

Abstract
SB-706375 potently inhibited [125I]hU-II binding to both mammalian recombinant and ‘native' UT receptors (Ki 4.7±1.5 to 20.7±3.6 nM at rodent, feline and primate recombinant UT receptors and Ki 5.4±0.4 nM at the endogenous UT receptor in SJRH30 cells).
Prior exposure to SB-706375 (1 μM, 30 min) did not alter [125I]hU-II binding affinity or density in recombinant cells (KD 3.1±0.4 vs 5.8±0.9 nM and Bmax 3.1±1.0 vs 2.8±0.8 pmol mg−1) consistent with a reversible mode of action.
The novel, nonpeptidic radioligand [3H]SB-657510, a close analogue of SB-706375, bound to the monkey UT receptor (KD 2.6±0.4 nM, Bmax 0.86±0.12 pmol mg−1) in a manner that was inhibited by both U-II isopeptides and SB-706375 (Ki 4.6±1.4 to 17.6±5.4 nM) consistent with the sulphonamides and native U-II ligands sharing a common UT receptor binding domain.
SB-706375 was a potent, competitive hU-II antagonist across species with pKb 7.29–8.00 in HEK293-UT receptor cells (inhibition of [Ca2+]i-mobilization) and pKb 7.47 in rat isolated aorta (inhibition of contraction). SB-706375 also reversed tone established in the rat aorta by prior exposure to hU-II (Kapp∼20 nM).
SB-706375 was a selective U-II antagonist with ⩾100-fold selectivity for the human UT receptor compared to 86 distinct receptors, ion channels, enzymes, transporters and nuclear hormones (Ki/IC50>1 μM). Accordingly, the contractile responses induced in isolated aortae by KCl, phenylephrine, angiotensin II and endothelin-1 were unaltered by SB-706375 (1 μM).
In summary, SB-706375 is a high-affinity, surmountable, reversible and selective nonpeptide UT receptor antagonist with cross-species activity that will assist in delineating the pathophysiological actions of U-II in mammals.
Keywords: Urotensin, UT receptor, SB-706375, SB-657510, G-protein-coupled receptor, vasoconstriction, GPR-14, hypertension, heart failure, radioligand binding
Introduction
Human urotensin-II (hU-II) induces profound cardiohaemodynamic effects upon systemic administration in the cat (Behm et al., 2004a), monkey (Ames et al., 1999) and in man (Böhm & Pernow, 2002; Lim et al., 2004). Not only does hU-II constitute ‘the most potent mammalian vasoconstrictor identified to date' (Ames et al., 1999), it also influences cardiorenal function by acting as a potent regulator of cardiac contractility (Russell et al., 2003; Kompa et al., 2004), a natriuretic factor (Song et al., 2003) and as a hypertrophic/proinflammatory factor (Watanabe et al., 2001; Zou et al., 2001; Tzanidis et al., 2003; Johns et al., 2004; Onan et al., 2004). As such, hU-II and its G-protein-coupled receptor, UT, are purported to be involved in the (dys)regulation of cardiorenal function (Douglas, 2003; Douglas et al., 2004a).
Based on a series of clinical observations, such as augmented plasma/tissue ‘U-II-like' activity and pharmacogenetic associations (i.e. SNP analysis), hU-II has been implicated recently in the aetiology of numerous cardiorenal and metabolic diseases including hypertension (Matsushita et al., 2001; Cheung et al., 2004), heart failure (Douglas et al., 2002; Ng et al., 2002; Richards et al., 2002; Russell et al., 2003; Lapp et al., 2004), atherosclerosis (Bousette et al., 2004; Maguire et al., 2004), renal failure (Totsune et al., 2001; Shenouda et al., 2002; Langham et al., 2004) and diabetes (Totsune et al., 2003; 2004; Wenyi et al., 2003). Unfortunately, however, the lack of suitable UT receptor antagonists (Dhanak et al., 2003; Douglas et al., 2004a) has, thus far, precluded a detailed investigation of the specific role of the U-II/UT receptor system in the pathogenesis of such disorders, either in preclinical species or in man. Although several novel peptidic and nonpeptidic UT receptor ligands have been described in the medical and patent literature, they are of limited functional utility due to poor potency, limited selectivity and/or retention of intrinsic activity/agonism, etc. To address these issues, the present report describes the identification and characterization of SB-706375 (Figure 1a), an arylsulphonamide developed from high-throughput screening leads originally identified in the legacy SmithKline Beecham compound collection. SB-706375 is a potent, surmountable, reversible and selective mammalian UT receptor antagonist. Since SB-706375 exhibits ‘pan-species' antagonism, it is proposed that this small molecule inhibitor will serve as a useful agent in delineating the (patho)physiological actions of U-II.
Figure 1.

Structures of the novel nonpeptide sulphonamide antagonists (a) SB-706375 (2-bromo-4,5-dimethoxy-N-[3-(R)-1-methyl-pyrrolidin-3-yloxy)-4-trifluromethyl-phenyl]-benzenesulphonamide HCl) and (b) SB-657510 (2-bromo-N-[4-chloro-3-((R)-1-methyl-pyrrolidin-3-yloxy)-phenyl]-4,5-dimethoxybenzenesulphonamide HCl).
Methods
Radioligand binding studies
The pharmacological properties of SB-706375 were assessed in a variety of radioligand binding studies (intact cells and membranes) using either (a) HEK293 cells stably expressing recombinant mammalian (mouse, rat, cat, monkey or human) UT receptors or (b) ‘native' human cells expressing endogenous UT receptors (human rhabdomyosarcoma SJRH30 cells; Douglas et al., 2004b).
Recombinant HEK293-UT receptor membrane preparation
HEK293 cells expressing the UT receptor were detached from 150 cm2 flasks with 1 mM EDTA in Ca2+/Mg2+-free Dulbecco's phosphate-buffered saline (DPBS), washed by centrifugation at 300 × g and stored as frozen pellets. Cell pellets were suspended in ice-cold buffer (10 mM Tris-HCl [pH 7.4], 5 mM Na-EDTA, 0.1 mM phenylmethylsulphonylfluoride [PMSF], 1.0 mg ml−1 bacitracin, 0.1 mg ml−1 aprotinin) and homogenized using a Dounce homogenizer (Bellco Glass, Inc., Vineland, NJ, U.S.A.). Homogenates were centrifuged at 47,000 × g for 20 min at 4°C. The pellets were washed twice by centrifugation in buffer (25 mM Tris-HCl [pH 7.4], 5 mM MgCl2, 2 mM Na-EGTA, 0.1 mg ml−1 bacitracin) and resuspended at 5 mg ml−1 for storage at −70°C. Protein concentration was measured by the Pierce (Rockford, IL, U.S.A.) bicinchoninic acid (BCA) method using bovine serum albumin (BSA) as a standard.
Saturation binding studies in recombinant HEK293-UT cell membranes
Saturation binding (KD, Bmax determination) was performed by scintillation proximity assay (SPA). Conditions were optimized for membrane protein concentration and amount of wheat germ agglutinin-coated (WGA) SPA beads (Amersham, Arlington Heights, IL, U.S.A.) and binding was carried out with 20–600 pM [125I]hU-II either in the absence (total binding) or presence (nonspecific binding) of 1 μM cold hU-II. The apparent equilibrium dissociation constants (KD) and the maximum binding sites (Bmax) from saturation binding experiments were calculated using the interactive nonlinear curve-fitting program of GraphPad Prism (San Diego, CA, U.S.A.).
Competition binding studies in recombinant HEK293-UT cell membranes
Competition binding was performed under similar conditions as those described above. [125I]hU-II (300 pM) was incubated with cell membranes in the presence of varying concentrations of U-II isoforms (1.0 pM–1.0 μM in 0.1% BSA) or SB-706375 (0.1 nM–10 μM in DMSO vehicle). WGA-SPA beads were suspended (50 mg ml−1) in binding buffer (25 mM Tris-HCl [pH 7.4], 5 mM MgCl2, 0.1% BSA) and stored at 4°C. At the time of the assay, WGA-SPA beads (12.5 μg ml−1) and recombinant UT receptor HEK293 membranes (50 μg well−1) were precoupled by gentle shaking (1 h, 25°C). Following preincubation, 100 μl of the complex, 10 μl of SB-706375 (0.1 nM–10 μM) and 50 μl [125I]hU-II (0.3 nM) were added to each well of a Packard OptiPlate™-96 microtitre plate (along with adequate binding buffer to bring the final volume of each well to 200 μl) using a Packard MultiProbe II EX robotic liquid handling system (Perkin-Elmer, Shelton, CT, U.S.A.). The assay plates were then sealed and shaken gently on an orbital shaker (1 h, 25°C). Finally, plates were spun at 1500 × g for 10 min and cell-bound radioactivity was determined (Packard Top Count). Specific binding was determined using cold hU-II (1 μM).
Reversibility of binding in HEK293 cells stably expressing the monkey recombinant UT receptor
Membranes from HEK293-monkey UT receptor cells were incubated with DMSO vehicle or SB-706375 (1 μM) for 30 min at 25°C following which KD and Bmax values were determined. Incubation mixtures were diluted with cold buffer consisting of 25 mM Tris-HCl, pH 7.4, 5 mM MgCl2, 2 mM Na-EGTA and 0.1 mg ml−1 bacitracin followed by centrifugation at 47,000 × g for 20 min at 4°C. Membrane pellets were then washed one more time and resuspended in 2 ml buffer and utilized for [125I]hU-II competition binding as detailed above.
Analysis of SB-706375 binding to native UT receptors in human intact SJRH30 cells
Binding of [125I]hU-II to native human UT receptors was studied according to Douglas et al. (2004b) using a whole-cell binding assay format in the human rhabdomyosarcoma cell line, SJRH30 (American Type Culture Collection number CRL-2061, ATCC, Manassas, VA, U.S.A.). Briefly, SJRH30 cells were washed with DPBS+ buffer (with 10 mM MgCl2, 0.7 mM CaCl2, 1.4 mM glucose, 0.2% BSA) immediately prior to exposure (37°C for 30 min in 1 ml DPBS+) to [125I]hU-II. After incubation, cells were washed four times with cold DPBS+ (1 ml), solubilized with 1 M NaOH (1 ml) and transferred to 12 × 75 mm glass tubes. Radioactivity was then measured in a Packard gamma counter (>85% efficiency). Saturation binding was carried out by incubating the cells with 5–300 pM [125I]hU-II in the absence (total binding) or presence of 1 μM hU-II (nonspecific binding). Competition binding was performed under similar conditions using 200 pM [125I]hU-II and different concentrations of competing ligands. All assays were performed in duplicate.
Inhibition of [3H]SB-657510 binding to recombinant HEK293-UT cell membranes
[3H]SB-657510 (specific activity 87 Ci mmol−1; Figure 1b) saturation binding to monkey recombinant UT-HEK293 cell membranes (10–15 μg) was initiated by the addition of increasing concentrations of radioligand in the absence (total binding) or presence (nonspecific binding) of hU-II (1 μM). Binding was performed at 25°C for 60 min (200 μl total volume of binding buffer; 20 mM Tris-HCl buffer at pH 7.4 containing 5 mM MgCl2, 0.2% BSA, 0.1 mg ml−1 bacitracin). Competition binding experiments were performed in duplicate using 10 nM [3H]SB-657510 and increasing concentrations of cold ligands. At the end of incubation, the reaction mixture was rapidly diluted with 2 ml cold wash buffer (0.9% NaCl w v−1) followed by rapid filtration over Skatron filtermates (Skatron Instruments, Norway). Radioactivity was counted in a beta liquid scintillation counter.
In vitro selectivity profile of SB-706375
The selectivity of SB-706375 for the UT receptor was assessed by examining the interaction between this sulphonamide ligand and a total of 86 distinct G-protein-coupled receptors, ion channels, enzymes, transporters and nuclear hormone receptor cross-screening assays using established protocols.
G-protein-coupled receptor targets screened included adenosine (A1/2A/3), adrenergic (α1A/1B/2B/2A/2C, β1/2/3), dopamine (D1/2/3/4), muscarinic (M1/2/3/4), serotonin (5-HT1A/B/D/E/F, HT2A/B/C, HT3, HT4, HT5A, HT6, HT7), histamine (H1/2), angiotensin II (AT1/2), ANP (GC-A), bradykinin (B1/2), calcitonin gene-related peptide (CGRP), melanocortin (MC4R), melatonin (ML1), neuropeptide Y (Y1/2), endothelin (ETA/B), neurotensin (NT1), cholecystokinin (CCKA), imidazoline (I2), neurokinin (NK2/3), benzodiazepine (BZD, central), cannabinoid (CB1/2), nicotinic (αBTX-sensitive, neuronal N-type) leukotriene (BLT1), galanin (GalR2), Il-8 (CXCR2), somatostatin (sst3/4), vasoactive intestinal peptide (PAC1, VPAC1), opioid (μ, δ, κ, σ), vasopressin (V1a), P2X, GABAA, AMPA, kainate and NMDA receptors. SB-706375 was also evaluated in a number of nuclear hormone receptor assays (glucocorticoid, estrogen (α,β), progesterone and testosterone) along with a variety of ion channel (including sodium [sites 1 and 2], calcium [L-type], potassium [SKCa, KV] and chloride [picrotoxinin-sensitive] channels), enzyme (including phosphodiesterases [isozymes I–V], phospholipase A2, eNOS, elastase, protein kinase C, Na+/K+-ATPase and EGF-tyrosine kinase) and biogenic amine uptake (noradrenaline and dopamine transporters) assays.
Evaluation of SB-706375 as a mammalian UT receptor antagonist
The ability of SB-706375 to function as a competitive U-II antagonist was assessed in (a) recombinant cells (inhibition of U-II-mediated [Ca2+]i-mobilization in HEK293 cells expressing mammalian UT receptors) and (b) native tissue (inhibition of U-II-induced contraction of rat aortae).
SB-706375 as a competitive, mammalian UT receptor antagonist in recombinant HEK293-UT cells
UT receptor-mediated Ca2+-mobilization studies were carried out in Fluo-3-loaded HEK293 cells stably expressing either the mouse, rat, monkey or human UT receptor using a microtitre plate-based fluorometric imaging plate reader (FLIPR, Molecular Devices, Sunnyvale, CA, U.S.A.) assay (Ames et al., 1999). On the day before the assay, cells expressing recombinant HEK293-UT were plated in 96-well black wall/clear bottom Biocoat plates (Becton Dickinson, San Jose, CA, U.S.A.) at ∼50,000 cells well−1 (producing ∼80–95% confluence on the day of assay). On the day of the experiment, growth media were aspirated and replaced with 100 μl ‘loading media' (Eagle's minimal essential media with Earl's salts, L-glutamine, 0.1% BSA, 2.5 mM probenecid, 4 μM Fluo-3-acetoxymethyl ester fluorescent indicator dye [2 mM stock solution in DMSO with 20% pluronic acid; Fluo-3AM, Molecular Probes, Eugene, OR, U.S.A.]). Cells were incubated for 1 h at 37°C at which point media were aspirated and replaced with identical media lacking Fluo-3AM. Cells were incubated for a further 10 min. Cells were then washed (three times with ‘assay buffer'; 120 mM NaCl, 4.6 mM KCl, 1.03 mM KH2PO4, 25 mM NaHCO3, 1.0 mM CaCl2, 1.1 mM MgCl2, 11 mM glucose, 20 mM HEPES [pH 7.4], 0.1% gelatin, 2.5 mM probenecid) at which point they were exposed to SB-706375 (0.1–30 μM) for 10 min prior to hU-II administration (10 pM–3.3 μM for the mouse and monkey UT receptor, 30 pM–10 μM for the rat and human UT receptor). The maximum change in agonist-induced fluorescence was quantified and antagonist pKb determined by Clark analysis (Lew & Angus, 1997).
Rat isolated aortae studies
The antagonistic properties of SB-706375 were assessed in rat isolated aortic contraction assays. All animal procedures were performed in accredited facilities in accordance with institutional guidelines (Animal Care and Use Committee, GlaxoSmithKline) and the Guide for the Care and Use of Laboratory Animals (DHSS #NIH 85-23). Male Sprague–Dawley rats (400 g; Charles River, Raleigh, NC, U.S.A.) were anaesthetized with 5% isoflurane in O2 and euthanized by exsanguination. Isolated aortae were cleaned of adherent tissue and denuded of endothelium using a pair of fine forceps (functional loss was confirmed using 10 μM carbachol). Vessel rings, approximately 2–3 mm in length, were suspended in 10 ml organ baths containing Krebs of the following composition (mM): NaCl, 112.0; KCl, 4.7; KH2PO4, 1.2; MgSO4, 1.2; CaCl2, 2.5; NaHCO3, 25.0; dextrose, 11.0; indomethacin, 0.01 (0.1% ethanol, v v−1). Krebs was maintained at 37±1°C and aerated with 95% O2 : 5% CO2 (pH 7.4). Changes in isometric force were measured under 1.0 g optimal resting tension using MLT0201/D force-displacement transducers (Letica Scientific Instruments, Barcelona, Spain) and recorded digitally using ADInstruments Chart 5.0 software (Colorado Springs, CO, U.S.A.). Following a 60 min equilibration period, the vessels were treated with standard concentrations of KCl (60 mM) and noradrenaline (10 μM). Subsequent agonist-induced responses were normalized to these responses. Each tissue was used to generate only one concentration–response curve.
Characterization of SB-706375-induced inhibition of hU-II contraction in rat isolated aortae (pKb determination): Rat isolated thoracic aortae were pretreated with either vehicle (0.1% DMSO) or SB-706375 (0.3, 1 or 3 μM) for 30 min following which cumulative concentration–response curves to hU-II (10 pM–10 μM) were constructed (pKb was determined by Clark analysis; Lew & Angus, 1997).
Selectivity of SB-706375 in rat isolated aortae: Paired thoracic aortae were pretreated with either vehicle (0.1% DMSO) or SB-706375 (1 μM, a concentration >30-fold greater than its pKb against hU-II in this tissue) for 30 min following which cumulative concentration–response curves to either KCl (5–90 mM), phenylephrine (0.1 nM–10 μM), angiotensin II (0.1 nM–1 μM) or endothelin-1 (0.1 nM–1 μM) were generated.
Reversibility of SB-706375-induced inhibition in rat isolated aortae: Following a 30-min pretreatment with either vehicle (0.1% DMSO) or 1 μM SB-706375, cumulative concentration–response curves to hU-II (0.1 nM–10 μM) were generated. Additional groups of tissues were washed (Krebs was replaced with fresh buffer, which did not contain any antagonist) repeatedly for either 5, 15, 30 or 60 min following which hU-II concentration–response curves were generated. For comparison, an identical washout study was performed in parallel using phenylephrine (1 nM–100 μM) and the competitive and reversible α1-adrenoceptor antagonist prazocin (3 nM; as with SB-706375 above, ∼30-fold above pKb determined in the rat isolated aorta; Aboud et al., 1993).
SB-706375-induced inhibition of hU-II established tone in rat isolated aortae: Paired aortae were contracted with 10 nM hU-II (∼EC80 concentration). Once the contractile response had reached a plateau (∼25 min), cumulative concentration–response curves to SB-706375 were generated by adding the antagonist to the organ bath in log unit intervals from 1 nM to 1 μM (DMSO vehicle was administered in equal volumes to a parallel set of aortae to serve as a ‘time control').
Drugs and materials
SB-706375 (2-bromo-4,5-dimethoxy-N-[3-(R)-1-methyl-pyrrolidin-3-yloxy)-4-trifluro-methyl-phenyl]-benzenesulphonamide HCl), SB-657510 (2-bromo-N-[4-chloro-3-((R)-1-methyl-pyrrolidin-3-yloxy)-phenyl]-4,5-dimethoxybenzenesulphonamide HCl) and [3H]SB-657510 (three [3H] incorporated into the pyrrolidine N-methyl group; specific activity 87 Ci mmol−1) were synthesized at GlaxoSmithKline (King of Prussia, PA, U.S.A.; Dhanak et al., 2002). The human isoform of U-II was synthesized by California Peptide Research Inc. (Napa, CA, U.S.A.) whereas goby, rat and mouse U-II were from Phoenix Pharmaceuticals Inc. (Mountain View, CA, U.S.A.). Porcine U-IIA/B isoforms were synthesized at GlaxoSmithKline. [125I]hU-II (Tyr9 monoiodinated) was custom-synthesized by Amersham (Arlington Heights, IL, U.S.A.; specific activity 2000 Ci mmol−1). Angiotensin II, carbachol, indomethacin, noradrenaline, phenylephrine and prazosin were from Sigma (St Louis, MO, U.S.A.). All other reagents used were of analytical grade unless otherwise stated. Reagents were made up freshly daily and stored in light-tight containers.
Data analysis
Unless stated to the contrary (pKb determinations, vide infra), all values are expressed as mean±standard error of the mean (s.e.m.). n represents the total number of individual experiments performed and statistical comparisons were made using paired, two-tailed t-tests (differences were considered significant when P⩽0.05) or ANOVA (Dunnett's multiple comparisons).
Equilibrium binding affinities (KD) and maximum binding site densities (Bmax) were determined by interactive nonlinear curve-fitting using GraphPad Prism (v3.0; GraphPad Software Inc., San Diego, CA, U.S.A.). Unless stated to the contrary (see section ‘Reversibility of binding in HEK293 cells stably expressing the monkey recombinant UT receptor'; vide infra), all KD estimates were determined by saturation binding analysis (estimated by exposing HEK293-UT cell membranes to increasing concentrations of radiolabel, that is [125I]hU-II or [3H]SB-657510). Hill coefficients (nH) were assumed to approximate unity (see Scatchard plot, Figure 4a; Ames et al., 1999; Elshourbagy et al., 2002; Aiyar et al., 2005):
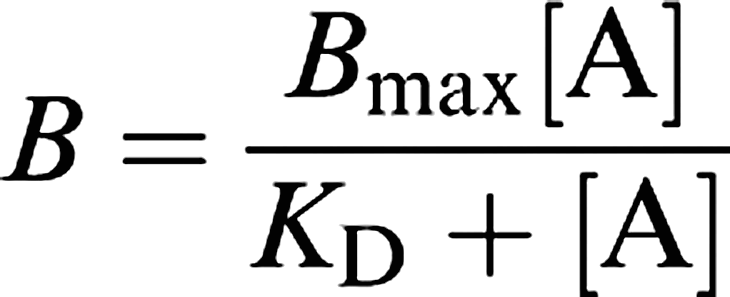 |
where B represents specific binding, [A] the concentration of ligand studied, Bmax the maximum number of binding sites and KD the equilibrium dissociation constant.
Figure 4.

(a) [3H]SB-657510 (0.2–25 nM) saturation binding to monkey UT-HEK293 cell membranes (nonspecific binding defined using 1 μM hU-II). Scatchard plot analysis (inset) revealed [3H]SB-657510 binding to a single class of receptors. (b) Inhibition of 10 nM [3H]SB-657510 binding (25°C for 30 min) to the monkey recombinant UT receptor by nonpeptidic (SB-706375 and SB-657510) and peptidic (human U-II) ligands. Each figure is representative of n=3 independent experiments run in duplicate. Binding parameters (KD, Bmax and Ki) were determined by nonlinear regression analysis (Table 4).
Kis were calculated by competition binding in recombinant HEK293 cell membranes (see section ‘Competition binding studies in recombinant HEK293-UT cell membranes' for [125I]hU-II binding and ‘Inhibition of [3H]SB-657510 binding to recombinant HEK293-UT cell membranes' for [3H]SB-657510 binding) and SJRH30 cells (see section ‘Analysis of SB-706375 binding to native UT receptors in human intact SJRH30 cells' for [125I]hU-II binding) using the equation by Cheng & Prusoff (1973):
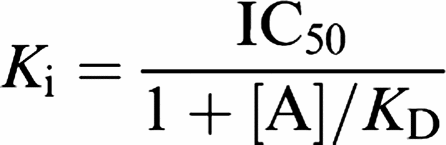 |
where [A] represents the concentration of competing ligand (nonpeptide antagonist or peptide agonist), IC50 the concentration of competing ligand that inhibits radiolabel binding by 50% and KD the equilibrium dissociation constant of the radioligand.
Agonist contractile response curves generated in rat isolated aortae and Ca2+-mobilization studies were analysed using the Hill equation (a logistic equation described previously; Douglas et al., 1995):
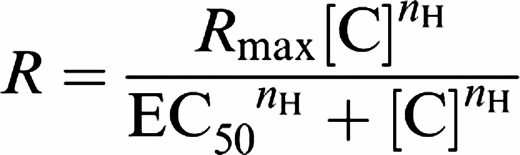 |
where R is the contractile response, [C] the concentration of agonist, EC50 the concentration of agonist required to produce a half contractile response, nH the Hill coefficient and Rmax the maximum contractile response.
Antagonist affinity determinations (pKb and associated 95% CI) for Ca2+-mobilization studies and isolated aortic contraction assays (see sections ‘SB-706375 as a competitive, mammalian UT receptor antagonist in recombinant HEK293-UT cells' and ‘Selectivity of SB-706375 in rat isolated aortae') were determined by nonlinear regression (Clark) analysis using the method of Lew & Angus (1997):
where [B] is the antagonist concentration and the constant log c is the difference between the antagonist pKb and the agonist control curve pEC50.
Finally, in one series of experiments (see section ‘Reversibility of binding in HEK293 cells stably expressing the monkey recombinant UT receptor'), KD was determined by homologous competition binding (whereby multiple concentrations of ‘cold' hU-II are used to compete for binding with a fixed concentration of [125I]hU-II). Data were analysed by LIGAND (assuming radioligand and competitor both bound reversibly to a single binding site; MacLigand, Version 4.97, NIH, Bethesda, MD, U.S.A.):
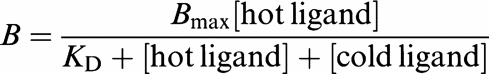 |
where B represents specific binding, [hot ligand] the single concentration of [125I]hU-II studied, [cold ligand] the concentration of unlabelled hU-II competing with the radiolabel for UT receptor binding, Bmax the maximum number of binding sites and KD the equilibrium dissociation constant (the equation was solved where the ‘cold ligand' IC50=[hot ligand]+KD).
Results
SB-706375 inhibits [125I]hU-II binding in mammalian recombinant HEK293-UT cells
[125I]hU-II (200 pM) binding to mouse, rat, cat, monkey and human recombinant UT receptors was specific (>90% total binding), saturable (Bmax∼325–1000 fmol mg−1 protein) and of high affinity (KD 0.2–0.6 nM). Binding was not evident in control (‘vector-transfected') HEK293 cell membranes. Goby, human, rat, mouse, pig (A/B isoforms) and human U-II all displaced radioligand from all five UT receptor isoforms with comparable affinities (Ki 0.5–5.7 nM; Table 1).
Table 1.
Pharmacological characterization of mammalian recombinant UT receptors with [125I]hU-II (saturation binding), SB-706375 and U-II isopeptides (competition binding)
| KD (pM) | Bmax (fmol mg−1) | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Mouse | Rat | Cat | Monkey | Human | Mouse | Rat | Cat | Monkey | Human | |
| Saturation binding | ||||||||||
| [125I]hU-II | 654±154a | 580±70b | 267±25 | 214±65a | 430±10b | 1011±125a | 325±80b | 790±60 | 497±68a | 447±58b |
| Ki (nM) | nH | |||||||||
| Mouse | Rat | Cat | Monkey | Human | Mouse | Rat | Cat | Monkey | Human | |
| Competition binding | ||||||||||
| SB-706375 | 19.1±1.0 | 20.7±3.6 | 4.7±1.5 | 5.3±1.5 | 9.3±1.0 | 0.91±0.12 | 1.75±0.46 | 1.02±0.01 | 0.99±0.04 | 0.97±0.06 |
| Human U-II | 5.7±2.8 | 2.1±0.2 | 2.3±0.1 | 2.5±0.7 | 2.5±0.3 | 1.10±0.04 | 0.94±0.02 | 1.05±0.06 | 0.77±0.06 | 0.84±0.04 |
| Goby U-II | 3.2±1.4 | 1.6±0.2 | 1.9±0.1 | 2.0±0.6 | 2.4±0.3 | 0.90±0.05 | 1.02±0.08 | 0.82±0.04 | 1.00±0.09 | 0.79±0.07 |
| Mouse U-II | 3.6±1.7 | 1.6±0.4 | 3.3±0.2 | 2.1±0.5 | 3.2±1.4 | 1.16±0.04 | 0.91±0.01 | 1.11±0.05 | 0.83±0.05 | 0.91±0.05 |
| Rat U-II | 2.5±0.8 | 1.4±0.2 | 2.7±0.6 | 1.8±0.4 | 2.9±1.2 | 0.89±0.05 | 0.91±0.03 | 0.79±0.03 | 0.86±0.03 | 0.85±0.02 |
| Porcine U-IIA | 2.4±1.1 | 0.8±0.3 | 5.0±1.8 | 1.5±0.4 | 1.6±0.2 | 1.16±0.17 | 0.82±0.04 | 1.41±0.38 | 0.75±0.07 | 0.95±0.06 |
| Porcine U-IIB | 1.9±1.0 | 0.5±0.2 | 1.8±0.6 | 1.3±0.3 | 0.9±0.1 | 1.09±0.07 | 0.92±0.05 | 1.19±0.23 | 0.84±0.09 | 1.10±0.08 |
All values, represented as mean±s.e.m. (n=3–6, duplicate determinations), were determined using membranes isolated from HEK293 cells stably transfected with the mouse, rat, cat, monkey or human UT receptor. U-II isopeptides displayed monophasic competition curves.
Data are from Elshourbagy et al. (2002) and are included for ease of comparison.
Data are from Ames et al. (1999) and are included for ease of comparison.
SB-706375 inhibited [125I]hU-II binding to recombinant UT receptors in a ‘pan-species' manner with inhibitory potencies (Ki) ranging from 4 to 20 nM across the five mammalian UT isoforms tested (Figure 2 and Table 1). Inhibition was concentration-dependent and the Hill slope (approximating unity) indicated the presence of a homogeneous, single population of binding sites.
Figure 2.

SB-706375 inhibits [125I]hU-II binding to HEK293 membranes stably transfected with mammalian recombinant UT receptors. Membranes isolated from HEK293 cells transfected with the mouse, rat, cat, monkey or human UT receptor were incubated with SB-706375 (0.1 nM–1 μM) and [125I]hU-II (0.3 nM) for 60 min at 25°C. Nonlinear regression analysis revealed high-affinity non-cooperative interactions with a single class of binding sites (each point represents the mean of duplicate determinations and is representative of three to six independent experiments; Kis are shown in Table 1).
Reversibility of SB-706375 binding in mammalian recombinant HEK293-UT cells
Relative to vehicle (DMSO)-treated control cells, prior exposure to 1 μM SB-706375 for 30 min did not alter [125I]hU-II binding affinity or density at the monkey recombinant UT receptor (Table 2) consistent with a reversible mode of SB-706375 binding.
Table 2.
Reversible nature of SB-706375 binding to monkey recombinant UT receptor
| KD (nM) | Bmax (pmol mg−1 protein) |
|
|---|---|---|
| Control (vehicle-treated) | 3.1±0.4 | 3.1±1.0 |
| Pretreated with SB-706375 | 5.8±0.9 | 2.8±0.8 |
Prior exposure to SB-706375 did not alter [125I]hU-II radioligand binding site density (Bmax) or affinity (KD). Competition binding was performed using membranes pretreated with either vehicle or SB-706375 (1 μM for 30 min at 25°C followed by extensive washing) from HEK293 cells stably transfected with the monkey UT receptor. Data are expressed as mean±s.e.m. (n=4, determined in duplicate).
High-affinity SB-706375 binding to native human UT receptors expressed in SJRH30 rhabdomyosarcoma cells
In accord with those Kis determined for the human UT receptor using recombinant HEK293 cell membranes, SB-706375 was a potent inhibitor (Ki 5 nM) of [125I]hU-II binding to native human UT receptors expressed by intact SJRH30 cells. Inhibition was monophasic, indicative of an interaction with a homogeneous population of binding sites. Both human and goby U-II also inhibited radioligand binding in this native human cell line in a concentration-dependent manner with Ki 0.5–0.6 nM (Figure 3 and Table 3).
Figure 3.

Inhibition of [125I]hU-II binding to native human UT receptors expressed by SJRH30 cells (200 pM at 37°C for 30 min) by goby and human U-II and SB-706375. Each point represents the mean of duplicate determinations and is representative of results from one of four independent experiments. Binding parameters (KD, Bmax and Ki) were determined by nonlinear regression analysis and were consistent with non-cooperative interaction with a single class of binding sites (Table 3).
Table 3.
SB-706375 inhibits [125I]hU-II binding at the native human UT receptor endogenously expressed in the rhabdomyosarcoma cell line SJRH30
| Ki (nM) | nH | |
|---|---|---|
| Competition binding | ||
| SB-706375 | 5.4±0.4 | 0.88±0.07 |
| Human U-II | 0.5±0.1 | 0.98±0.16 |
| Goby U-II | 0.6±0.2 | 0.89±0.08 |
All values are represented as mean±s.e.m. (n=4, duplicate determinations).
In vitro selectivity profile of SB-706375
SB-706375 was inactive when cross-screened against a diverse range of 86 distinct (see section ‘In vitro selectivity profile of SB-706375' mentioned earlier) G-protein-coupled receptors, ion channels, enzymes, transporters and nuclear hormone receptor assays (Ki/IC50 were all >1 μM). As such, SB-706375 functioned as a selective UT receptor antagonist with ⩾100-fold selectivity for the human UT receptor isoform (Ki 9 nM).
Inhibition of [3H]SB-657510 binding to recombinant HEK293-UT cell membranes
Specific [3H]SB-657510 (10 nM) binding (95±4% total binding) rapidly reached steady state (⩽10 min at 25°C) in monkey UT-HEK293 membranes and was maintained for ⩾2 h. The rate of association and dissociation of [3H]SB-657510 binding was fast, reversible and specific for UT-HEK293 membranes. [3H]SB-657510 kinetic rate constants for both association (for a single concentration of 10 nM [3H]SB-657510) and dissociation (made by adding excess cold ligand at equilibrium [30 min]) were estimated to be t1/2 2.0 and 1.2 min, respectively. [3H]SB-657510 binding was saturable, and Scatchard analysis suggested the presence of a single class of high-affinity binding sites (KD∼2 nM and Bmax∼0.9 pmol mg−1; Figure 4a and Table 4). No specific binding was observed to other GPCRs, and no displacement of [3H]SB-657510 from the UT receptor was evident following exposure to endothelin-1, neuromedin-U, somatostatin or CGRP. [3H]SB-657510 binding was inhibited by SB-706375 and ‘cold' SB-657510 and by human and goby U-II with Kis ranging from 4 to 17 nM (Figure 4b and Table 4), values consistent with those generated previously using [125I]hU-II as the radioligand (Table 1).
Table 4.
Inhibition of [3H]SB-657510 binding to monkey UT-HEK293 cell membranes by nonpeptide sulphonamide antagonists (SB-706375 and SB-657510) and native U-II isopeptides
| KD (nM) | Bmax (fmol mg−1 protein) | |
|---|---|---|
| Saturation binding | ||
| [3H]SB-657510 | 2.6±0.4 | 860±120 |
| Ki (nM) | nH | |
| Competition binding | ||
| SB-706375 | 4.6±1.3 | 0.99±0.05 |
| SB-657510 Ki | 17.6±5.4 | 0.89±0.03 |
| Human U-II Ki | 11.4±3.3 | 1.16±0.15 |
| Goby U-II Ki | 14.6±4.3 | 0.96±0.12 |
All values are represented as mean±s.e.m. (n=3–4, duplicate determinations).
Competitive, mammalian UT receptor antagonist in recombinant HEK293-UT cells
As previously reported (Ames et al., 1999; Elshourbagy et al., 2002), hU-II was a potent agonist at mouse, rat, monkey and human UT receptors stably expressed in HEK293 cells ([Ca2+]i-mobilization EC50s 0.10±0.00, 1.33±0.41, 0.13±0.04 and 0.50±0.14 nM, respectively; n=3). SB-706375 (0.1–30 μM) produced a concentration-dependent inhibition of hU-II-induced Ca2+-mobilization in all four recombinant cell lines tested (Figure 5). The resultant rightward, parallel shifts in the hU-II concentration–response curves were consistent with a competitive, surmountable mode of action. In agreement with this, Clark analysis revealed pKbs for SB-706375 that ranged from 7.29 to 8.00 (∼10–50 nM; Figure 6 and Table 5). Exposure to SB-706375 did not result in any change in basal [Ca2+]i levels in HEK293 cells expressing mammalian recombinant UT receptors.
Figure 5.

SB-706375 is a competitive antagonist of hU-II-induced [Ca2+]i-mobilization (FLIPR) in HEK293 cells stably expressing the (a) mouse, (b) rat, (c) monkey or (d) human UT receptor. Concentration–response curves to hU-II (10 pM–3.3 μM for mouse and monkey UT receptors, 30 pM–10 μM for rat and human UT receptors) were rightward-shifted in a parallel manner by SB-706375 (0.1–30 μM) without any significant suppression of the maximal response to hU-II consistent with competitive antagonism (n=3).
Figure 6.

SB-706375 is a competitive antagonist of hU-II-induced [Ca2+]i-mobilization in HEK293 cells stably expressing the (a) mouse, (b) rat, (c) monkey or (d) human UT receptor. Clark plot analysis (global nonlinear regression) revealed pKb values of 7.29, 7.94, 7.81 and 8.00 (7.20–7.38, 7.51–8.38, 7.64–7.97 and 7.83–8.17, 95% CI). The unity lines shown through the data are included for display purposes only.
Table 5.
Potency determination (pKb) for SB-706375 in intact HEK293 cells stably expressing mammalian recombinant UT receptors using an hU-II-induced [Ca2+]i-mobilization (FLIPR) assay
| UT receptor isoform | pKb |
|---|---|
| Mouse | 7.29 (7.20–7.38) |
| Rat | 7.94 (7.51–8.38) |
| Cata | — |
| Monkey | 7.81 (7.64–7.97) |
| Human | 8.00 (7.83–8.17) |
pKb values were determined by Clark analysis (global nonlinear regression; Lew & Angus, 1997) and are represented as mean values with 95% confidence intervals in parentheses (n=3).
Antagonism was not determined at the cat UT receptor.
SB-706375 pKb determination in the rat isolated aorta
Exposure of rat isolated aortic rings to SB-706375 (0.3–3.0 μM) resulted in concentration-dependent, rightward, parallel shifts in the hU-II concentration–response curves (Figure 7). Inhibition was surmountable inasmuch as the maximal contractile responses (Rmax) to hU-II remained unaltered (97–122% KCl response; Table 6). Global nonlinear regression (Clark) analysis revealed a pKb of 7.47 (7.25–7.69 [95% confidence interval]), a value corresponding to an affinity of ∼33 nM. Pretreatment of rat isolated aortae with SB-706375 did not result in any change in basal tone, that is, SB-706375 was devoid of any intrinsic activity and did not induce a contractile response in this tissue.
Figure 7.

SB-706375 is a competitive inhibitor of hU-II-induced contraction in the rat isolated aorta. (a) SB-706375 (0.3–3.0 μM) produces concentration-dependent rightward, parallel shifts in the hU-II concentration–response curve with no suppression of the maximal response to hU-II consistent with a competitive mode of antagonism (n=12). (b) Clark plot (global nonlinear regression analysis) revealed 7.47 pKb (7.25–7.69 95% CI). The unity line shown through the data is included for display purposes only.
Table 6.
SB-706375 is a competitive antagonist of hU-II-induced contraction of the rat isolated aorta
| Treatment | EC50 (nM) | Rmax (% 60 mM KCl) |
nH | Dose ratio |
|---|---|---|---|---|
| Vehicle | 2.3±0.4 | 97±10 | 1.90±0.15 | — |
| 0.3 μM SB-706375 | 24.0±7.7 | 122±9 | 1.93±0.20 | 13.1±3.5 |
| 1.0 μM SB-706375 | 79.3±15.1 | 122±7 | 1.97±0.19 | 47.6±9.2 |
| 3.0 μM SB-706375 | 182.6±43.4 | 114±8 | 1.68±0.18 | 166.2±66.3 |
SB-706375 (0.3–3.0 μM) caused concentration-dependent, parallel rightward shifts in the hU-II concentration–response curve. Values are expressed as mean±s.e.m. (n=12). Neither the maximum contractility (Rmax) nor Hill slope (nH) was altered by prior exposure to SB-706375 (parallel rightward shifts in the U-II concentration–response curve) as assessed by ANOVA (analysis for repeated measures; Dunnett's multiple comparisons post-test) consistent with surmountable, competitive mode of action in the rat isolated aorta.
Selective inhibition of hU-II-induced contraction in the rat isolated aorta
Exposure to 1 μM SB-706375, a concentration ∼30-fold over the pKb for hU-II in this vessel, did not alter the contractile responses induced by either (a) membrane depolarization (with KCl) or (b) contractile mechanisms involving ‘non-U-II' G-protein-coupled receptors, for example, phenylephrine (α1-adrenoceptor), angiotensin II (AT1) and endothelin-1 (ETA; Table 7 and Figure 8).
Table 7.
SB-706375 (1 μM) is a selective hU-II antagonist in the rat isolated aorta
| Treatment | EC50 (nM) | Rmax (% 60 mM KCl) |
nH |
|---|---|---|---|
| Phenylephrine | |||
| Vehicle-treated | 19.8±3.1 | 146±6 | 2.19±0.41 |
| SB-706375-treated | 23.6±2.7 | 138±7 | 2.30±0.35 |
| Angiotensin II | |||
| Vehicle-treated | 6.3±1.5 | 38±6 | 2.52±1.17 |
| SB-706375-treated | 8.9±1.9 | 40±12 | 1.85±0.53 |
| Endothelin-1 | |||
| Vehicle-treated | 8.5±1.1 | 153±6 | 2.28±0.35 |
| SB-706375-treated | 8.6±1.3 | 151±7 | 1.84±0.10 |
| KCla | |||
| Vehicle-treated | 15.4±0.9 | 101±7 | 8.27±1.73 |
| SB-706375-treated | 16.9±0.9 | 95±6 | 10.51±1.09 |
Pretreatment of rat isolated aortae with SB-706375 (1 μM) does not affect contractions invoked in the rat isolated aorta by KCl, endothelin-1, angiotensin II or phenylephrine. Values are expressed as mean±s.e.m. (n=4).
Contractile responses to KCl are expressed as mM (EC50) and % response to 10 μM noradrenaline (Rmax). SB-706375 treatment did not alter EC50, Rmax or nH values of any spasmogen studied (paired t-test).
Figure 8.

SB-706375 (1 μM) was a selective hU-II antagonist in the rat aorta since 1 μM concentration of the ligand (a concentration some 30-fold above its pKb against hU-II in this tissue) failed to inhibit the contractile actions of (a) KCl, (b) phenylephrine, (c) angiotensin II or (d) endothelin-1 (n=4) in the same tissue.
Reversibility of SB-706375-induced inhibition in the rat isolated aorta
The degree of SB-706375-induced inhibition of hU-II-mediated contraction was attenuated by removal of SB-706375 from the organ bath by repeated washing (Figure 9a and Table 8), that is, the inhibitory effects of SB-706375 were reversible. Relative to vehicle-treated tissues, a 30 min preincubation with 1 μM SB-706375 shifted the hU-II concentration–response curve ∼30-fold to the right provided SB-706375 was left in contact with the tissues (Table 8). However, repeatedly rinsing the organ baths with normal, ‘antagonist-free' Krebs solution for 5–60 min following an initial 30 min preincubation period with 1 μM SB-706375 resulted in time-dependent, leftward shifts of the hU-II concentration–response curves back towards the control (vehicle-treated) hU-II concentration–response curve. Similar data were obtained with phenylephrine/prazosin (Figure 9b and Table 8). The ‘reversibility/washout' rate constants for SB-706375 and prazosin were similar, estimated to be 0.022 min−1 (r=0.998) and 0.029 min−1 (r=0.883), respectively (Figure 9c).
Figure 9.

The inhibitory properties of (a) SB-706375 (hU-II-induced contraction) and (b) prazosin (phenylephrine-induced contraction) are reversible by ‘washout' in the rat isolated aorta. Following a 30 min preincubation with antagonist (1 μM SB-706375 or 3 nM prazosin) or vehicle, cumulative concentration–response curves to agonists (hU-II or phenylephrine) were generated. Repeatedly washing tissues to remove antagonist from the organ bath for 5, 15, 30 or 60 min prior to initiating the hU-II or phenylephrine concentration–response curves attenuated the degree of antagonism (i.e. rightward shift) observed (n=5). (c) The rate at which SB-706375 and prazosin were washed out of the isolated aorta (0.022 and 0.029 min−1, respectively) was estimated by plotting the degree of antagonism observed (‘dose ratio', based on agonist potency in vehicle-treated tissues; see Table 8) against the duration of repeated washing (5–60 min of ‘washout').
Table 8.
Inhibitory actions of 1 μM SB-706375 (hU-II-induced contraction) and 3 nM prazosin (phenylephrine-induced contraction) are reversible by repeated washing (5–60 min) of the rat isolated aorta
| Treatment | Washout time (min) | hU-II EC50 (nM) | hU-II Rmax (% 60 mM KCl) | Dose ratio | nH |
|---|---|---|---|---|---|
| Vehicle | — | 6±2 | 113±15 | — | 1.47±0.21 |
| SB-706375 | 0 | 148±21 | 90±12 | 32.3±6.6 | 1.68±0.08 |
| SB-706375 | 5 | 40±11 | 109±15 | 10.9±4.5 | 1.69±0.12 |
| SB-706375 | 15 | 38±9 | 122±11 | 9.2±2.9 | 1.36±0.13 |
| SB-706375 | 30 | 29±8 | 116±12 | 6.4±1.9 | 1.29±0.10 |
| SB-706375 | 60 | 14±3 | 115±14 | 3.3±1.0 | 1.66±0.24 |
| Treatment | Washout time (min) | Phenylephrine EC50 (nM) | Phenylephrine Rmax (% 60 mM KCl) | Dose ratio | nH |
|---|---|---|---|---|---|
| Vehicle | — | 13±2 | 146±8 | — | 2.03±0.18 |
| Prazosin | 0 | 868±319 | 120±9 | 70.1±26.6 | 1.48±0.24 |
| Prazosin | 5 | 243±52 | 140±3 | 22.8±6.4 | 1.68±0.20 |
| Prazosin | 15 | 257±74 | 130±8 | 25.1±9.9 | 2.08±0.36 |
| Prazosin | 30 | 82±13 | 149±5 | 7.3±1.0 | 1.43±0.12 |
| Prazosin | 60 | 60±10 | 137±5 | 5.5±1.0 | 2.14±0.22 |
Following a 30 min preincubation period with either vehicle or SB-706375 (1 μM), vessels were exposed to increasing concentrations of hU-II. Removal of SB-706375 from the organ bath by repeated washing of the vessels for 5–60 min significantly attenuated the parallel rightward shift seen in the hU-II concentration–response curve, indicating that the inhibitory effects of SB-706375 were reversible. Data were similar to those obtained with an unrelated, pharmacologically distinct antagonist, 3 nM prazosin (phenylephrine [α1-adrenoceptor]-induced contraction). All values are expressed as mean±s.e.m. (n=5). Neither the maximum contractility (Rmax) nor Hill slope (nH) determined for U-II and phenylephrine was altered by prior exposure to either SB-706375 or prazosin (parallel rightward shifts in the agonist concentration–response curves) as assessed by ANOVA (analysis for repeated measures) consistent with surmountable, competitive modes of action for both antagonists in the rat isolated aorta.
SB-706375-induced inhibition of hU-II established tone in rat isolated aortae
SB-706375 readily attenuated (100% reversal) tone established in the rat aorta by prior exposure to hU-II with 88.6±10.2 nM IC50 (Figure 10). Application of the Cheng–Prusoff equation (assuming a 3 nM EC50 for hU-II) yielded an estimated Kapp of ∼20 nM, consistent with the 7.47 pKb (33 nM) reported above (see section ‘SB-706375 pKb determination in the rat isolated aorta'). Established hU-II-induced tone was reduced by 50% within 12.1±1.7 min of exposure to 10 nM SB-706375.
Figure 10.

SB-706375 reverses hU-II-induced tone established in the rat aorta. Tissues were contracted with 10 nM hU-II and, once induced tone had reached a plateau, aortae were exposed to either vehicle or SB-706375 added to the organ bath in a cumulative manner. SB-706375 reversed the established tone to baseline (100% reversal; n=4).
Discussion
In recent years, U-II has emerged as a putative cardiorenal target of considerable interest within the pharmaceutical and medical communities (Douglas & Ohlstein, 2000; Maguire & Davenport, 2002; Russell, 2004). This interest has been driven, in large part, by the pronounced vasopressor activities exerted by U-II in intact mammals such as the cat (Behm et al., 2004a), monkey (Ames et al., 1999) and man (Böhm & Pernow, 2002; Lim et al., 2004). Further to this, several clinical studies have noted an emerging association between elevated U-II levels or genotype and cardiorenal diseases such as hypertension, heart failure, atherosclerosis, renal failure and diabetes (see Gilbert et al., 2004; Richards & Charles, 2004). Nevertheless, the precise (patho)physiological significance of U-II remains ambiguous given the fact that the haemodynamic actions of U-II vary significantly between species (in contrast to the cat and primate, bolus i.v. U-II lacks any haemodynamic activity in sheep and induces a ‘paradoxical' vasodilation in the rat; Hasegawa et al., 1992; Watson et al., 2003; Gardiner et al., 2004).
Although plethysmographic (Böhm & Pernow, 2002), iontophoretic (Lim et al., 2004) and direct intradermal injection (Leslie et al., 2000) studies demonstrate that local U-II administration elevates regional vascular resistance in healthy humans and heart failure patients, at least one well-respected group has failed to demonstrate any such response using almost identical techniques (Affolter et al., 2002; Wilkinson et al., 2002). Further, and in contrast to several other reports (MacLean et al., 2000; Maguire et al., 2000; 2004; Russell et al., 2001; Paysant et al., 2001; Camarda et al., 2002b), Hillier et al. (2001) failed to observe any significant contractile activity with U-II in human isolated arteries in a series of carefully controlled in vitro experiments. Clearly, the need for additional studies to help address these inconsistencies is evident. While observations of SNP associations and elevated plasma U-II levels might be suggestive of a causative role for U-II in the pathogenesis of cardiorenal diseases, such a proposition lacks the affirmation afforded by specific UT receptor antagonists. However, the dearth of such inhibitors has, until now, been prohibitive. It is hoped that the findings of the present study, which details the pharmacodynamic characterization of the novel UT receptor antagonist SB-706375 (see Table 9), will mitigate this issue and facilitate the delineation of the (patho)physiological actions of U-II.
Table 9.
Synopsis of affinity estimates for SB-706375 at mammalian (mouse, rat, cat, monkey and human) UT receptors as determined in radioligand binding studies (native and recombinant UT receptor assays) and in functional assays using both intact cells (recombinant UT receptor) and isolated arteries (native UT receptor)
| Mouse | Rat | Cat | Monkey | Human | |
|---|---|---|---|---|---|
| Radioligand binding Ki (nM) estimates (competition binding) | |||||
| HEK293-UT membranes ([125I]hU-II) | 19.1 | 20.7 | 4.7 | 5.3 | 9.3 |
| HEK293-UT membranes ([3H]SB-657510) | — | — | — | 4.6 | — |
| Intact human SJRH30 cells ([125I]hU-II) | — | — | — | — | 5.4 |
| Functional pKb estimates | |||||
| Intact HEK293-UT cells (inhibition of [Ca2+]i-mobilization) | 7.29 | 7.94 | — | 7.81 | 8.00 |
| Isolated aortic rings (inhibition of contraction) | — | 7.47 | — | — | — |
The existing UT receptor ligands described in the literature (Dhanak et al., 2003; Douglas et al., 2004a) are of limited utility as ‘tool antagonists' due to a combination of pharmacodynamic and pharmacokinetic considerations. Firstly, many exhibit poor affinities for UT receptor isoforms from non-human, preclinical species for example, SB-710411 lacks potency at the rat UT receptor (Behm et al., 2002). Indeed, the affinities of the majority of such putative UT receptor antagonists are rarely (if ever) reported at non-rat/non-human UT receptor isoforms (i.e. cat and mouse UT receptors). This is an important consideration since, in addition to the rat, the mouse (Vergura et al., 2004) and cat (Behm et al., 2004a) represent two important species for studying the pharmacodynamic actions of U-II. Further, with the exception of the quinolinylurea palosuran (ACT-058362; Clozel et al., 2004), all UT receptor ligands published to date are peptidic and, as such, are inherently unsuitable for chronic enteric administration. In addition, several rat UT receptor antagonists (e.g. lanreotide, SB-710411, [Orn8]hU-II and urantide) possess intrinsic activity in mouse (partial agonism) and primate (full agonism) UT receptor systems, thus further complicating the interpretation of the pharmacological actions and hence restricting their use (Camarda et al., 2002a; 2004; Herold et al., 2002; Behm et al., 2004b). Finally, relatively little is known about the selectivity profiles of such putative antagonists at present (significant ‘secondary interactions' have been established for SB-710411, BIM-23127 and lanreotide/octreotide; Behm et al., 2002; Herold et al., 2002; 2003). As will be detailed below, the limitations described above do not apply to SB-706375, a potent and selective UT receptor antagonist active across mammalian species. As such, the discovery of SB-706375 represents a significant advance in the U-II field.
SB-706375 possesses high affinity for all five of the mammalian UT receptors that have been cloned, including the recently identified cat receptor (Aiyar et al., 2005). In addition to being a potent ligand at recombinant rodent, feline and primate UT receptors (4–20 nM Ki), SB-706375 also inhibits [125I]hU-II binding with high affinity at endogenous human UT receptors expressed in native human SJRH30 cells (5 nM Ki). As such, SB-706375 is approximately an order of magnitude more potent than the other known nonpeptide U-II antagonist at endogenous human UT receptors (46 nM IC50 in human rhabdomyosarcoma TE671 cells; Clozel et al., 2004). Therefore, SB-706375 is suitable for studying the actions of U-II at both recombinant and native UT receptors across species.
In accord with the radioligand binding studies, SB-706375 acted as a surmountable antagonist of hU-II across all species tested, antagonizing hU-II-induced [Ca2+]i-mobilization in intact HEK293 cells with pKbs ranging from around 10 to 50 nM (Hill slopes approximating unity). Such activity is also evident in native tissues (competitive, surmountable inhibition in rat aortae with pKb 33 nM). Thus, it is concluded that SB-706375 constitutes an agent suitable for attenuating the actions of U-II in cells and tissues from a diverse range of mammalian species.
The ‘pan-species' antagonistic properties of SB-706375, coupled with the associated low nanomolar affinity for rodent, cat and primate UT receptors, clearly differentiate this arylsulphonamide from other putative UT receptor ligands. To date, no other antagonists (e.g. BIM-23127, [Orn8]hU-II, urantide, etc.) have been profiled against monkey or cat UT receptors. Indeed, of this list, only [Orn8]hU-II has been studied in the mouse (Vergura et al., 2004). The potency of SB-706375 is also striking. Relative to SB-706375 (9 nM rat UT receptor Ki), a compound such as palosuran lacks appreciable affinity at the rat recombinant UT receptor (IC50 cited as 1500 and >10,000 nM at the rat UT receptor depending on whether CHO cell membranes or intact cells were used; Clozel et al., 2004). Based on these values, SB-706375 is in excess of 100- to >1000-fold more potent than palosuran as a rat UT receptor ligand. While SB-706375 is only four- to seven-fold more potent at the rat UT receptor than the peptides SB-710411 and BIM-23127 (32 and 63 nM rat UT receptor Ki, respectively; Herold et al., 2003; Behm et al., 2004b), it is distinguished from these somatostatin/neuromedin B analogues by superior pharmacodynamic selectivity (vide infra).
Although SB-706375 and urantide (5 nM rat UT receptor Ki; Patacchini et al., 2003) are essentially equipotent human UT receptor ligands, only the former functions as an antagonist at the human UT receptor. A recent study has revealed that urantide is actually a potent partial agonist at the human UT receptor (Camarda et al., 2004) with pEC50 8.11 and an intrinsic activity [α]∼0.8 relative to hU-II for [Ca2+]i-mobilization (consistent with these findings, urantide is also a potent and efficacious cat and monkey artery spasmogen with EC50 ∼10 nM and intrinsic activities [α]∼0.5–0.8; D. Behm & S. Douglas, unpublished observation). Similar observations have also been made with other peptidic moieties such as SB-710411, lanreotide, BIM-23127 and [Orn8]hU-II, which all behave as partial agonists at the mouse UT receptor and/or full agonists at monkey and human UT receptors (Camarda et al., 2002a; 2004; Herold et al., 2002). SB-710411, for example, is an efficacious antagonist at the rat UT receptor, yet it promotes phosphoinositide hydrolysis in monkey UT-HEK293 cells and induces contraction of the monkey isolated carotid artery (Behm et al., 2004b). Such liabilities are not, however, a characteristic shared by SB-706375, which does not exhibit any intrinsic activity at any of the mammalian recombinant UT receptor isoforms tested or in isolated blood vessels.
Ultimately, it will be necessary to study the pharmacodynamic effects of UT receptor antagonists in appropriate cell, tissue and whole animal systems before the true (patho)physiological significance of U-II in mammals becomes apparent. In order to be able to interpret data generated under such conditions unambiguously, a compound must be selective for the UT receptor and without any appreciable ‘off target' effects. Regrettably, relatively little is known about the selectivity and specificity of existing putative UT receptor antagonists. Indeed, the limited selectivity information that is available suggests that caution should be used when interpreting the observed biological consequences of antagonist administration. For example, BIM-23127 is a relatively potent neuromedin B ligand (Herold et al., 2002; 2003), whereas lanreotide, octreotide and SB-710411 all exhibit direct/indirect interactions with somatostatin and endothelin-1 receptors at reasonable concentrations (Behm et al., 2002; Herold et al., 2002). Although a handful of antagonists (e.g. [Pen5, Orn8]hU-II[4-11], urantide and palosuran) have been shown to have no effect on contractile and relaxant responses elicited in the rat isolated aorta by noradrenaline, 5-HT, endothelin-1 and acetylcholine (at 10 μM, <2-fold greater than the pD2′ of palosuran for U-II in the rat aorta), such studies are not in themselves rigorous tests of pharmacodynamic selectivity. As such, knowledge of the specificity of such agents is currently incomplete. Consequently, it becomes difficult to interpret data generated in vivo with such agents with any degree of certainty. However, compare the selectivity window defined for palosuran with that established for SB-706375, for example. Based on the ratio of an antagonist's potency in the rat isolated aorta against U-II (6 μM and 33 nM, respectively) compared to the highest concentration of antagonist studied (1 or 10 μM) that is known not to inhibit ‘non-U-II' vasoactive factors (e.g. endothelin-1) in this same tissue, palosuran's selectivity is defined as ⩾1.6-fold in rat isolated aorta (Clozel et al., 2004) vs ⩾30-fold for SB-706375 in the same tissue. Not only does SB-706375 have a larger selectivity window, this parameter has been defined against a much larger list of diverse molecular targets (>1 μM IC50 defined against a total of 86 distinct G-protein-coupled receptors, hormone receptors, tyrosine kinase receptors, ion channels, enzymes and neurotransmitter re-uptake systems). While not exhaustive, this level of selectivity profiling for SB-706375 is extensive relative to the lack of comparable data generated with the other available UT receptor antagonists. This should enable SB-706375 to be used in vivo in a more meaningful and less ambiguous manner than existing tool compounds.
The ambiguity that poor or incompletely defined selectivity profiling creates can be seen with studies describing the purported renoprotective effects of systemic palosuran administration (10 mg kg−1 h−1 i.v. infusion) in a rat renal artery ligation–reperfusion model. In this study, Clozel et al. (2004) failed to demonstrate equivocally that this palosuran dosage regimen blocks the pharmacodynamic actions of U-II (i.e. it is unknown if this dosage regimen of palosuran inhibits the haemodynamic response evoked by an exogenous U-II challenge). Indeed, the dose used in vivo was selected on the basis of palosuran's ability to augment the reactive hyperemia observed upon release of the renal artery clamp, an effect that might well have little to do with UT receptor occupancy. As such, it is difficult to associate the renoprotective effects reported with UT receptor blockade in an equivocal manner. This is of particular concern since plasma palosuran levels attained during this study (∼5 μM) fell below the affinity of this compound for the rat UT receptor expressed in either the rat isolated aorta or in intact rat recombinant UT-CHO cells (pD2′ 6 μM and IC50 >10 μM, respectively; Clozel et al., 2004). Although such considerations might be less of an issue when palosuran is studied in man (due to increased potency at the human UT receptor), it is an issue when using this compound in the rat.
SB-706375 functioned as a reversible UT receptor antagonist. The inhibition observed in the rat isolated aorta could be readily ‘washed out', an action consistent with a competitive mode of antagonism. The reversibility seen with SB-706375 was similar to that seen with the α1-adrenoceptor antagonist prazosin in this preparation where washout rate constants were in the order of ∼2–3% min−1 for both agents. In agreement with this, the reversibility of UT receptor/SB-706375 binding was also demonstrated using monkey UT-HEK293 membranes. Exposure to 1 μM SB-706375 for 30 min immediately followed by washout did not alter [125I]U-II binding affinity or density.
hU-II is classified as a ‘pseudo-irreversible' ligand at the UT receptor, that is to say that it is has an extremely slow dissociation rate from native UT receptors (<15% at 90 min from native hUT receptors expressed in SJRH30 cells; Douglas et al., 2004b). Similar radioligand binding data have been recorded at the recombinant UT receptor (Onan, 2004). Consistent with this observation, the contractile actions of U-II are extremely ‘resistant to washout' in mammalian isolated arteries (the time required for a contractile response to return to baseline values approaches ∼1 h in the rat and ⩾8 h in the monkey; Douglas et al., 2000). Despite this profile of U-II, SB-706375 was readily able to reverse ‘established hU-II-induced tone' in the rat aorta with Kapp∼20 nM. This observation is critical as it indicates that it is not necessary to use SB-706375 in a ‘prophylactic' manner in order to antagonize the actions of hU-II in an in vivo model. To date, such an action has not been confirmed for other UT receptor antagonists (with the exception of BIM-23127; Herold et al., 2003).
The present report also describes for the first time the characterization of a nonpeptidic UT receptor radiolabel, namely the tritiated radiotracer [3H]SB-657510. The development of a high-affinity nonpeptide radioligand such as [3H]SB-657510 offers several distinct advantages over currently available UT receptor radiotracers, that is, iodinated fish, amphibian and mammalian U-II isoforms. Antagonists such as [3H]SB-657510 are generally preferred as radiolabels over agonists such as [125I]hU-II for the characterization of receptors for several reasons. Firstly, antagonist binding does not result in receptor activation and subsequent desensitization/downregulation (SB-657510 is devoid of any intrinsic activity at the UT receptor [pKb 7.25 in rat isolated aorta for ‘cold' SB-657510; S. Douglas & D. Behm, unpublished data]). Further, antagonist : receptor binding is insensitive to guanine nucleotides and, as such, radiolabels are able to recognize multiple receptor affinity states (note that, to date, the regulation of U-II agonist binding to the UT receptor by guanine nucleotides has not been demonstrated) and, by virtue of its nonpeptidic nature, [3H]SB-657510 is refractory to protease degradation. The present study demonstrated that [3H]SB-657510 binding was specific (95% of total) and of high affinity (KD∼2 nM). The rapid dissociation of [3H]SB-657510 binding from the UT receptor (upon addition of cold hU-II) was consistent with a reversible mode of binding. SB-706375 and human and goby hU-II interacted with a common binding site labelled by [3H]SB-657510. Consequently, [3H]SB-657510 can be considered a useful alternative to [125I]hU-II for the pharmacological characterization of the U-II receptor.
In summary, the present report describes the identification and characterization of the arylsulphonamide SB-706375 as a potent, surmountable, reversible and selective mammalian UT receptor antagonist (Table 9). This compound offers several distinct advantages over currently available peptidic and nonpeptidic UT receptor antagonists. Since SB-706375 exhibits high-affinity cross-species antagonism, this nonpeptidic, small molecule inhibitor will serve as a useful agent in delineating the (patho)physiological actions of U-II in both preclinical species and man.
Acknowledgments
We thank L. Gordon, V. Holland, A. Garner, L. Campbell and K. Winborn (Assay Development and Compound Profiling, GlaxoSmithKline, Harlow, U.K.) for their expert technical assistance in assessing the selectivity of SB-706375 in a series of radioligand binding studies described herein.
Abbreviations
- ATCC
American Type Culture Collection
- BCA
bicinchoninic acid
- BSA
bovine serum albumin
- [Ca2+]i
intracellular calcium
- CHO cell
Chinese hamster ovary cell
- DMSO
dimethylsulphoxide
- DPBS+
Dulbecco's phosphate-buffered saline
- EDTA
ethylenediaminetetraacetic acid
- FLIPR
fluorometric imaging plate reader
- HEK293 cells
human embryonic kidney cells
- SB-706375
2-bromo-4,5-dimethoxy-N-[3-(R)-1-methyl-pyrrolidin-3-yloxy)-4-trifluro-methyl-phenyl]-benzenesulphonamide HCl
- SB-657510
2-bromo-N-[4-chloro-3-((R)-1-methyl-pyrrolidin-3-yloxy)-phenyl]-4,5-dimethoxybenzenesulphonamide HCl
- s.e.m.
standard error of the mean
- SNP
single nucleotide polymorphism
- SPA
scintillation proximity assay
- (h)U-II
(human) urotensin-II
- (h)UT receptor
(human) urotensin-II receptor
- WGA
wheat germ agglutinin-coated
References
- ABOUD R., SHAFII M., DOCHERTY J.R. Investigation of the subtypes of alpha 1-adrenoceptor mediating contractions of rat aorta, vas deferens and spleen. Br. J. Pharmacol. 1993;109:80–87. doi: 10.1111/j.1476-5381.1993.tb13534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AFFOLTER J.T., NEWBY D.E., WILKINSON I.B., WINTER M.J., BALMENT R.J., WEBB D.J. No effect on central or peripheral blood pressure of systemic urotensin II infusion in humans. Br. J. Clin. Pharmacol. 2002;54:617–621. doi: 10.1046/j.1365-2125.2002.t01-1-01704.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- AIYAR N.V., JOHNS D.G., AO Z., DISA J., BEHM D.J., FOLEY J.J., BUCKLEY P.T., SARAU H.M., VAN DER KEYL H.K., ELSHOURBAGY N.A., DOUGLAS S.A. Cloning and pharmacological characterization of the cat urotensin-II receptor (UT) Biochem. Pharmacol. 2005;69:1069–1079. doi: 10.1016/j.bcp.2004.12.016. [DOI] [PubMed] [Google Scholar]
- AMES R.S., SARAU H.M., CHAMBERS J.K., WILLETTE R.N., AIYAR N.V., ROMANIC A.M., LOUDEN C.S., FOLEY J.J., SAUERMELCH C.F., COATNEY R.W., AO Z., DISA J., HOLMES S.D., STADEL J.M., MARTIN J.D., LIU W.S., GLOVER G.I., WILSON S., MCNULTY D.E., ELLIS C.E., ELSHOURBAGY N.A., SHABON U., TRILL J.J., HAY D.W., OHLSTEIN E.H., BERGSMA D.J., DOUGLAS S.A. Human urotensin-II is a potent vasoconstrictor and agonist for the orphan receptor GPR14. Nature. 1999;401:282–286. doi: 10.1038/45809. [DOI] [PubMed] [Google Scholar]
- BEHM D.G., HEROLD C.L., OHLSTEIN E.H., KNIGHT S.D., DHANAK D., DOUGLAS S.A. Pharmacological characterization of SB-710411 (Cap-c[D-Cys-Pal-D-Trp-Lys-Val-Cys]-Cpa-amide), a novel peptidic urotensin-II receptor antagonist. Br. J. Pharmacol. 2002;137:449–458. doi: 10.1038/sj.bjp.0704887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BEHM D.J., DOE C.P., JOHNS D.G., MANISCALCO K., STANKUS G.P., WIBBERLEY A., WILLETTE R.N., DOUGLAS S.A. Urotensin-II: a novel systemic hypertensive factor in the cat. Naunyn Schmiedeberg's Arch. Pharmacol. 2004a;369:274–280. doi: 10.1007/s00210-004-0873-1. [DOI] [PubMed] [Google Scholar]
- BEHM D.J., HEROLD C.L., CAMARDA V., AIYAR N.V., DOUGLAS S.A. Differential agonistic and antagonistic effects of the urotensin-II ligand SB-710411 at rodent and primate UT receptors. Eur. J. Pharmacol. 2004b;25:113–116. doi: 10.1016/j.ejphar.2004.03.059. [DOI] [PubMed] [Google Scholar]
- BÖHM F., PERNOW J. Urotensin II evokes potent vasoconstriction in humans in vivo. Br. J. Pharmacol. 2002;135:25–27. doi: 10.1038/sj.bjp.0704448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BOUSETTE N., PATEL L., DOUGLAS S.A., OHLSTEIN E.H., GIAID A. Increased expression of urotensin II and its cognate receptor GPR14 in atherosclerotic lesions of the human aorta. Atherosclerosis. 2004;176:117–123. doi: 10.1016/j.atherosclerosis.2004.03.023. [DOI] [PubMed] [Google Scholar]
- CAMARDA V., GUERRINI R., KOSTENIS E., RIZZI A., CALO' G., HATTENBURGER A., ZUCCHINI M., SALVADORI S., REGOLI D. A new ligand for the urotensin II receptor. Br. J. Pharmacol. 2002a;137:311–314. doi: 10.1038/sj.bjp.0704895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CAMARDA V., RIZZI A., CALO' G., GENDRON G., PERRON S.I., KOSTENIS E., ZAMBONI P., MASCOLI F., REGOLI D. Effects of human urotensin II in isolated vessels of various species: comparison with other vasoactive agents. Naunyn Schmiedeberg's Arch. Pharmacol. 2002b;365:141–149. doi: 10.1007/s00210-001-0503-0. [DOI] [PubMed] [Google Scholar]
- CAMARDA V., SONG W., MARZOLA E., SPAGNOL M., GUERRINI R., SALVADORI S., REGOLI D., THOMPSON J.P., ROWBOTHAM D.J., BEHM D.J., DOUGLAS S., CALO' G., LAMBERT D.G. Urantide mimics urotensin-II induced calcium release in cells expressing recombinant UT receptors. Eur. J. Pharmacol. 2004;498:83–86. doi: 10.1016/j.ejphar.2004.07.089. [DOI] [PubMed] [Google Scholar]
- CHEUNG B.M., LEUNG R., MAN Y.B., WONG L.Y. Plasma concentration of urotensin II is raised in hypertension. J. Hypertens. 2004;22:1341–1344. doi: 10.1097/01.hjh.0000125452.28861.f1. [DOI] [PubMed] [Google Scholar]
- CHENG Y.C., PRUSOFF W.H. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzymatic reaction. Biochem. Pharmacol. 1973;22:357–366. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- CLOZEL M., BINKERT C., BIRKER-ROBACZEWSKA M., BOUKHADRA C., DING S.S., FISCHLI W., HESS P., MATHYS B., MORRISON K., MULLER C., MULLER C., NAYLER O., QIU C., REY M., SCHERZ M.W., VELKER J., WELLER T., XI J.F., ZILTENER P. Pharmacology of the urotensin-II receptor antagonist palosuran (ACT-058362: 1-[2-(4-benzyl-4-hydroxy-piperidin-1-yl)-ethyl]-3-(2-methyl-quinolin-4-yl)-urea sulfate salt): first demonstration of a pathophysiological role of the urotensin system. J. Pharmacol. Exp. Ther. 2004;311:204–212. doi: 10.1124/jpet.104.068320. [DOI] [PubMed] [Google Scholar]
- DHANAK D., GALLAGHER T.F., KNIGHT S.D.Preparation of sulfonamides as antagonists of urotensin II PCT Int. Appl. 2002. WO 2002089792
- DHANAK D., NEEB M.J., DOUGLAS S.A. Urotensin-II receptor modulators. Ann. Rep. Med. Chem. 2003;38:99–110. [Google Scholar]
- DOUGLAS S.A. Human urotensin-II as a novel cardiovascular target: ‘heart' of the matter or simply a fishy ‘tail'. Curr. Opin. Pharmacol. 2003;3:159–167. doi: 10.1016/s1471-4892(03)00012-2. [DOI] [PubMed] [Google Scholar]
- DOUGLAS S.A., BECK G.R., ELLIOTT J.D., OHLSTEIN E.H. Pharmacological evidence for the presence of three distinct functional endothelin receptor subtypes in the rabbit lateral saphenous vein. Br. J. Pharmacol. 1995;114:1529–1540. doi: 10.1111/j.1476-5381.1995.tb14936.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGLAS S.A., DHANAK D., JOHNS D.G. From ‘gills to pills': urotensin-II as a regulator of mammalian cardiorenal function. Trends Pharmacol. Sci. 2004a;25:76–85. doi: 10.1016/j.tips.2003.12.005. [DOI] [PubMed] [Google Scholar]
- DOUGLAS S.A., NASELSKY D., AO Z., DISA J., HEROLD C.L., LYNCH F., AIYAR N.V. Identification and pharmacological characterization of native, functional human urotensin-II receptors in rhabdomyosarcoma cell lines. Br. J. Pharmacol. 2004b;142:921–932. doi: 10.1038/sj.bjp.0705743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGLAS S.A., OHLSTEIN E.H. Human urotensin-II, the most potent mammalian vasoconstrictor identified to date, as a therapeutic target for the management of cardiovascular disease. Trends Cardiovasc. Med. 2000;10:229–237. doi: 10.1016/s1050-1738(00)00069-4. [DOI] [PubMed] [Google Scholar]
- DOUGLAS S.A., SULPIZIO A.C., PIERCY V., SARAU H.M., AMES R.S., AIYAR N.V., OHLSTEIN E.H., WILLETTE R.N. Differential vasoconstrictor activity of human urotensin-II in vascular tissue isolated from the rat, mouse, dog, pig, marmoset and cynomolgus monkey. Br. J. Pharmacol. 2000;131:1262–1274. doi: 10.1038/sj.bjp.0703690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOUGLAS S.A., TAYARA L., OHLSTEIN E.H., HALAWA N., GIAID A. Congestive heart failure and expression of myocardial urotensin II. Lancet. 2002;359:1990–1997. doi: 10.1016/S0140-6736(02)08831-1. [DOI] [PubMed] [Google Scholar]
- ELSHOURBAGY N.A., DOUGLAS S.A., SHABON U., HARRISON S., DUDDY G., SECHLER J.L., AO Z., MALEEFF B.E., NASELSKY D., DISA J., AIYAR N.V. Molecular and pharmacological characterization of genes encoding urotensin-II peptides and their cognate G-protein-coupled receptors from the mouse and monkey. Br. J. Pharmacol. 2002;136:9–22. doi: 10.1038/sj.bjp.0704671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GARDINER S.M., MARCH J.E., KEMP P.A., BENNETT T. Bolus injection of human UII in conscious rats evokes a biphasic haemodynamic response. Br. J. Pharmacol. 2004;143:422–430. doi: 10.1038/sj.bjp.0705954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GILBERT R.E., DOUGLAS S.A., KRUM H. Urotensin-II as a novel therapeutic target in the clinical management of cardiorenal disease. Curr. Opin. Invest. Drugs. 2004;5:276–282. [PubMed] [Google Scholar]
- HASEGAWA K., KOBAYASHI Y., KOBAYASHI H. Vasodepressor effects of urotensin II in rats. Neuroendocrinol. Lett. 1992;14:357–363. [Google Scholar]
- HEROLD C.L., BEHM D.J., BUCKLEY P.T., FOLEY J.J., DOUGLAS S.A. The peptidic somatostatin analogs lanreotide, BIM-23127 and BIM-23042 are urotensin-II receptor ligands. Pharmacologist. 2002;44:170–171. [Google Scholar]
- HEROLD C.L., BEHM D.J., BUCKLEY P.T., FOLEY J.J., WIXTED W.E., SARAU H.M., DOUGLAS S.A. The neuromedin B receptor antagonist, BIM-23127, is a potent antagonist at human and rat urotensin-II receptors. Br. J. Pharmacol. 2003;139:203–207. doi: 10.1038/sj.bjp.0705251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HILLIER C., BERRY C., PETRIE M.C., O'DWYER P.J., HAMILTON C., BROWN A., MCMURRAY J. Effects of urotensin II in human arteries and veins of varying caliber. Circulation. 2001;103:1378–1381. doi: 10.1161/01.cir.103.10.1378. [DOI] [PubMed] [Google Scholar]
- JOHNS D.G., AO Z., NASELSKY D., HEROLD C.L., MANISCALCO K., SAROV-BLAT L., STEPLEWSKI K., AIYAR N., DOUGLAS S.A. Urotensin-II-mediated cardiomyocyte hypertrophy: effect of receptor antagonism and role of inflammatory mediators. Naunyn Schmiedeberg's Arch. Pharmacol. 2004;370:238–250. doi: 10.1007/s00210-004-0980-z. [DOI] [PubMed] [Google Scholar]
- KOMPA A.R., THOMAS W.G., SEE F., TZANIDIS A., HANNAN R.D., KRUM H. Cardiovascular role of urotensin II: effect of chronic infusion in the rat. Peptides. 2004;25:1783–1788. doi: 10.1016/j.peptides.2004.03.029. [DOI] [PubMed] [Google Scholar]
- LANGHAM R.G., KELLY D.J., GOW R.M., ZHANG Y., DOWLING J.K., THOMSON N.M., GILBERT R.E. Increased expression of urotensin II and urotensin II receptor in human diabetic nephropathy. Am. J. Kidney Dis. 2004;44:826–831. [PubMed] [Google Scholar]
- LAPP H., BOERRIGTER G., COSTELLO-BOERRIGTER L.C., JAEKEL K., SCHEFFOLD T., KRAKAU I., SCHRAMM M., GUELKER H., STASCH J.P. Elevated plasma human urotensin-II-like immunoreactivity in ischemic cardiomyopathy. Int. J. Cardiol. 2004;94:93–97. doi: 10.1016/j.ijcard.2003.05.008. [DOI] [PubMed] [Google Scholar]
- LESLIE S.J., DENVIR M., WEBB D.J.Human urotensin II causes vasoconstriction in the human skin microcirculation Circulation 2000102II-113 [Google Scholar]
- LEW M.J., ANGUS J.A. An improved method for analysis of competitive agonist/antagonist interactions by non-linear regression. Ann. N.Y. Acad. Sci. 1997;812:179–181. doi: 10.1111/j.1749-6632.1997.tb48166.x. [DOI] [PubMed] [Google Scholar]
- LIM M., HONISETT S., SPARKES C.D., KOMESAROFF P., KOMPA A., KRUM H. Differential effect of urotensin II on vascular tone in normal subjects and patients with chronic heart failure. Circulation. 2004;109:1212–1214. doi: 10.1161/01.CIR.0000121326.69153.98. [DOI] [PubMed] [Google Scholar]
- MACLEAN M.R., ALEXANDER D., STIRRAT A., GALLAGHER M., DOUGLAS S.A., OHLSTEIN E.H., MORECROFT I., POLLAND K. Contractile responses to human urotensin-II in rat and human pulmonary arteries: effect of endothelial factors and chronic hypoxia in the rat. Br. J. Pharmacol. 2000;130:201–204. doi: 10.1038/sj.bjp.0703314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGUIRE J.J., DAVENPORT A.P. Is urotensin-II the new endothelin. Br. J. Pharmacol. 2002;137:579–588. doi: 10.1038/sj.bjp.0704924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGUIRE J.J., KUC R.E., DAVENPORT A.P. Orphan-receptor ligand human urotensin II: receptor localization in human tissues and comparison of vasoconstrictor responses with endothelin-1. Br. J. Pharmacol. 2000;131:441–446. doi: 10.1038/sj.bjp.0703601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAGUIRE J.J., KUC R.E., WILEY K.E., KLEINZ M.J., DAVENPORT A.P. Cellular distribution of immunoreactive urotensin-II in human tissues with evidence of increased expression in atherosclerosis and greater constrictor response of small compared to large coronary arteries. Peptides. 2004;25:1767–1774. doi: 10.1016/j.peptides.2004.01.028. [DOI] [PubMed] [Google Scholar]
- MATSUSHITA M., SHICHIRI M., IMAI T., IWASHINA M., TANAKA H., TAKASU N., HIRATA Y. Co-expression of urotensin II and its receptor (GPR14) in human cardiovascular and renal tissues. J. Hypertens. 2001;19:2185–2190. doi: 10.1097/00004872-200112000-00011. [DOI] [PubMed] [Google Scholar]
- NG L.L., LOKE I., O'BRIEN R.J., SQUIRE I.B., DAVIES J.E. Plasma urotensin in human systolic heart failure. Circulation. 2002;106:2877–2880. doi: 10.1161/01.cir.0000044388.19119.02. [DOI] [PubMed] [Google Scholar]
- ONAN D.Cardiovascular and regulatory aspects of the urotensin-II receptor 2004. PhD Thesis, Monash University, Melbourne, Australia
- ONAN D., PIPOLO L., YANG E., HANNAN R.D., THOMAS W.G. Urotensin II promotes hypertrophy of cardiac myocytes via mitogen-activated protein kinases. Mol. Endocrinol. 2004;18:2344–2354. doi: 10.1210/me.2003-0309. [DOI] [PubMed] [Google Scholar]
- PATACCHINI R., SANTICIOLI P., GIULIANI S., GRIECO P., NOVELLINO E., ROVERO P., MAGGI C.A. Urantide: an ultrapotent urotensin II antagonist peptide in the rat aorta. Br. J. Pharmacol. 2003;140:1155–1158. doi: 10.1038/sj.bjp.0705555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PAYSANT J., RUPIN A., SIMONET S., FABIANI J.N., VERBEUREN T.J. Comparison of the contractile responses of human coronary bypass grafts and monkey arteries to human urotensin-II. Fund. Clin. Pharmacol. 2001;15:227–231. doi: 10.1046/j.1472-8206.2001.00032.x. [DOI] [PubMed] [Google Scholar]
- RICHARDS A.M., CHARLES C. Urotensin II in the cardiovascular system. Peptides. 2004;25:1795–1802. doi: 10.1016/j.peptides.2004.04.017. [DOI] [PubMed] [Google Scholar]
- RICHARDS A.M., NICHOLLS M.G., LAINCHBURY J.G., FISHER S., YANDLE T.G. Plasma urotensin II in heart failure. Lancet. 2002;360:545–546. doi: 10.1016/s0140-6736(02)09709-x. [DOI] [PubMed] [Google Scholar]
- RUSSELL F.D. Emerging roles of urotensin-II in cardiovascular disease. Pharmacol. Ther. 2004;103:223–243. doi: 10.1016/j.pharmthera.2004.07.004. [DOI] [PubMed] [Google Scholar]
- RUSSELL F.D., MEYERS D., GALBRAITH A.J., BETT N., TOTH I., KEARNS P., MOLENAAR P. Elevated plasma levels of human urotensin-II immunoreactivity in congestive heart failure. Am. J. Physiol. 2003;285:H1576–H1581. doi: 10.1152/ajpheart.00217.2003. [DOI] [PubMed] [Google Scholar]
- RUSSELL F.D., MOLENAAR P., O'BRIEN D.M. Cardiostimulant effects of urotensin-II in human heart in vitro. Br. J. Pharmacol. 2001;132:5–9. doi: 10.1038/sj.bjp.0703811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SHENOUDA A., DOUGLAS S.A., OHLSTEIN E.H., GIAID A. Localization of urotensin-II immunoreactivity in normal human kidneys and renal carcinoma. J. Histochem. Cytochem. 2002;50:885–889. doi: 10.1177/002215540205000702. [DOI] [PubMed] [Google Scholar]
- SONG W., ASHTON N., BALMENT R.J. Effects of single bolus urotensin II injection in anaesthetised rats. J. Physiol. 2003;552P:P107. [Google Scholar]
- TOTSUNE K., TAKAHASHI K., ARIHARA Z., SONE M., ITO S., MURAKAMI O. Increased plasma urotensin II levels in patients with diabetes mellitus. Clin. Sci. 2003;104:1–5. doi: 10.1042/. [DOI] [PubMed] [Google Scholar]
- TOTSUNE K., TAKAHASHI K., ARIHARA Z., SONE M., MURAKAMI O., ITO S., KIKUYA M., OHKUBO T., HASHIMOTO J., IMAI Y. Elevated plasma levels of immunoreactive urotensin II and its increased urinary excretion in patients with Type 2 diabetes mellitus: association with progress of diabetic nephropathy. Peptides. 2004;25:1809–1814. doi: 10.1016/j.peptides.2004.06.024. [DOI] [PubMed] [Google Scholar]
- TOTSUNE K., TAKAHASHI K., ARIHARA Z., SONE M., SATOH F., ITO S., KIMURA Y., SASANO H., MURAKAMI O. Role of urotensin II in patients on dialysis. Lancet. 2001;358:810–811. doi: 10.1016/S0140-6736(01)06002-0. [DOI] [PubMed] [Google Scholar]
- TZANIDIS A., HANNAN R.D., THOMAS W.G., ONAN D., AUTELITANO D.J., SEE F., KELLY D.J., GILBERT R.E., KRUM H. Direct actions of urotensin II on the heart: implications for cardiac fibrosis and hypertrophy. Circ. Res. 2003;93:246–253. doi: 10.1161/01.RES.0000084382.64418.BC. [DOI] [PubMed] [Google Scholar]
- VERGURA R., CAMARDA V., RIZZI A., SPAGNOL M., GUERRINI R., CALO' G., SALVADORI S., REGOLI D. Urotensin II stimulates plasma extravasation in mice via UT receptor activation. Naunyn Schmiedeberg's Arch. Pharmacol. 2004;370:347–352. doi: 10.1007/s00210-004-0991-9. [DOI] [PubMed] [Google Scholar]
- WATANABE T., PAKALA R., KATAGIRI T., BENEDICT C.R. Synergistic effect of urotensin II with mildly oxidized LDL on DNA synthesis in vascular smooth muscle cells. Circulation. 2001;104:16–18. doi: 10.1161/hc2601.092848. [DOI] [PubMed] [Google Scholar]
- WATSON A.M.D., LAMBERT G.W., SMITH K.J., MAY C.N. Urotensin II acts centrally to increase epinephrine and ACTH release and cause potent inotropic and chronotropic actions. Hypertension. 2003;42:373–379. doi: 10.1161/01.HYP.0000084633.85427.E6. [DOI] [PubMed] [Google Scholar]
- WENYI Z., SUZUKI S., HIRAI M., HINOKIO Y., TANIZAWA Y., MATSUTANI A., SATOH J., OKA Y. Role of urotensin II gene in genetic susceptibility to Type 2 diabetes mellitus in Japanese subjects. Diabetologia. 2003;46:972–976. doi: 10.1007/s00125-003-1145-1. [DOI] [PubMed] [Google Scholar]
- WILKINSON I.B., AFFOLTER J.T., DE HAAS S.L., PELLEGRINI M.P., BOYD J., WINTER M.J., BALMENT R.J., WEBB D.J. High plasma concentrations of human urotensin II do not alter local or systemic hemodynamics in man. Cardiovasc. Res. 2002;53:341–347. doi: 10.1016/s0008-6363(01)00485-0. [DOI] [PubMed] [Google Scholar]
- ZOU Y., NAGAI R., YAMAAKI T. Urotensin II induces hypertrophic responses in cultured cardiomyocytes from neonatal rats. FEBS Lett. 2001;508:57–60. doi: 10.1016/s0014-5793(01)03015-0. [DOI] [PubMed] [Google Scholar]