curve
- k+
pseudo-first-order rate constant (for binding to inactivated channels)
- LTG
lamotrigine
- PHT
phenytoin
- rNav1.2
rat brain type IIa Na+ channel
- TTX
tetrodotoxin
- τrepr
time constant of repriming (recovery from inactivation)
- V102862
4-(4-fluorophenoxy)benzaldehyde semicarbazone
- Vhalf
Boltzmann parameter, voltage of half-maximum channel availability
References
- BEAN B.P. Nitrendipine block of cardiac calcium channels: high-affinity binding to the inactivated state. Proc. Natl. Acad. Sci. U.S.A. 1984;81:6388–6392. doi: 10.1073/pnas.81.20.6388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEAN B.P., COHEN C.J., TSIEN R.W. Lidocaine block of cardiac sodium channels. J. Gen. Physiol. 1983;81:613–642. doi: 10.1085/jgp.81.5.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BUTTERWORTH J.F., STRICHARTZ G.R. Molecular mechanisms of local anesthesia: a review. Anesthesiology. 1990;72:711–734. doi: 10.1097/00000542-199004000-00022. [DOI] [PubMed] [Google Scholar]
- CARTER R.B., VANOVER K.E., WHITE H.S., WOLF H.H., PUTHUCODE R.N., DIMMOCK J.R. Characterization of the anticonvulsant properties of Co 102862, a novel blocker of voltage-gated sodium channels. Soc. Neurosci. Abstr. 1997;23:2163. [Google Scholar]
- CARTER R.B., VANOVER K.E., WILENT W., XU Z., WOODWARD R.M., ILYIN V.I.Anti-allodynic and anti-hyperalgesic effects of the novel voltage-dependent sodium channel blocker Co 102862 in a rat model of peripheral neuropathy Proceedings of the International Symposium on ‘Ion channels in Pain and Neuroprotection' 199919San Francisco, March 14–17, 1999, p
- CATTERALL W.A. Common modes of drug action on Na+ channels: local anesthetics, antiarrhythmics and anticonvulsants. Trends Pharmacol. Sci. 1987;8:57–65. [Google Scholar]
- CATTERALL W.A. Cellular and molecular biology of voltage-gated sodium channels. Physiol. Rev. 1992;72:S15–S48. doi: 10.1152/physrev.1992.72.suppl_4.S15. [DOI] [PubMed] [Google Scholar]
- CATTERALL W.A. From ion currents to molecular mechanisms: the structure and function of voltage-gated sodium channels. Neuron. 2000;26:13–25. doi: 10.1016/s0896-6273(00)81133-2. [DOI] [PubMed] [Google Scholar]
- DICHTER M.A., AYALA G.F. Cellular mechanisms of epilepsy: a status report. Science. 1987;237:157–237. doi: 10.1126/science.3037700. [DOI] [PubMed] [Google Scholar]
- DIMMOCK J.R., PUTHUCODE R.N., SMITH J.M., HETHERINGTON M., QUAIL J.W., PUGAZHENTHI U., LECHLER T., STABLES J.P. (Aryloxy)aryl semicarbazones and related compounds: a novel class of anticonvulsant agents possessing high activity in the maximal electroshock screen. J. Med. Chem. 1996;39:3984–3998. doi: 10.1021/jm9603025. [DOI] [PubMed] [Google Scholar]
- DIMMOCK J.R., SIDHU K.K., THAYER R.S., MACK P., DUFFY M.J., REID R.S., QUAIL J.W., PUGAZHENTHI U., ONG A., BIKKER J.A., WEAVER D. Anticonvulsant activities of some arylsemicarbazones displaying potent oral activity in the maximal electroshock screen in rats accompanied by high protection indices. J. Med. Chem. 1993;36:2243–2252. doi: 10.1021/jm00068a001. [DOI] [PubMed] [Google Scholar]
- GOLDIN A.L. Evolution of voltage-gated Na+ channels. J. Exp. Biol. 2002;205:575–584. doi: 10.1242/jeb.205.5.575. [DOI] [PubMed] [Google Scholar]
- GOLDIN A.L., BARCHI R.L., CALDWELL J.H., HOFMANN F., HOWE J.R., HUNTER J.C., KALLEN R.G., MANDEL G., MEISLER M.H., NETTER Y.B., NODA M., TAMKUN M.M., WAXMAN S.G., WOOD J.N., CATTERALL W.A. Nomenclature of voltage-gated sodium channels. Neuron. 2000;28:365–368. doi: 10.1016/s0896-6273(00)00116-1. [DOI] [PubMed] [Google Scholar]
- GORDON D.D., MERRICK V., AULD V., DUNN R., GOLDIN A.L., DAVIDSON N., CATTERALL W.A. Tissue-specific expression of the RI and RII sodium channel subtypes. Proc. Natl. Acad. Sci. U.S.A. 1987;84:8682–8686. doi: 10.1073/pnas.84.23.8682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HAMILL O.P., MARTY A., NEHER E., SAKMANN B., SIGWORTH F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pfluegers Arch. Eur. J. Physiol. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- HILLE B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug–receptor reaction. J. Gen. Physiol. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HONDEGHEM L.M., KATZUNG B.G. Antiarrhythmic agents: the modulated receptor mechanism of action of sodium and calcium channel blocking drugs. Annu. Rev. Pharmacol. Toxicol. 1984;24:387–423. doi: 10.1146/annurev.pa.24.040184.002131. [DOI] [PubMed] [Google Scholar]
- ILYIN V.I., HODGES D.D. Inhibition of rBIIa sodium channels in HEK-293 cells by Co 102862: kinetic and steady-state parameters. Soc. Neurosci. Abstr. 1999;25:1868. [Google Scholar]
- ILYIN V.I., WHITTEMORE E.R., PUTHUCODE R.N., DIMMOCK J.R., WOODWARD R.M. Co 102862, a novel anticonvulsant, is a potent, voltage-dependent blocker of voltage-gated Na+ channels in rat hippocampal neurons. Soc. Neurosci. Abstr. 1997;23:2163. [Google Scholar]
- ILYIN V.I., WOODWARD R.M. Kinetic and steady-state parameters for inhibition of hSkM1 sodium channels by Co 102862, phenytoin, carbamazepine and lamotrigine. Soc. Neurosci. Abstr. 1998;24:1939. [Google Scholar]
- ISOM L.L., DE JONGH K.S., PATTON D.E., REBER B.F.X., OFFORD J., CHARBONNEAU H., WALSH K., GOLDIN A.L., CATTERALL W.A. Primary structure and functional expression of the Beta1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- JOHANNESSEN S.I., STRANDJORD R.E. Concentration of carbamazepine (Tegretol) in serum and in cerebrospinal fluid in patients with epilepsy. Epilepsia. 1973;14:373–379. doi: 10.1111/j.1528-1157.1973.tb03976.x. [DOI] [PubMed] [Google Scholar]
- KUO C.-C., BEAN B.P. Slow binding of phenytoin to inactivated sodium channels in rat hippocampal neurons. Mol. Pharmacol. 1994;46:716–725. [PubMed] [Google Scholar]
- KUO C.-C., CHEN R.-C., LOU B.-S. Inhibition of Na+ current by diphenhydramine and other diphenyl compounds: molecular determinants of selective binding to the inactivated channels. Mol. Pharmacol. 2000;57:135–143. [PubMed] [Google Scholar]
- KUO C.-C., HUANG R.-S., LU L., CHEN R.C. Carbamazepine inhibition of neuronal Na+ currents: quantitative distinction from phenytoin and possible therapeutic implications. Mol. Pharmacol. 1997;51:1077–1083. doi: 10.1124/mol.51.6.1077. [DOI] [PubMed] [Google Scholar]
- KUO C.-C., LU L. Characterization of lamotrigine inhibition of Na+ channels in rat hippocampal neurons. Br. J. Pharmacol. 1997;121:1231–1238. doi: 10.1038/sj.bjp.0701221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PATTON D.E., ISOM L.L., CATTERAL W.A., GOLDIN A.L. The adult rat brain beta 1 subunit modifies activation and inactivation gating of multiple sodium channel alpha subunits. J. Biol. Chem. 1994;269:17649–17655. [PubMed] [Google Scholar]
- RAGSDALE D.S., SCHEUER T., CATTERALL W.A. Frequency and voltage-dependent inhibition of type IIA Na+ channels, expressed in a mammalian cell line, by local anesthetic, antiarrhythmic, and anticonvulsant drugs. Mol. Pharmacol. 1991;40:756–765. [PubMed] [Google Scholar]
- ROGAWSKI M.A., PORTER R.J. Antiepileptic drugs: pharmacological mechanisms and clinical efficacy with consideration of promising developmental stage compounds. Pharmacol. Rev. 1990;42:223–286. [PubMed] [Google Scholar]
- SAMBROOK J., FRITSCH E.F., MANIATIS T. Molecular Cloning, A Laboratory Manual 1989Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; Vols. 1–3 [Google Scholar]
- SATO C., UENO Y., ASAI K., TAKAHASHI K., SATO M., ENGEL A., FUJIYOSHI Y. The voltage-sensitive sodium channel is a bell-shaped molecule with several cavities. Nature. 2001;409:1047–1051. doi: 10.1038/35059098. [DOI] [PubMed] [Google Scholar]
- SCHEUER T., AULD V.J., BOYD S., OFFORD J., DUNN R., CATTERALL W.A. Functional properties of rat brain sodium channels expressed in a somatic cell line. Science. 1990;247:854–858. doi: 10.1126/science.2154850. [DOI] [PubMed] [Google Scholar]
- SHERWIN A.L., EISEN A.A., SOKOLOWSKI C.D. Anticonvulsant drugs in human epileptogenic brain. Arch. Neurol. 1973;29:73–77. doi: 10.1001/archneur.1973.00490260017001. [DOI] [PubMed] [Google Scholar]
- VERDOORN T.A., DRAGUHN A., YMER S., SEEBURG P.H., SAKMANN B. Functional properties of recombinant rat GABAA receptors depend upon subunit composition. Neuron. 1990;4:919–928. doi: 10.1016/0896-6273(90)90145-6. [DOI] [PubMed] [Google Scholar]
- WEST J.W., SCHEUER T., MAECHLER L., CATTERALL W.A. Efficient expression of rat brain type IIA Na+ channel α subunits in a somatic cell line. Neuron. 1992;8:59–70. doi: 10.1016/0896-6273(92)90108-p. [DOI] [PubMed] [Google Scholar]
- WESTENBROEK R.E., MERRICK D.K., CATTERALL W.A. Differential subcellular localization of the RI and RII Na+ channel subtypes in central neurons. Neuron. 1989;3:695–704. doi: 10.1016/0896-6273(89)90238-9. [DOI] [PubMed] [Google Scholar]
- WHITTEMORE E.R., KOERNER J.F. Pre-exposure to L-homocysteinsulfinic acid blocks quisqualate-induced sensitization to L-2-amino-4-phosphonobutanoic acid (L-AP4) Eur. J. Pharmacol. 1991;192:435–438. doi: 10.1016/0014-2999(91)90237-k. [DOI] [PubMed] [Google Scholar]
- XIE X.M., LANCASTER B., PEAKMAN T., GARTHWAITE J. Interaction of the antiepileptic drug lamotrigine with recombinant rat brain type IIA Na+ channels and with native Na+ channels in rat hippocampal neurons. Pfluegers Arch. Eur. J. Physiol. 1995;430:437–446. doi: 10.1007/BF00373920. [DOI] [PubMed] [Google Scholar]
- YANG Y.-C., KUO C.-C. Inhibition of Na+ current by imipramine and related compounds: different binding kinetics as an inactivation stabilizer and as an open channel blocker. Mol. Pharmacol. 2002;62:1228–1237. doi: 10.1124/mol.62.5.1228. [DOI] [PubMed] [Google Scholar]