

Abstract
GW274150 ([2-[(1-iminoethyl) amino]ethyl]-L-homocysteine) and GW273629 (3-[[2-[(1-iminoethyl)amino]ethyl]sulphonyl]-L-alanine) are potent, time-dependent, highly selective inhibitors of human inducible nitric oxide synthase (iNOS) vs endothelial NOS (eNOS) (>100-fold) or neuronal NOS (nNOS) (>80-fold). GW274150 and GW273629 are arginine competitive, NADPH-dependent inhibitors of human iNOS with steady state Kd values of <40 and <90 nM, respectively.
GW274150 and GW273629 inhibited intracellular iNOS in J774 cells in a time-dependent manner, reaching IC50 values of 0.2±0.04 and 1.3±0.16 μM, respectively. They were also acutely selective in intact rat tissues: GW274150 was >260-fold and 219-fold selective for iNOS against eNOS and nNOS, respectively, while GW273629 was >150-fold and 365-fold selective for iNOS against eNOS and nNOS, respectively.
The pharmacokinetic profile of GW274150 was biphasic in healthy rats and mice with a terminal half-life of ∼6 h. That of GW273629 was also biphasic in rats, producing a terminal half-life of ∼3 h. In mice however, elimination of GW273629 appeared monophasic and more rapid (∼10 min). Both compounds show a high oral bioavailability (>90%) in rats and mice.
GW274150 was effective in inhibiting LPS-induced plasma NOx levels in mice with an ED50 of 3.2±0.7 mg kg−1 after 14 h intraperitoneally (i.p.) and 3.8±1.5 mg kg−1 after 14 h when administered orally. GW273629 showed shorter-lived effects on plasma NOx and an ED50 of 9±2 mg kg−1 after 2 h when administered i.p.
The effects of GW274150 and GW273629 in vivo were consistent with high selectivity for iNOS, as these inhibitors were of low potency against nNOS in the rat cerebellum and did not cause significant effects on blood pressure in instrumented mice.
Keywords: Nitric oxide synthase, NOS2, NOS-II, GW274150, GW273629
Introduction
Nitric oxide synthases (NOS; EC 1.14.13.39) are a family of enzymes that catalyse the formation of nitric oxide (NO) from L-arginine, O2 and NADPH with the production of citrulline and NADP+. The family of enzymes consists of two calcium-calmodulin-dependent, constitutive isoforms, neuronal NOS (nNOS, NOS-I) and endothelial NOS (eNOS, NOS-III), and a calcium-independent, inducible NOS (iNOS, NOS-II) (Knowles & Moncada, 1994; Alderton et al., 2001). NO is an important physiological mediator that plays a role in the regulation of blood pressure and blood flow, as a neurotransmitter in the central and peripheral systems and in the immune system (Rodeberg et al., 1995). However, the overproduction of NO by iNOS is implicated in the pathophysiology of several disease states such as septic and endotoxic shock, neurodegenerative disorders, inflammatory diseases such as asthma, rheumatoid arthritis and multiple sclerosis (reviewed in Whittle, 1997; Hobbs et al., 1999; Folkerts et al., 2001; Vallance & Leiper, 2002). Since inhibition of the endothelial isoform produces hypertension (Moncada & Higgs, 1995), it is desirable to develop inhibitors of the inducible isoform for therapeutic use with a high degree of selectivity over the endothelial isoform. Inhibition of eNOS and nNOS may also have a range of other undesirable effects as a consequence of their widespread physiological roles, especially in normal vascular endothelial and gastrointestinal function. The acetamidine-containing analogues of arginine, N5-(1-iminoethyl)-L-ornithine (L-NIO) and N6-(1-iminoethyl)-L-lysine (L-NIL) have been known for some time to be partially selective inhibitors of iNOS vs eNOS and nNOS (McCall et al., 1991; Moore et al., 1994). They are both 30–50-fold selective for iNOS vs eNOS, and approximately 20-fold vs nNOS. The first highly selective inhibitors of iNOS vs eNOS were the bis-isothioureas reported by Garvey et al. (1994). For example S,S′-(1,3-phenylenebis(1,2-ethanediyl) bis-isothiourea is an L-arginine-competitive, rapidly reversible inhibitor of human iNOS with a selectivity of 190-fold vs eNOS, although it is only five-fold selective vs nNOS. Unfortunately, the utility of this series of compounds as pharmacological agents is limited by the poor cellular and tissue penetration of the more selective compounds, as well as by significant acute toxicity. 1400W (N-(3-(amino methyl) benzyl) acetamidine, also identified by Garvey et al. (1997), proved to be a further step forward, since it is not only highly selective as an iNOS inhibitor vs both eNOS and nNOS (>5000-fold and >250-fold, respectively) but also penetrates cells and tissues. However, 1400W has to be used with caution because large bolus doses have toxic effects: intravenous doses >10 mg kg−1 caused immediate death, presumably from cardiac toxicity, although the mechanism of this has not been identified. GW274150 ([2-[(1-iminoethyl) amino] ethyl]-L-homocysteine) and its close structural homologue GW273629 (3-[[2-[(1-iminoethyl) amino] ethyl] sulphonyl]-L-alanine) are novel NOS inhibitors that have been identified from a series of acetamidine amino acids (Young et al., 2000), which, like 1400W, have a very high degree of selectivity for iNOS vs both eNOS and nNOS. This paper provides the biochemical and pharmacological characterisation of GW274150 and GW273629, which will underpin the use of these compounds as selective pharmacological tools and potentially as therapeutic agents. These two compounds are currently in clinical development for inflammatory conditions including asthma (www.gsk.com). Some of these data have been reported previously in oral presentations (Alderton et al., 2000; Knowles et al., 2000).
Methods
Measurements of IC50 values for inhibition of human and rodent NOS isoforms by the oxyhaemoglobin assay
NOS activity was measured by the conversion of oxyhaemoglobin to methaemoglobin by NO in a microtitre plate assay as described in Dawson & Knowles (1998). The final concentrations of components in the assay were HEPES 100 mM, dithiothreitol 100 μM, MgCl2 0.8 mM, oxyhaemoglobin 5 μM, L-arginine 30 μM, NADPH 100 μM, FAD 1 μM, FMN 1 μM, calmodulin 100 nM, tetrahydrobiopterin (BH4) 10 μM. The IC50 values were measured at 37°C over the periods 0–10 and 15–30 min and the results shown are the mean of three experiments. The human iNOS, eNOS and nNOS isoforms, and rat nNOS were expressed in Spodoptera frugiperda cells using the baculovirus expression system (Charles et al., 1996). Murine iNOS was obtained from J774 cells activated with murine interferon γ and LPS (LPS) from Salmonella typhosa for 24 h. Cell lysates were used as the enzyme source after treating with Dowex-50 (Na+ form) ion-exchange resin to remove endogenous arginine.
Purification of human iNOS expressed in Escherichia coli
Compounds were first identified and characterised on baculovirus-expressed human NOS isoforms as described above. For subsequent studies recombinant human iNOS was coexpressed in Escherichia coli (Fosetta et al., 1996) and purified by a combination of methods (Fosetta et al., 1996; Rodrigeuz-Crespo & Oritz de Montellano, 1996). The cell paste was resuspended in an equal volume of disruption buffer: 20 mM HEPES pH 7.4, 2 μM BH4, 1 mM DTT, 1 mM EDTA, 10 μM FAD, 10 μM FMN, 50 mM NaCl, 0.6% (v v−1) CHAPS detergent and the following protease inhibitors were added: 20 μM leupeptin, 2 μM 3,4–dichloroisocoumarin (3,4-DCI), 2 μM trans-epoxysuccinyl-L-leucylamido-(4-guanidino)-butane (E-64) and 2 μM pepstatin A. The homogenate was sonicated on ice for 3 × 30 s with 2 min delays, then centrifuged at 100,000 × g for 30 min at 4°C. The supernatant was first adjusted to 20% (w v−1) ammonium sulphate and centrifuged. This supernatant was adjusted to 40% (w v−1) ammonium sulphate and the resulting protein pellet was resuspended and then dialysed against 25 mM HEPES pH 7.5, 10% (v v−1) glycerol, 1 mM DTT, 2 μM FAD, 2 μM FMN, 1 mM L-arginine, 10 μM BH4 and 100 mM NaCl (buffer A). The protein was loaded onto a 2′-5′-ADP-Sepharose column, the column was washed with five column volumes of Buffer A, then iNOS was reverse eluted with Buffer A plus10 mM NADPH. The purified iNOS was dialysed against 25 mM HEPES pH 7.5, 10% (v v−1) glycerol, 1 mM DTT and stored at −80°C. The final protein was estimated to be 30% pure by densitometry on SDS–PAGE and had a specific activity of 150 nmol NO produced min−1 mg protein−1.
Enzyme kinetics experiments with GW274150
Detailed enzyme kinetic experiments were performed with purified human NOS. For Ki determinations of GW274150 with eNOS, nNOS and iNOS, initial rates were measured over 0–10, 0–10 and 0–5 min, respectively, in the oxyhaemoglobin assay over a range of arginine and GW274150 concentrations. For Km determinations, rates were measured over 0–10 min in the oxyhaemoglobin assay over a range of arginine concentrations. Data were fitted to the following equation using Grafit (Erithacus Software Ltd):
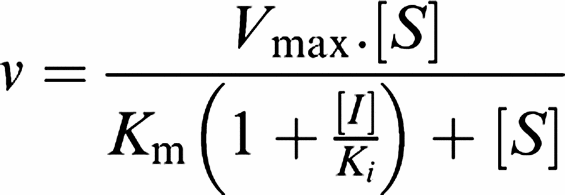 |
The reversibility of inhibition was studied as follows: purified iNOS (4.3 mg ml−1 diluted 1 in 100–43 ng ml−1) was incubated at 37°C in HEPES buffer (100 mM pH 7.4) containing DTT (5 mM) and BH4 (5 μM). The preincubations were set up ±GW274150 (200 μM) or (20 μM), ±NADPH (5 mM). Activities of the preincubation mixtures were monitored for various times up to 3 h by the removal of samples and dilution (200-fold) into assay reagent. The activity was measured by a modification of the oxyhaemoglobin procedure in 1 ml quartz cuvettes at 37°C in the Hewlett Packard diode array spectrophotometer. The final concentrations of components in the assay were HEPES 100 mM, DTT 100 μM, MgCl2 0.8 mM, oxyhaemoglobin 5 μM, L-arginine 2 mM, NADPH 100 μM, FAD 1 μM, FMN 1 μM, CaM 100 nM and BH4 10 μM.
To follow the time course of the onset of iNOS inhibition by GW274150, rates were measured and analysed using the integrated rate equation modified such that the steady-state rate is constrained to zero:
where v0=initial rate, k=transition rate constant, A0=initial reading (Morrison & Walsh, 1988) by a modification of the oxyhaemoglobin procedure described above over a range of GW274150 concentrations and at 30 μM L-arginine. In experiments where the progress plots were run over longer time periods (2000 s), the steady-state rate was zero at GW274150 concentrations >20 μM (data not shown); therefore, the steady-state rate can justifiably be fixed at zero for experiments run over shorter time periods (1400 s) where the rates have not reached the steady state. The activities were measured in 1 ml quartz cuvettes in a Hewlett Packard diode array spectrophotometer at 37°C. The difference in absorbance at 420 and 405 nm was recorded every 30 s for 10 min.
The final concentrations of components in the assay were HEPES 100 mM, DTT 100 μM, MgCl2 0.8 mM, oxyhaemoglobin 5 μM, L-arginine 30 μM, NADPH 100 μM, FAD 1 μM, FMN 1 μM, calmodulin 100 nM and BH4 10 μM.
Reversed phase chromatography of [14C]GW274150 and [14C]GW273629
[U-14C]GW274150 (100 μM) or [U-14C]GW273629 (100 μM) were incubated with purified human iNOS (at a concentration that would convert 10 nmol of L-arginine to citrulline min−1) for 180 min at 37°C in the following buffer: HEPES pH 7.4 100 mM, DTT 100 μM, MgCl2 0.8 mM, L-arginine 5 μM, NADPH 10 μM, FAD 1 μM, FMN 1 μM, calmodulin 100 nM and BH4 10 μM. The experiment was performed in duplicate. Enzyme was replaced with buffer for the control reactions. Protein was removed by precipitation with 0.1% (v v−1) formic acid and centrifugation. Aliquots (50 μl) were analysed on a Zorbax Si HPLC column and eluted by a gradient over 20 min at 1 ml min−1 from 0.1% formic acid in acetronitrile to 0.1% (v v−1) formic acid in water. GW274150 was eluted from the column at 12.6 min and GW273629 was eluted from the column at 12.0 min.
Inhibition of iNOS in J774 cells
The cell permeable diacetate derivative DAF-2DA is hydrolysed to DAF-2 by cytosolic esterases. DAF-2 then reacts with O2 and NO to produce a fluorescent derivative DAF-2T, which is detected by excitation at 492 nm and emission at 515 nm (Kojima et al., 1998). Murine monocytic leukaemia J774 cells were dispensed into 96-well fluorescent plates at a density of 30,000 cells per well in RPMI medium supplemented with 10% (v v−1) fetal calf serum (FCS) and 1% penicillin/streptomycin. After 24 h, the medium was changed to DMEM supplemented with 10% FCS, 1% penicillin/streptomycin, murine interferon γ (5 U ml−1), LPS (1 μg ml−1) and incubated for a further 24 h. The plates were then washed with 200 μl of phosphate-buffered saline (PBS) per well, and the medium was replaced with 100 μl of compound diluted in PBS and 100 μl of PBS containing L-arginine (200 μM), 20% FCS, murine interferon γ (10 U ml−1), LPS (2 μg ml−1) and DAF-2DA (20 μM). The cells were incubated for a further 4 h before detection of NO at 0.5, 1, 2, 4 and 24 h later. The detection of NO produced in J774 cells not treated with inhibitor increased linearly to 200,000 fluorescence units. The mean of quadruplicate data for inhibition by GW274150 and GW273629 was fitted to the equation Y=Y0+Ae−x/t using Origin 5.0.
Inhibition of eNOS and iNOS in rat aortic rings
All animal studies described in this paper were carried out in accordance with current U.K. Home Office procedural requirements. The preparation and procedures were modified from that of Russell (1998). Male Wistar rats were killed by cervical dislocation and the thoracic aorta was excised, washed in a modified Krebs' bicarbonate buffer (containing, in mM, NaCl, 118.5; KCl, 4.7; CaCl2, 2.5; KH2PO4, 1.2; MgSO4, 1.1; NaHCO3, 25.0; glucose, 11.1 and 5 μM indomethacin, hereafter referred to as ‘Krebs buffer'), cleared of adhering fat and connective tissue and the vessels were then cut transversely into 4 mm rings. For endothelium-denuded rings, the aortae were cut into 6–8 mm sections and the intimal surface rubbed with a pipe cleaner before the rings were trimmed to their final length of 4 mm. The rings were mounted on two L-shaped titanium hooks, the upper hook being connected to an isometric transducer. They were lowered into 25 ml organ baths containing Krebs buffer at 37°C and were gassed continuously with 95% O2/5% CO2. The resting tension was set to 2 g and recorded on a chart recorder via the isometric transducers. The rings were equilibrated for 45 min, washing every 15 min with fresh Krebs buffer, allowing the rings to relax and a new baseline was established by setting all the rings to 1 g tension. A cumulative concentration contraction curve to phenylephrine (1–10,000 nM) was then obtained. From these data the EC90 for phenylephrine was determined. The rings were washed every 15 min with fresh Krebs buffer to allow them to relax back to baseline tension. The EC90 concentration of phenylephrine was added back to the organ baths to contract the tissues. At this point, a cumulative concentration relaxation curve to acetylcholine (5–2560 nM) was obtained for each ring in order to assess the integrity of the endothelium. Relaxation of greater than 60% was taken as an indicator of an intact endothelium. Once more the rings were allowed to re-equilibrate for 45 min by washing every 15 min with fresh Krebs buffer, and brought back to baseline tension. The rings that had been assessed as having intact endothelium (>60% relaxation to acetylcholine) were then contracted with a small amount of phenylephrine (EC10). Cumulative concentration contraction curves were then obtained for the NO synthase inhibitors (0.1–300 μM) with 15 min intervals between doses. Under these circumstances, inhibition of eNOS by an NOS inhibitor resulted in loss of tone exerted by NO, that is, resulting in a contraction of the tissues.
Using endothelial denuded aortic rings (set at 1 g baseline), a cumulative concentration contraction curve to phenylephrine (1–10,000 nM) was obtained. From these data, the EC90 for phenylephrine was determined for each ring separately. The rings were re-equilibrated for 45 min by washing every 15 min with fresh Krebs buffer to allow them to relax back to baseline tension. LPS (100 ng ml−1) was added to the organ baths 15 min prior to contracting the tissues submaximally with phenylephrine (EC90), determined from the dose–response curves above. The rings were left for a period of 6 h during which time induction of iNOS in the vessel wall resulted in a gradual time-dependant vasorelaxation, despite the continued presence of phenylephrine; subsequent inhibition of the iNOS resulted in recontraction of the rings. In rings that demonstrated greater than 30% relaxation 6 h after addition of LPS, indicating a substantial induction of iNOS, NOS inhibitors were added (0.01–300 μM) to the baths with 15 min intervals between doses to obtain cumulative concentration–contraction curves.
Inhibition of nNOS in rat brain slices
Male Wistar rats were killed by cervical dislocation, the brains rapidly excised and placed on filter paper moistened with artificial CSF (ACSF), on a Petri dish cooled on ice. The cortex was separated from the rest of the brain, weighed and chopped into 0.2 mm × 0.2 mm × ∼2 mm slices. The slices were suspended in 6 ml ACSF and left for ∼10 min. Tissue was transferred with 40 ml ACSF into 100 ml conical flasks, placed in an orbital shaking water bath at 37°C and gassed with O2/CO2 for 45–60 min. The contents of the flasks were centrifuged at 1000 r.p.m. for 4–5 min at room temperature, supernatant was removed and the slices resuspended in 35 ml high potassium ACSF. The centrifugation step was repeated, the tissues were pooled and resuspended in 10 volumes high potassium ACSF. NO synthesis by nNOS in the presence and absence of GW274150 and GW273629 was assessed in rat cortical slices by the Ng-nitro-L-arginine-inhibitable conversion of L-[U-14C]arginine to [14C] citrulline as described in Lizasoain et al. (1995).
Pharmacokinetic evaluation of GW274150 and GW273629
Healthy rats and mice were dosed intravenously or orally (1 or 10 mg kg−1) with GW274150 or GW273629 during separate pharmacokinetic studies and plasma samples were collected. In brief, plasma was assayed for parent compound by fluorescence derivatisation with a quaternary ammonium reagent (or by solid-phase extraction for GW274150 in rat plasma) followed by HPLC with mass spectrometric detection. Plasma samples were also collected from GW273629-treated mice (30 mg kg−1) administered as a 48 h infusion immediately following injection of endotoxin and prior to onset of shock.
The plasma pharmacokinetics were generated using noncompartmental analysis with commercial software. The areas under the plasma concentration–time curves (AUC) were estimated using the linear trapezoidal rule and extrapolated to infinity by addition of the product of the last measured concentration divided by the respective terminal phase rate constant. The terminal phase rate constants were estimated by linear regression of logarithmic transformed terminal plasma concentration–time data providing values for half-life (T1/2) by division from ln 2. The maximum plasma concentrations (Cmax) and the time at which they were measured (Tmax) were read directly from the data. Clearance (CL) was estimated as the dose divided by the AUC, and the volume of distribution at steady state (Vss) was estimated from the clearance multiplied by the mean residence time (MRT).
Inhibition of iNOS activity in vivo as assessed by nitrate and nitrite levels in CD-1 mouse plasma following LPS challenge
Adult male CD-1 mice, 20–30 g, were injected intravenously (i.v.) at time zero with LPS prewarmed to 37°C at 1 mg ml−1 and 3 ml kg−1 body weight. At 4 h, plasma from four LPS-dosed control mice was sampled. Three groups of 12 mice were also treated with either GW274150 or GW273629 at 30 mg kg−1 i.p. or 100 mg kg−1 i.p. dissolved in injection saline, or vehicle control, and injected at 4 ml kg−1 body weight, 4 h after the LPS dose. A time course for the inhibition of LPS-induced elevation of plasma NOx was investigated by sampling plasma from three treated individuals from each group at 6, 12, 18 and 24 h post LPS treatment for GW274150 and at 6, 8 and 12 h post LPS treatment for GW273629. Plasma was sampled under anaesthetic (oxygen/nitrous oxide/isoflurane) by cardiac puncture through the diaphragm and removal of whole blood by syringe. Once sampled, the mice were killed by cervical dislocation. Blood was then transferred to heparinised sample tubes prior to centrifugation at 6000 g and room temperature for 3 min in a bench centrifuge and subsequent removal of plasma to separate prelabelled, MilliQ-deionised-water-washed tubes. The time course was studied on two separate occasions.
Dose–response studies were carried out essentially as above. Dosing of animal groups with GW274150 or GW273629 dissolved in saline for injection or vehicle control was carried out 4 h post LPS dosing at a range of dosing levels administered i.p. at 4 ml kg−1 by syringe. Plasma was collected at 18 h post LPS dosing for GW274150 and 6 h post LPS dosing for GW273629. Results from two separate experiments were combined for each compound. The NOx levels were measured according to the methods of Verdon et al. (1995) as follows:
plasma samples were diluted 2.5-fold with 14 mM sodium phosphate pH 7.4 (assay buffer). Separate series of nitrate and nitrite standards with concentrations from 200 μM with six serial two-fold dilutions from this were prepared in assay buffer. A blank containing only assay buffer was also included. Duplicate 50 μl samples of diluted plasma, standards or blank were added to separate wells of a 96-well plate. To one of these samples, a further 40 μl assay buffer was added. The second sample was treated with 40 μl of assay buffer containing glucose-6-phosphate (2.5 mM), NADPH-dependent nitrate reductase (200 U l−1) and glucose-6-phosphate dehydrogenase (400 U l−1). A further 10 μl assay buffer containing NADPH (10 μM) was added to initiate the conversion of nitrate to nitrite. The conversion was allowed to run to completion over 1 h at room temperature. Griess reagent was prepared just before use by mixing equal volumes of N-(1-naphthyl) ethylenediamine (0.1% w v−1 in MilliQ water) and sulphanilamide (1% w /v−1 in 5% v v−1 concentrated orthophosphoric acid). Griess reagent was added as a 100 μl aliquot to each sample following its 1 h incubation. After a further 15 min incubation, the microplate was read in an absorbance reader with a 550 nm filter and a 630 nm reference filter. Standard curves for conversion of nitrate to nitrite were linear between 1.5 and 100 μM.
Blood pressure measurement methods
Conscious mice were chronically cannulated to enable administration of drug and measurement of blood pressure as previously described (Rees et al., 1998).
Inhibition of brain NOx in rats
At time zero, adult male Wistar rats were injected intravenously via the tail vein, with either saline or 20, 50, 100 or 200 mg kg−1 iNOS inhibitor at 1 ml kg−1 body weight. Animals were killed after 30 min by cervical dislocation and the head was removed. Brains were removed intact and the cerebellum immediately freeze-clamped in liquid nitrogen. These tissue samples were stored on dry ice until the end of the experiment when they were transferred to a −80°C freezer for later analysis. Total nitrate and nitrite (NOx) levels in rat cerebellum were measured according to a method modified from Salter et al. (1996). Cerebellar samples were thawed, homogenised on ice twice for 10 s, and then centrifuged at 20,000 × g at 4°C for 10 min. For the nitrate-nitrite conversion, 100 μl of the supernatant was incubated with 20 μl of Reagent B (50 μl of 120 mM FAD, 50 μl of 14.4 mM NADPH, 120 μl of 10 U ml−1 nitrate reductase and 984 μl of 14 mM sodium phosphate buffer pH 7.4) for 1 h at 37°C. Nitrite was then detected using a chemiluminescence detector as follows: the samples or sodium nitrite standards were injected as 10 μl aliquots into a 3 : 1 mix of glacial acetic acid and sodium iodide solution (3 g per 50 ml water) under reflux. The nitrite formed by the enzymatic reduction step above was converted to NO and carried to a chemiluminescence detector (CLD 77AM, Eco Physics) under a constant stream of nitrogen.
Materials
GW274150 and GW273629 were synthesised as the hydrochloride salts as described (Young et al., 2000). BH4 was from Dr B Schirks Labs, Switzerland. Other materials were from sources described above or from standard suppliers (Sigma, Fischer).
Results
NOS enzyme potency, selectivity and mechanism of action
Human iNOS (baculovirus expressed, recombinant) was inhibited by GW274150, GW273629 and L-NMMA in a concentration-dependent manner, when assessed over a 15 min incubation after a 15 min preincubation at 30 μM L-arginine, with IC50 values in the micromolar range (Table 1). When tested against human NOS isoforms (baculovirus expressed, recombinant), GW274150 was a highly selective inhibitor of human iNOS with 248-fold selectivity vs human eNOS and 81-fold selectivity vs human nNOS (Table 1). GW274150 was of similar potency against rodent iNOS and human iNOS and was 46-fold selective for rodent iNOS vs rodent nNOS (Table 1). GW273629 was also selective for iNOS with >110-fold selectivity vs eNOS, and was also >210-fold selective for rodent iNOS vs rodent nNOS (Table 1). In contrast, L-NMMA, a widely used nonselective NOS inhibitor, was of similar potency against all the NOS isoforms. When inhibition of human iNOS was assessed without preincubation, over 10 min, both GW274150 and GW273629 were less potent (6.9±0.9 and 20.1±5.0 μM, respectively), showing that they are both progressive inhibitors of this isoform.
Table 1.
Comparison of IC50 values for inhibition of recombinant human and rodent NOS isoforms by GW274150, GW273629 and L-NMMA measured over 15 min after a 15 min preincubation, in the presence of 30 μM L-arginine

Detailed enzyme kinetics were performed with human iNOS (E. coli expressed, recombinant) purified on ADP-sepharose. The substrate L-arginine inhibited the binding of GW274150 to human iNOS with an apparent Ki of 1.12±0.1 μM (Figure 1), which is similar to the measured Km value for arginine and human iNOS of 3.38±0.58 μM (n=4, data not shown). Thus, GW274150 is competitive with L-arginine for binding to human iNOS. The progress plots for inhibition of purified human iNOS by GW274150 with time were curved (Figure 2a) showing time-dependent inhibition of iNOS by GW274150. The data were fitted to the general integrated rate equation modified such that the steady-state rate is constrained to zero. The resulting Ktrans values were plotted against inhibitor concentration (Figure 2b), which gave hyperbolic plots intercepting very close to the origin, suggesting that GW274150 is an irreversible inhibitor of human iNOS. The on rate (kon) for GW274150 and human iNOS was determined from the asymptote of Figure 2b to be 0.00706±0.00033 s−1 (0.42 min−1). The concentration of GW274150 giving half maximal transition rate was 64±5.9 μM at 30 μM L-arginine; the apparent Ki for the initial EI complex formation was calculated to be 6.4 μM. Inhibition of human iNOS by GW274150 did not reverse when iNOS was diluted into a reaction assay at an excess (2 mM) L-arginine after a 1 h preincubation with GW274150 and NADPH. No evidence for reversal was seen over the 3 h that the dilution reaction was followed. However, when NADPH was omitted from the preincubation, GW274150 inhibition could be reversed and approximately 90% of the iNOS activity was recovered in the dilution assay. Therefore, the irreversible nature of the inhibition of iNOS by GW274150 is dependent on NADPH. Although no significant reversal of inhibition by GW274150 was evident from the data obtained, a small degree of reversal (<20% at 4000 s) would not be readily detectable, and longer assays to assess this more definitively were not possible because of the instability of the isolated enzyme over extended periods. From this estimate of the maximum reversal it can be calculated that the koff for a full reversal of inhibition must be less than 0.5 × 10−4 s−1, and the overall steady-state binding constant Kd=Ki (koff/kon) of GW274150 for human iNOS is <40 nM (assuming linearity of the control and blank rates in the incubation with NADPH and assuming first-order recovery). In contrast to the inhibition of iNOS, GW271450 did not show time-dependent inhibition of human eNOS or human nNOS and Ki values were determined using 0–10 min initial rates as 185±32 μM (n=3) and 4.57±0.23 μM (n=3), respectively. Therefore, the steady state selectivity of GW274150 for human iNOS is >5800-fold vs human eNOS and >114-fold vs human nNOS.
Figure 1.

Measurement of the Ks for GW274150 and purified human iNOS in the oxyhaemoglobin assay.
Figure 2.

Time-dependent inactivation of purified human iNOS by GW274150. (a) Progress plots of iNOS inhibition by GW27410 in the oxyhaemoglobin assay. (b) A plot of ktrans values against GW274150 concentration obtained from fitting the data for purified human iNOS inhibition by GW274150 in (a) to the integrated rate equation for slow binding inhibitors.
Studies with GW273629 showed similar behaviour, with a maximal on rate (kon) for GW273629 and human iNOS of 0.0066±0.0003s−1, an apparent Ki of 5.54±1.39 μM for arginine inhibition of GW273629 binding and an apparent Ki for the initial EI complex formation of 15 μM (data not shown). The overall steady-state binding constant of GW273629 for human iNOS is <90 nM when estimated in the same way as for GW274150. Preliminary experiments showed that inhibition by GW273629 of human endothelial NOS (heNOS) and human neuronal NOS (hnNOS) was rapidly reversible, and that the Ki values for GW273629 were 18±1 and 85±7 μM against hnNOS and heNOS, respectively (Garvey, E.P., unpublished data). Therefore, the steady-state selectivity of GW273629 for human iNOS is >944-fold vs human eNOS and >200-fold vs human nNOS.
Neither GW274150 nor GW273629 were substrates for iNOS. Incubation of an amount of purified human iNOS that would convert 1 nmol of L-arginine to citrulline min−1 with either 10 nmol [14C]GW274150 or GW273629 was carried out for 180 min at 37°C. The enzyme was removed by precipitation and centrifugation, and the supernatant was analysed by high-performance liquid chromatography. No new peaks were observed and [14C]GW274150 or GW273629 were quantitatively recovered, consistent with an absence of chemical modification of inhibitor during the incubation.
Cellular and tissue efficacy and selectivity
An assay of inhibition of intracellular murine iNOS was carried out by measurement of DAF fluorescence in IFNγ- and LPS-stimulated intact J774 cells, with a concentration of 100 μM L-arginine in the medium, which is the concentration present in human plasma (Lentner, 1984). GW274150 became progressively more potent over 24 h, with a predicted steady-state IC50 of 0.2±0.04 μM (Figure 3a), consistent with the time-dependent inhibition of the isolated enzyme. Similarly, GW273629 also became more potent with time (Figure 3b), reaching a steady-state IC50 of 1.3±0.16 μM.
Figure 3.

Increased potency of inhibition of iNOS in murine J774 cells by (a) GW274150 and (b) GW273629 over time. J774 cells were induced to express iNOS with LPS/IFNγ before addition of DAF-2DA and a range of inhibitor concentrations, and the fluorescence read at times indicated. Mean±s.d. from one experiment, representative of six.
The potency and selectivity of GW274150 and GW273629 against iNOS, eNOS and nNOS were also assessed in intact rat tissues. Exposure of endothelium-denuded aortic rings to LPS for 6 h, to induce iNOS activity, resulted in a profound NOS-dependent relaxation; iNOS inhibitory activity of compounds was then assessed by the contraction caused by inhibiting NO synthesis. Aortic ring preparations with intact endothelium were used to determine eNOS inhibitory activity of compounds as assessed by the contraction caused by inhibiting NO synthesis. Synthesis of NO by nNOS activity in intact cortical slice preparations from rat brain was assessed from the NOS-dependent conversion of 14C-L-arginine to 14C-citrulline. GW274150 was a potent inhibitor of iNOS in rat aortic rings, with an ED50 of 1.15±0.6 μM and was more potent on iNOS than either L-NMMA or GW273629 (Table 2). In contrast, GW274150 was a very weak inhibitor of eNOS in aortic rings and of nNOS in cortical slices. GW274150 was therefore approximately 250-fold and 200-fold selective for iNOS against eNOS and nNOS, respectively, and GW273629 was approximately 150- and 300-fold selective for iNOS against eNOS and nNOS, respectively. Both these compounds contrast with L-NMMA, which had a similar potency against all three NOS isoforms.
Table 2.
Potency and selectivity of GW274150 compared with GW273629 and L-NMMA on iNOS vs eNOS and nNOS in intact rat tissue assays
| Compound | Vascular ring NO synthesis ED50 (μM) (selectivity for iNOS) | Brain slice NO synthesis IC50 (μM) (selectivity for iNOS) | |
|---|---|---|---|
| iNOS | eNOS | nNOS | |
| GW274150 | 1.15±0.56 | 43% inhibition at 300 μM (>260) | 252±1.1 (219) |
| GW273629 | 2.0±0.6 | 45% inhibition at 300 μM (>150) | 729±181 (365) |
| L-NMMA | 5.2±0.8 | 16.3±1.5 (3.1) | 12.6±0.5 (2.4) |
Fold selectivities for iNOS are shown in brackets.
Pharmacokinetics
The pharmacokinetic profile of GW274150 was biphasic in healthy rats and mice with a terminal half-life of ∼6 h (Table 3). The pharmacokinetics of GW273629 were also biphasic in healthy rats producing a terminal half-life of ∼3 h. In healthy mice however, the pharmacokinetics appeared monophasic with a much more rapid elimination (approximately 10 min), but a terminal phase may have been disguised by the limit of quantification. GW274150 had a greater volume of distribution and a lower maximum observed plasma concentration than those for GW273629 at comparable dose levels. The slower clearance of GW274150 compared to GW273629 results in a greater systemic exposure as measured by the AUC. Therefore, in vivo, it would be expected that GW274150 would produce a more prolonged effect on iNOS inhibition than GW273629 by virtue of its longer half-life. Both compounds show a high oral bioavailability in the preclinical species tested including rats and mice (90%) (Schwartz, S.I., unpublished data).
Table 3.
Pharmacokinetics of GW274150 and GW273629 in healthy rats and mice following intravenous dosing
| Compound | Species | Dose (mg kg−1) | AUC∞ (h*μg ml−1) | Cmax (μg ml−1) | Tmax (h) | T1/2 (h) | CL (l h−1 kg−1) | Vss (l kg−1) |
|---|---|---|---|---|---|---|---|---|
| GW274150 | Mouse | 1 | 6.0 | 1.6 | 0.08 | 5.7 | 0.17 | 1.32 |
| Rat | 10 | 74.2 | 13.4 | 0.08 | 6.5 | 0.14 | 1.04 | |
| GW273629 | Mouse | 10 | 15.5 | 49.1 | 0.08 | 0.14 | 0.65 | 0.11 |
| Rat | 10 | 25.9 | 36.7 | 0.08 | 3.0 | 0.39 | 0.65 | |
n=3 mice per time point (males) and n=4 rats per time point (two males and two females).
Efficacy and selectivity in vivo
It has previously been shown that administration of LPS to rodents causes a substantial and widespread induction of the calcium-independent form of NO synthase, iNOS (Salter et al., 1991); this results in substantial increases in the plasma of the major breakdown products of NO, nitrate and nitrite (collectively termed NOx). The effects of GW274150 and GW273629 on LPS-induced increases in plasma NOx were measured as a means of determining the efficacy and potency of these compounds in inhibiting iNOS in vivo. LPS administration to mice resulted in an increase in synthesis of NO as assessed by an increase in plasma NOx, from basal levels of 16±6 μM (mean±s.e.m., n=4), following a lag phase of 2–3 h. The increase in plasma NOx was substantial by 6 h and peaked at approximately 12–18 h (Figure 4a). GW274150 100 mg kg−1 i.p. caused inhibition of the elevation in plasma NOx, with 92% inhibition at 2 h, 100% inhibition at 8 h, 100% inhibition at 14 h and 98% inhibition at 20 h after its administration. Administration of GW274150 at 30 mg kg−1 inhibited the rise in plasma NOx with a similar time course but to a lesser extent (Figure 4a). GW274150 had an ED50 of 3.2±0.7 mg kg−1 after 14 h (18 h post LPS dosing) when administered intraperitoneally (Figure 4b). A very similar ED50 of 3.8±1.5 mg kg−1 was obtained when the dosing route was oral, consistent with a high oral bioavailability. GW273629 dosed i.p. also suppressed the endotoxin-induced increase in plasma NOx in mice with an ED50 of 9±2 mg kg−1 over 2 h (Figure 5a). The inhibition of the increase in plasma NOx declined over the subsequent 6 h, consistent with the relatively short plasma half-life of this compound in mice (Figure 5b, Table 3). GW273629 does not show any evidence of causing a rebound in NO synthesis after its clearance, since plasma NOx concentrations in GW273629-treated mice remain lower than in controls even at 8 h when only low GW273629 concentrations would remain in the plasma.
Figure 4.

Effect of GW274150 i.v. on LPS-induced elevation of mouse plasma NOx. (a) The time course of mouse plasma NOx inhibition by GW274150 over 24 h. LPS was dosed at zero time and GW274150 was dosed at 4 h. (b) Dose response to GW274150 on plasma NOx. LPS was dosed at zero time and GW274150 was dosed at 4 h. Plasma was sampled at 18 h. Data represent means±s.e.m. for 4–6 animals. The 18 h LPS control NOx (for convenience of presentation plotted at 0.01 mg kg−1) was 1467±57 μM and the 4 h LPS control value (for convenience of presentation plotted at 1000 mg kg−1) was 170±26 μM.
Figure 5.

Effect of GW273629 i.v. on LPS-induced elevation of mouse plasma NOx. (a) The time course of effects of GW273629 i.v. on LPS-induced elevation of mouse plasma NOx over 12 h. LPS was dosed at zero time and GW273629 was dosed at 4 h. (b) Dose response to GW273629 on plasma NOx. LPS was dosed at zero time and GW273629 was dosed at 4 h. Plasma was sampled at 6 h. The data shown are means±s.e.m. from four mice except 6 h LPS control which is an average and a range for two values. The 6 h LPS control NOx (for convenience of presentation plotted at 0.001 mg kg−1) was 335±20 μM and the 4 h LPS control value (for convenience of presentation plotted at 400 mg kg−1) was 157±17 μM.
In short-term studies in conscious, chronically instrumented mice, blood pressure in untreated mice was stable over a 7 h observation period, whereas endotoxin administration (E. coli O26:B6 by i.v. bolus) caused a progressive fall in blood pressure from a basal mean arterial blood pressure of 97±3 mmHg down to 83±5 mmHg at 7 h (Figure 6a), GW273629 (1–1000 mg kg−1 i.v.) selectively raised blood pressure in a dose-dependent manner in these endotoxin-shocked mice, to a mean arterial blood pressure of 110±6 mmHg (Figure 6a). GW273629 over the same dose range had no significant effect on mean arterial blood pressure in normal mice (Figure 6b). This selectivity of effect is unlike that of the nonselective iNOS inhibitor L-NMMA that increased mean arterial blood pressure in both normal and endotoxin-shocked mice to levels substantially above normal (approximately 150 mmHg, Rees et al., 1998). The pharmacokinetics of GW273629 in these LPS-infected mice showed a longer half-life (approximately 2 h) and a much greater volume of distribution (up to 2 L kg−1). The increase in volume (and resulting increase in half-life) may have been a result of the surgical procedure of infusing the mice with saline to prevent hypovolemia induced by the shock state.
Figure 6.

The effect of GW273629 on the blood pressure of endotoxin-shocked and normal mice. (a) GW273629 increases blood pressure in endotoxin-shocked mice. Left panel: the progressive fall in mean arterial blood pressure (MABP) over a 7 h period following endotoxin administration (E. coli, 026:B6, 12.5 mg kg−1 i.v. bolus). Right panel: effect of GW273629 (1–1000 mg kg−1 i.v. bolus) on MABP in the conscious mouse following endotoxin. The maximum increase in MABP was measured over the 20 min period between successive doses of GW273629. Each point is the mean±s.e.m. of six animals. (b) GW273629 has no effect on blood pressure in normal mice. Left panel: basal MABP over a 7 h period prior to GW273629 administration. Right panel: effect of GW273629 (1–1000 mg kg−1 i.v. bolus) on MABP in the conscious mouse. The maximum increase in MABP was measured over a 20 min period between successive doses of GW273629. Each point is the mean±s.e.m. of six animals.
GW274150 was found to be a low potency inhibitor of nNOS in the rat cerebellum in vivo with no significant effects observed at doses up to 50 mg kg−1 (13±14% inhibition, n=5 per group). Higher doses of GW274150 did cause significant inhibition: 47±5% inhibition at 100 mg kg−1, 63±4% inhibition at 200 mg kg−1 (n=5 per group). GW273629 had no significant effect on rat cerebellum nNOS at all doses up to 1000 mg kg−1 (7±2% inhibition, n=4 per group).
Discussion
GW274150 and GW273629 are potent, highly selective inhibitors of both human and rodent iNOS
The therapeutic use of iNOS inhibitors is likely to be critically dependent on candidate medicines being potent and highly selective against iNOS vs the other NOS isoforms, as well as having appropriate pharmacokinetic properties. The data presented show that both GW273629 and GW274150 are highly selective, time-dependent inhibitors of isolated iNOS vs eNOS or nNOS. The kinetic studies suggest that they are arginine site, mechanism-based inhibitors of iNOS, requiring active enzyme and NADPH substrate to permit inhibition to proceed from the initial relatively weak binding to the enzyme, E+I↔EI, to more potent inhibition/inactivation, EI → EI* (where E is iNOS, I is the inhibitor, EI is the initial noncovalent complex and EI* is a modified complex), either with a conformational change to tight binding or with covalent changes to the enzyme, inhibitor or both. Previously published work on the partially selective iNOS inhibitors L-NIO and L-NIL suggest that they have a similar mechanism of action and that this may involve reaction with and the loss of haem from the enzyme, perhaps by forming an unstable haem adduct which breaks down to biliverdin (Bryk & Wolff, 1998; Wolff et al., 1998). Further investigation would be needed to confirm whether this was the case for inhibition of iNOS by GW274150 and GW273629. In contrast, these compounds are rapidly acting, simple competitive, low potency inhibitors of eNOS and nNOS.
The observed characteristics of inhibition of iNOS by these compounds are likely to have important implications in their experimental and clinical use:
The steady-state potency and selectivity of both compounds for iNOS over eNOS and nNOS are higher than indicated by the initial short-term (up to 30 min) inhibition experiments.
Inhibition will develop relatively slowly. For example at an arginine concentration of 30 μM, which is in the physiological range, 1 μM (∼0.2 μg ml−1), GW274150 would take ∼100 min to reach half the maximum level of inhibition of iNOS. However, inhibition will be achieved more rapidly at higher concentrations of the compounds, up to a maximum rate of 0.42 min−1 with either compound, such that maximum inhibition would be achieved within 10 min.
The duration of inhibition in vivo is predicted from these data to be longer than the pharmacokinetic half-life of the compounds. Given the lack of significant reversal of inhibition over 1 h, the duration of inhibition in vivo may be determined as much by the rate of iNOS turnover/resynthesis as by the pharmacokinetic half-life.
GW274150 and GW273629 are potent, highly selective iNOS inhibitors in cells and tissues
Consistent with the inhibition of iNOS isolated enzyme, inhibition of iNOS in intact J774 cells also proved to be time-dependent, reaching steady-state IC50 values of 0.2 μM (GW274150) and 1.3 μM (GW273629). Selective inhibition of iNOS vs eNOS and nNOS was observed in rat tissue preparations; however, because the iNOS rat aortic ring experiments were of rather short duration (15 min), these are likely to have underestimated the steady-state selectivity over eNOS and nNOS.
GW274150 and GW273629 are highly selective iNOS inhibitors in vivo
The pharmacokinetics of GW273629 and GW274150 in the rat showed that these compounds are cleared relatively slowly (terminal T1/2 3–6 h) and are orally bioavailable; GW273629 was however, more rapidly cleared in mice. Understanding of these properties is important for the experimental use of these compounds in animals, and could be consistent with use as oral or parenteral medicines if these properties scale normally to humans. Inhibition of iNOS in vivo, as assessed by inhibition of the LPS-provoked rise in plasma NOx in mice, was consistent with the in vitro potency on iNOS and the pharmacokinetics, with prolonged inhibition by GW274150 (ED50∼3.5 mg kg−1) and more transient inhibition by GW273629 (ED50 9 mg kg−1). The effects of GW273629 were also determined on mouse blood pressure, in both normal and LPS-shocked mice; the data were consistent with iNOS efficacy and selectivity, although again these are likely to have been underestimated because of the short duration of the exposure of iNOS to the compound (15 min at each dose level). Others have also reported that these compounds have pharmacological effects in rodents consistent with iNOS inhibition in vivo, at 5 mg kg−1 (Cuzzocrea et al., 2002; McDonald et al., 2002; Chatterjee et al., 2003; Evans & Whittle, 2003; Dugo et al., 2004). The two compounds were either ineffective (GW273629 no effect at up to 1000 mg kg−1) or of very low potency (GW274150 no effect at up to 50 mg kg−1) on nNOS in the brain as assessed by rat cerebellum NOx levels, in contrast to N-nitro-L-arginine (Salter et al., 1996). The low potencies of GW274150 and GW273629 on nNOS in the brain in vivo are consistent with results from the in vitro rat cortical slice assay and the recombinant rodent nNOS assay.
GW274150 and GW273629 are selective pharmacological tools and potential therapeutic agents
The data presented in this paper show that GW274150 and GW273629 are potent, highly selective inhibitors of both human and rodent iNOS in vitro against the isolated enzymes, in intact rat tissue assays and in vivo in rodent models. GW274150 and GW273629 will therefore be useful pharmacological tools in the investigation of the role of iNOS in a variety of animal models and do not possess the acute toxicity previously reported for 1400W (Garvey et al., 1997). The high selectivity, demonstrated here both in vitro and in vivo by the low potency of GW274150 and GW273629 on eNOS and nNOS, will also be of critical importance in the clinical use of these compounds, since they will avoid undesirable effects on the control of normal blood pressure and flow and on the vascular-protective effects attributed to eNOS, as well as any undesirable effects on nitrergic nerve function in the peripheral and central nervous systems which could result from inhibition of nNOS. GW274150 has potential to form the basis for therapies for the treatment of a range of inflammatory disorders of the lung, joint and gastrointestinal tract and in neuropathic pain (Whittle, 1997; Hobbs et al., 1999; Alderton et al., 2000; Folkerts et al., 2001; Cuzzocrea et al., 2002; Dugo et al., 2004), while GW273629, with a shorter pharmacokinetic half-life, has potential for the treatment of more acute conditions (Knowles et al., 2000; McDonald et al., 2002; Chatterjee et al., 2003).
Acknowledgments
We thank Jenny Lee for assistance with animal studies, Simon Parry, Tony Frend and Pam Vince for contributions to the pharmacokinetic studies, Ian Fellowes for provision of 14C-labelled compounds and Graham Hart for advice and discussions on the enzyme kinetic studies.
Abbreviations
References
- ALDERTON W., ANGELL A., CLAYTON N., CRAIG C., DAWSON J., FREND A., MCGILL J., MANGEL A., MONCADA S., REES D., RUSSELL L., SCHWARTZ S., WASLIDGE N., KNOWLES R.GW274150 is a potent, long-acting, highly selective inhibitor of inducible nitric oxide synthase with therapeutic potential in post-operative ileus Biology of Nitric Oxide 20007London: Portland Press; 22ed. Moncada, S., Gustafsson, L.E., Wiklund, N.P., & Higgs, E.A., pp [Google Scholar]
- ALDERTON W.K., COOPER C.C., KNOWLES R.G. Nitric oxide synthases: structure, function and inhibition. Biochem. J. 2001;357:593–615. doi: 10.1042/0264-6021:3570593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BRYK R., WOLFF D.J. Mechanism of inducible nitric oxide synthase inactivation by aminoguanidine and L-N6-(1-iminoethyl)lysine. Biochemistry. 1998;37:4844–4852. doi: 10.1021/bi972065t. [DOI] [PubMed] [Google Scholar]
- CHARLES I.G., SCORER C.A., MORO M.A., FERNANDEZ C., CHUBB A., DAWSON J., FOXWELL N., KNOWLES R.G., BAYLIS S.A. Expression of human nitric oxide synthase isozymes. Methods Enzymol. 1996;268:449–460. doi: 10.1016/s0076-6879(96)68047-1. [DOI] [PubMed] [Google Scholar]
- CHATTERJEE P.K., PATEL N.S., SIVARAJAH A., KVALE E.O., DUGO L., CUZZOCREA S., BROWN P.A., STEWART K.N., MOTA-FILIPE H., BRITTI D., YAQOOB M.M., THIEMERMANN C. GW274150, a potent and highly selective inhibitor of iNOS, reduces experimental renal ischemia/reperfusion injury. Kidney Int. 2003;63:853–865. doi: 10.1046/j.1523-1755.2003.00802.x. [DOI] [PubMed] [Google Scholar]
- CUZZOCREA S., CHATTERJEE P.K., MAZZON E., MCDONALD M.C., DUGO L., DI PAOLA R., SERRAINO I., BRITTI D., CAPUTI A.P., THIEMERMANN C. Beneficial effects of GW274150, a novel, potent and selective inhibitor of iNOS activity, in a rodent model of collagen-induced arthritis. Eur. J. Pharmacol. 2002;453:119–129. doi: 10.1016/s0014-2999(02)02338-5. [DOI] [PubMed] [Google Scholar]
- DAWSON J., KNOWLES R.G.A microtitre plate assay of human NOS isoforms Meth. Molec. Biol. 100, Nitric Oxide Protocols 1998Totowa NJ: Humana Press; 237–242.ed. Titheradge, M.A. pp [DOI] [PubMed] [Google Scholar]
- DUGO L., MARZOCCO S., MAZZON E., DI PAOLO R., GENOVESE T., CAPUTI A.P., CUZZOCREA S. Effects of GW274150, a novel and selective inhibitor of iNOS activity, in acute lung inflammation. Br. J. Pharmacol. 2004;141:979–987. doi: 10.1038/sj.bjp.0705683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS S.M., WHITTLE B.J. Role of bacteria and inducible nitric oxide synthase activity in the systemic inflammatory microvascular response provoked by indomethacin in the rat. Eur. J. Pharmacol. 2003;461:63–71. doi: 10.1016/s0014-2999(02)02959-x. [DOI] [PubMed] [Google Scholar]
- FOLKERTS G., KLOEK J., MUIJSERS R.B.R., NIJKAMP F.P. Reactive nitrogen and oxygen species in airway inflammation. Eur. J. Pharmacol. 2001;429:251–262. doi: 10.1016/s0014-2999(01)01324-3. [DOI] [PubMed] [Google Scholar]
- FOSETTA J.D., NIU X.D., LUNN C.A., ZAVODNY P.J., NARULA S.K., LUNDELL D. Expression of human inducible nitric oxide synthase in Escherichia coli. FEBS Lett. 1996;379:135–138. doi: 10.1016/0014-5793(95)01496-9. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., FURFINE E.S., KIFF R.J., LASZLO F., WHITTLE B.J.R., KNOWLES R.G. 1400W is a slow-, tight-binding and highly selective inhibitor of inducible nitric oxide synthase in vitro and in vivo. J. Biol. Chem. 1997;272:4959–4963. doi: 10.1074/jbc.272.8.4959. [DOI] [PubMed] [Google Scholar]
- GARVEY E.P., OPLINGER J.A., TANOURY G.J., SHERMANN P.A., FOWLER M., MARSHALL S., HARMON M.F., PAITH J.E., FURFINE E.S. Potent and selective inhibition of human nitric oxide synthases. J. Biol. Chem. 1994;269:26669–26676. [PubMed] [Google Scholar]
- HOBBS A.J., HIGGS A., MONCADA S. Inhibition of nitric oxide synthase as a potential therapeutic target. Ann. Rev. Pharmacol. Toxicol. 1999;39:191–220. doi: 10.1146/annurev.pharmtox.39.1.191. [DOI] [PubMed] [Google Scholar]
- KOJIMA H., NAKATSUBO N., KIKUCHI K., KAWAHARA S., KIRINO Y., NAGOSHI H., HIRATA Y., NAGANO T. Detection and imaging of nitric oxide with novel fluorescent indicators: diaminofluoresceins. Anal. Chem. 1998;70:2446–2453. doi: 10.1021/ac9801723. [DOI] [PubMed] [Google Scholar]
- KNOWLES R., DAWSON J., WASLIDGE N., RUSSELL R., ANGELL A., CRAIG C., SCHWARTZ S., EVANS S., WHITTLE B., GARVEY E., MONKHOUSE J., MONCADA S., REES D.GW273629 is a highly-selective, short-acting iNOS inhibitor both in vitro and in vivo; beneficial effects in endotoxin shock Biology of Nitric Oxide 20007London: Portland Press; ed. Moncada, S., Gustafsson, L. E., Wiklund, N. P, & Higgs, E. A., pp. 21 [Google Scholar]
- KNOWLES R.G., MONCADA S. Nitric oxide synthases in mammals. Biochem. J. 1994;298:249–258. doi: 10.1042/bj2980249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LENTNER C.Geigy Scientific Tables 19843(ed)Composition of Blood. Basle, Switzerland: International Medical and Pharmaceutical Information, Ciba Geigy Limited [Google Scholar]
- LIZASOAIN I., KNOWLES R.G., MONCADA S. Inhibition by Lamotrigine of the generation of nitric oxide in rat forebrain slices. J. Neurochem. 1995;64:636–642. doi: 10.1046/j.1471-4159.1995.64020636.x. [DOI] [PubMed] [Google Scholar]
- MCCALL T.B., FEELISCH M., PALMER R.M., MONCADA S. Identification of N-iminoethyl-L-ornithine as an irreversible inhibitor of nitric oxide synthase in phagocytic cells. Br. J. Pharmacol. 1991;102:234–238. doi: 10.1111/j.1476-5381.1991.tb12159.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCDONALD M.C., IZUMI M., CUZZOCREA S., THIEMERMANN C. A novel, potent and selective inhibitor of the activity of inducible nitric oxide synthase ( GW274150) reduces the organ injury in hemorrhagic shock. J. Physiol. Pharmacol. 2002;53:555–569. [PubMed] [Google Scholar]
- MONCADA S., HIGGS E.A. Molecular mechanisms and therapeutic strategies related to nitric oxide. FASEB J. 1995;9:1319–1330. [PubMed] [Google Scholar]
- MOORE W.M., WEBBER R.K., JEROME G.M., TJOENG F.S., MISKO T.P., CURRIE M.G. l-N6-(1-iminethyl)lysine: a selective inhibitor of inducible nitric oxide synthase. J Med. Chem. 1994;37:3886–3888. doi: 10.1021/jm00049a007. [DOI] [PubMed] [Google Scholar]
- MORRISON J.F., WALSH C.T. The behaviour and significance of slow-binding enzyme inhibitors. Adv. Enzymol. Relat. Areas Mol. Biol. 1988;61:201–301. doi: 10.1002/9780470123072.ch5. [DOI] [PubMed] [Google Scholar]
- REES D.D., MONKHOUSE J.E., CAMBRIDGE D., MONCADA S. Nitric oxide and the haemodynamic profile of endotoxin shock in the conscious mouse. Br. J. Pharmacol. 1998;124:540–546. doi: 10.1038/sj.bjp.0701815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RODEBERG D.A., CHAET M.S., BASS R.C., ARKOVITZ M.S., GARCIA V.F. Nitric oxide: an overview. Am. J. Surg. 1995;170:292–303. doi: 10.1016/s0002-9610(05)80017-0. [DOI] [PubMed] [Google Scholar]
- RODRIGEUZ-CRESPO I., ORITZ de MONTELLANO P.R. Human endothelial nitric oxide synthase: expression in Escherichia coli. Arch. Biochem. Biophys. 1996;336:151–156. doi: 10.1006/abbi.1996.0543. [DOI] [PubMed] [Google Scholar]
- RUSSELL R.J.Use of arginine analogs as inhibitors of nitric oxide synthase in rat aortic rings Methods in Molecular Biology 1998100Totowa, New Jersey: Humana Press; 243–250.Nitric Oxide Protocols ed. Titheradge, M.A., pp [DOI] [PubMed] [Google Scholar]
- SALTER M., DUFFY C., GARTHWAITE J., STRIJBOS P.J.L.M. Ex vivo measurement of brain tissue nitrite and nitrate accurately reflects nitric oxide synthase activity in vivo. J. Neurochem. 1996;66:1683–1690. doi: 10.1046/j.1471-4159.1996.66041683.x. [DOI] [PubMed] [Google Scholar]
- SALTER M., KNOWLES R.G., MONCADA S. Widespread tissue distribution, species distribution and changes in activity of Ca(2+)-dependent and Ca(2+)-independent nitric oxide synthases. FEBS Lett. 1991;291:145–149. doi: 10.1016/0014-5793(91)81123-p. [DOI] [PubMed] [Google Scholar]
- VALLANCE P., LEIPER J. Blocking NO synthesis: how, where and why. Nat. Rev. Drug Disc. 2002;1:939–950. doi: 10.1038/nrd960. [DOI] [PubMed] [Google Scholar]
- VERDON C.P., BURTON B.A., PRIOIR R.L. Sample pretreatment with nitrate reductase and glucose-6-phosphate dehydrogenase quantitatively reduces nitrate while avoiding interference by NADP(+) when the Griess reaction is used to assay for nitrite. Anal. Biochem. 1995;224:502–508. doi: 10.1006/abio.1995.1079. [DOI] [PubMed] [Google Scholar]
- WHITTLE B.J. Nitric oxide: a mediator of inflammation or mucosal defence. Eur. J. Gastroenterol. Hepatol. 1997;9:1026–1032. doi: 10.1097/00042737-199711000-00002. [DOI] [PubMed] [Google Scholar]
- WOLFF D.J., LUBESKIE A., GAULD D.S., NEULANDER M.J. Inactivation of nitric oxide synthases and cellular nitric oxide formation by N6-iminoethyl-L-lysine and N5-iminoethyl-L-ornithine. Eur. J. Pharmacol. 1998;350:325–334. doi: 10.1016/s0014-2999(98)00267-2. [DOI] [PubMed] [Google Scholar]
- YOUNG R.J., BEAMS R.M., CARTER K., CLARK H.A., COE D.M., CHAMBERS C.L., DAVIES P.I., DAWSON J., DRYSDALE M.J., FRANZMAN K.W., FRENCH C., HODGSON S.T., HODSON H.F., KLEANTHOUS S., RIDER P., SANDERS D., SAWYER D.A., SCOTT K.J., SHEARER B.G., STOCKER R., SMITH S., TACKLEY M.C., KNOWLES R.G. Inhibition of inducible nitric oxide synthase by acetamidine derivatives of hetero-substituted lysine and homolysine. Bioorg. Med. Chem. Lett. 2000;10:597–600. doi: 10.1016/s0960-894x(00)00055-x. [DOI] [PubMed] [Google Scholar]