

Abstract
Intestinal inflammation enhances the potency of μ-opioid receptor (MOR) agonists inhibiting gastrointestinal transit and increases the expression of MOR in mice intestine. The precise mechanisms implicated in the increased expression of MOR during intestinal inflammation are not known. The aim of the study is to evaluate if nitric oxide released during intestinal inflammation could modulate MOR gene expression and affect gastrointestinal transit.
Intestinal inflammation was induced by the intragastric administration of croton oil. In CD-1 mice, with and without inflammation, we evaluated the antitransit effects of morphine in animals treated with NOS inhibitors (L-NAME and L-NIL) and the intestinal levels of iNOS enzyme mRNA. The antitransit effects of morphine and the expression of MOR mRNA in the gut of wild-type (WT) and iNOS−/− mice were also assessed. Gastrointestinal transit was measured with charcoal meal and mRNA levels determined by real-time PCR.
In CD-1 mice, inflammation induced a 10-fold increase (P<0.0001) in iNOS mRNA levels in the gut. The absence of iNOS gene and treatment of CD-1 mice with L-NAME or L-NIL abolished the increased antitransit effects of morphine observed during inflammation. Moreover, although the basal levels of MOR mRNA were similar in WT and iNOS animals (−/−), intestinal inflammation only increased the MOR expression in the gut of WT (P<0.01) but not in iNOS−/− mice.
The results suggest that nitric oxide derived from the increased expression of iNOS is implicated in the enhanced effects of morphine and in the upregulation of MOR gene transcription observed during intestinal inflammation.
Keywords: Inflammation, intestine, iNOS knockout mice, nitric oxide, opioid receptor and transcription
Introduction
μ-Opioid receptor (MOR) a member of the G-protein-linked receptor superfamily is found in the central (Mansour et al., 1995) and peripheral nervous system (Coggeshall et al., 1997; Sternini 2001). This receptor is expressed in various tissues including the gut, with particular abundance in the myenteric and submucosal plexi (Bagnol et al., 1997) and modulates several intestinal functions such as motility and secretion (Valle et al., 2000; Pol & Puig, 2004).
We have shown previously that intestinal inflammation induced by croton oil enhances the potency of MOR agonists inhibiting gastrointestinal transit and increases the transcription and expression of MOR in CD-1 mice intestine (Puig & Pol, 1998; Pol et al., 2001). It has been demonstrated that cytokines may modulate the expression of the MOR gene. Thus, studies performed with interleukin-6 (IL-6) knockout mice demonstrated decreased levels of MOR in the brain compared with wild-type (WT) animals, suggesting a positive regulation of this receptor by IL-6 (Bianchi et al., 1999). In addition, studies in vitro have shown that IL-1, IL-4, IL-6 and TNFα enhance the MOR gene transcription in various immune, endothelial and neuronal cells (Vidal et al., 1998; Kraus et al., 2001, 2003; Börner et al., 2004). However, the precise mechanisms implicated in the increased expression of MOR during intestinal inflammation are not yet elucidated.
Nitric oxide is a ubiquitous mediator of numerous physiological processes. In the gastrointestinal tract, it regulates motility, secretion and blood flow (Esplugues, 2002). The expression of the inducible isoform of nitric oxide synthase (iNOS) as well as nitric oxide synthesis is enhanced in inflammatory bowel diseases (Kimura et al., 1998; McCafferty et al., 1999; Yue et al., 2001). However, the role that iNOS-derived nitric oxide plays in the pathophysiology of this condition is controversial. Different authors have postulated that both proinflammatory and protective properties of nitric oxide are produced by iNOS (Dikopoulos et al., 2001; Krieglstein et al., 2001; Grisham et al., 2002). In the gut, studies performed in iNOS-deficient mice show that they develop a more severe colitis that WT in response to different inflammatory agents, supporting the fact that nitric oxide may play a potential protective role in the inflammatory response of bowel diseases (McCafferty et al., 1997, 1999). In contrast, other studies in iNOS knockout mice showed resistance to trinitrobenzene sulfonic acid (TNBS) as well as to dextran sulphate sodium-induced colitis, indicating that iNOS plays a critical role in the onset of bowel inflammation (Hokari et al., 2001; Krieglstein et al., 2001; Beck et al., 2004).
Several studies have shown that nitric oxide can modulate the effects of MOR agonists during acute peripheral inflammation. In this experimental condition, the antinociceptive effects of μ-opioids were enhanced or diminished by the peripheral administration of nitric oxide donors or NOS inhibitors, respectively (Nozaki-Taguchi & Yamamoto, 1998; Tasatargil & Sadan, 2004). No studies have been carried out to evaluate the effects of nitric oxide in the antitransit effects of morphine during intestinal inflammation.
The aim of the present study is to evaluate if nitric oxide released during inflammation could modulate the MOR gene expression and gastrointestinal transit. Our hypothesis was that peripheral inflammation would induce an increase in nitric oxide levels, which could enhance the expression of MOR in the gut and impair gastrointestinal transit. We evaluated in CD-1 mice, following the induction of intestinal inflammation, the antitransit effects of morphine in animals treated with NOS inhibitors ((NG-nitro-L-arginine-methyl ester hydrochloride (L-NAME)) and (L-N6-(1-iminoethyl)-lysine-hydrochloride (L-NIL)) and the intestinal levels of iNOS mRNA. In addition, the antitransit effects of morphine and the expression of MOR mRNA were also evaluated in the gut from WT and iNOS−/− mice, with and without intestinal inflammation.
Methods
Animals
In this study, male Swiss CD-1 mice (Charles River Laboratories, Montpelier, France), iNOS-deficient mice (C57BL/6-NOS2tm1Lau; Jackson Laboratories, Bar Harbor, ME, U.S.A.) and their WT littermates (C57BL/6; Charles River Laboratories, Montpelier, France) weighing 25–30 g were used. The study protocol was approved by the local Committee of Animal Use and Care of our Institution, in accordance with the International Association for the Study of Pain guidelines on ethical standards for investigations in animals. Mice were housed under 12-h/12-h light/dark conditions in a room with controlled temperature (22°C) and humidity (66%). Animals had free access to food and water and were used after a minimum of 4 days acclimatization to the housing conditions. All experiments were conducted between 09:00 and 17:00.
Induction of inflammation
Intestinal inflammation was induced by the intragastric administration of two doses (0.05 ml) of croton oil (Sigma-Aldrich, St Louis, MO, U.S.A.) diluted in olive oil (1 : 1) and administered 24 h apart; control animals received the same volume of intragastric saline. In both instances, before the administration of croton oil or saline, animals were fasted for 18 h, except for free access to water. Morphological changes induced by croton oil, 5 days after the first dose administration, have been reported previously by our group (Puig & Pol, 1998) and were established by optical microscopy. In brief, a clear disruption of the mucosa with a massive infiltration of lymphocytes within the submucosa was observed in animals treated with croton oil. The greatest morphological inflammatory changes after treatment with croton oil were observed in the jejunum.
Gastrointestinal transit
Gastrointestinal transit was measured according to the procedure used in our laboratory (Pol et al., 1999). Briefly, food was removed 18 h before the experiment, but animals had free access to water. After fasting for 18 h, animals received intragastric charcoal meal (0.25 ml of a suspension of 10% vegetable charcoal in 5% gum acacia), and 20 min later, the gastrointestinal transit was evaluated. Animals were killed by cervical dislocation, and the small intestine was separated from the omentum to avoid stretching. The length of the intestine from the pyloric sphincter to the ileocecal junction and the distance traveled by the charcoal meal were measured. For each animal, gastrointestinal transit was calculated as the percent of the distance traveled by the charcoal, relative to the total length of the small intestine (% of gastrointestinal transit).
The antitransit effects of subcutaneous morphine (MOR agonist) were evaluated in CD-1, WT and iNOS−/− mice, treated with saline or croton oil. The effects of morphine on the inhibition of gastrointestinal transit, in CD-1 mice treated with NOS inhibitors, were also evaluated. We used L-NAME as a nonselective NOS inhibitor, L-NIL as a selective iNOS inhibitor and NG-nitro-D-arginine-methyl ester hydrochloride (D-NAME) as an inactive isomer of NAME. The NOS inhibitors were administered intraperitoneally at 10 mg kg–1 for 4 consecutive days starting at the time of the second administration of croton oil. In all instances, gastrointestinal transit was measured 5 days after the administration of the first dose of croton oil (or saline) and 4 days after the first dose of NOS inhibitors (or vehicle). Morphine and vehicle were given at day 5, 30 min before the marker.
Morphine hydrochloride was purchased from Alcaiber S.A. (Madrid, Spain), L-NAME and L-NIL from Tocris (Ellisville, U.S.A.) and D-NAME from Sigma-Aldrich (St Louis, MO, U.S.A.). All drugs were dissolved in sterile pyrogen-free 0.9% sodium chloride just before use and injected in a final volume of 10 ml kg−1.
Myeloperoxidase (MPO) activity
MPO activity was measured as an indicator of granulocyte (primarily neutrophil) infiltration into the tissue using a modification of the method of Krawisz et al. (1984). Briefly, 5 days after the first dose administration of croton oil or saline, segments of jejunum weighing 50–150 mg from animals treated with and without NOS inhibitors were removed. They were homogenized (50 mg ml−1) in ice-cold 50 mM potassium phosphate buffer containing 5 g l−1 of hexadecyltrimethylammonium bromide. The homogenates were frozen and thawed three times, and then centrifuged at 13,000 × g for 15 min at 4°C. Aliquots (7 μl) of the supernatant of each sample were added to a 96-well microtiter plate and 200 μl of potassium phosphate buffer (50 mM, pH 6.0) containing O-dianisidine dihydrochloride (0.167 mg ml−1) and hydrogen peroxide (0.0005%) was added to each well. Absorbance was read at 450 nm using a 96-well microplate reader and MPO activities were expressed as units per g of wet weight of tissue.
Tissue isolation and total RNA extraction
Due to the fact that maximal inflammation in the gut induced by croton oil was observed in the jejunum, all experiments were performed in this section (Puig & Pol, 1998). At 5 days after the first dose of croton oil administration, jejunums from CD-1, WT and iNOS−/− mice were excised, placed in sterile microfuge tubes, snap frozen in liquid nitrogen and stored at −80°C until assay. For the isolation of jejunum, we collected 10 cm of the small intestine, starting 2 cm distal to the ligament of Treitz. Jejunal preparations were homogenized in ice cold with a homogenizer (Ultra-Turf, T8; Ika Werke, Staufen, Germany) and their total RNA was extracted with TRIzol (Invitrogen, Renfrewshire, England). The amount of the purified RNA (A260/A280 ratio was ⩾1.9) was determined by spectrophotometry.
Reverse transcription
In all experiments, 1 μg of total RNA was reverse transcribed into cDNA using SuperScript II RNAse H− reverse transcriptase (RT) (Invitrogen, Renfrewshire, U.K.) in a final volume of 10 μl. Negative controls were performed in which all of the components were included except RT.
TaqMan probe real-time polymerase chain reaction (PCR)
A predeveloped assay and a designed mice TaqMan assay were used to quantify iNOS (ID:Mm00440485_m1) and MOR gene expression, respectively (Assays-on-Demand and Design Gene Expression Products, Applied Biosystems, CA, U.S.A.). Phosphoglycerate kinase-1 (PGK-1) RNA gene was used as an endogenous control (ID:Mm00435617_m1). TaqMan probes were labeled at the 5′-end with the reporter dye molecule FAM (6-carboxy-fluorescein) and at the 3′-end with the quencher dye molecule TAMRA (6-carboxy tetramethyl rhodamine). All TaqMan PCR primers were located in two different exons of each gene to avoid amplification of any contaminating genomic DNA.
The 2 × universal master mix (Applied Biosystems, CA, U.S.A.) containing PCR buffer, MgCl2, dNTPs and the thermal stable AmpliTaq Gold® DNA polymerase was used in the PCR reactions. The PCR reaction mixture also contained forward and reverse primers at 900 nM, 250 nM TaqMan probe and 1 μl of the cDNA. RNAse/DNAse-free water was added to the master mix to obtain a final volume of 20 μl. The PCR reaction mixture was transferred to a MicroAmp optical 384-well reaction plate and incubated at 95°C for 10 min to activate the Amplitaq Gold DNA polymerase, and then run for 50 cycles at 95°C for 30 s and 60°C for 1 min on the Applied Biosystems ABI PRISM 7900HT Sequence Detection System. All samples were assayed in duplicate. To validate the specificity of a primer set, negative controls were analyzed in duplicate to confirm that there was no fluorescence resulting from genomic DNA contamination.
The PCR results were analyzed with the Sequence Detector Software version 2.1 (Applied Biosystems, CA, U.S.A.). The standardized target gene was compared with an external reference (a cDNA that was used in every assay). According to the comparative threshold cycle (CT) method (Livak & Schmittgen, 2001), the average CT values for target genes were normalized with respect to the average CT values for an endogenous reference (PGK-1 gene), to yield the ΔCT. The average ΔCT value obtained from an external reference was then subtracted from the average ΔCT value acquired for each target sample, to give the ΔΔCT. The relative copy number was calculated by the expression 2−(ΔΔCT).
Data analysis
Data are expressed as groups mean±s.e.m. The inhibitory effects of the morphine on gastrointestinal transit are expressed as a percentage of inhibition of the gastrointestinal transit in a drug-treated animal (test gastrointestinal transit) when compared with the mean of gastrointestinal transit measured in a group of vehicle-treated mice (vehicle gastrointestinal transit; n=10)
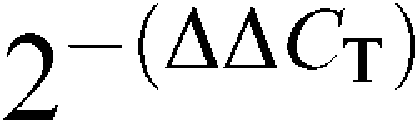 |
ED50±s.e.m. (dose which produced 50% of maximal effect) values were determined by linear regression analysis of dose–response relations based on at least six animals per dose (Tallarida & Murray, 1986). Statistical analysis for significant differences between two groups was obtained by Student's t-test. When multiple groups were compared, two- or one-way analysis of variance (ANOVA) was used, followed by a Tukey post hoc test. A value of P<0.05 was considered significant. For evaluating the correlation between MPO activity and the morphine antitransit effects in animals treated with and without NOS inhibitors, a Pearson test was used.
Results
Gastrointestinal transit
Gastrointestinal transit was evaluated in CD-1, iNOS-deficient mice and their littermates at 5 days after treatment with saline or croton oil. The two-way ANOVA showed a significant effect of genotype (P<0.001), croton oil treatment (P<0.001) and their interaction (P<0.013). The administration of croton oil significantly enhanced gastrointestinal transit in CD-1 and WT (P<0.02; Tukey test) but not in iNOS−/− mice (Figure 1). In saline-treated animals, no significant differences in gastrointestinal transit were observed between genotypes.
Figure 1.

Percent of gastrointestinal transit in CD-1, WT and iNOS−/−-deficient mice treated with saline (SS) or croton oil (CO). Each column represents the mean±s.e.m. of six to eight animals. *Compares CO with SS, P<0.02 and #compares iNOS-CO with CD-1-CO and WT-CO group, P<0.001 (Tukey's test).
In saline- and croton oil-treated CD-1 mice, the effects of two NOS inhibitors (L-NAME and L-NIL) on the gastrointestinal transit were evaluated and the results are shown in Table 1. The two-way ANOVA demonstrated a significant effect of croton oil (P<0.001), NOS inhibitor treatment (P<0.004) and their interaction (P<0.001). The administration of croton oil significantly enhanced gastrointestinal transit in vehicle- (54.7%) and D-NAME- (52.2%) treated animals (P<0.001, Tukey's test). This increased gastrointestinal transit induced by croton oil was completely (P<0.001) and significantly reduced (14.7%; P<0.03, Tukey's test) by cotreatment of the animals with L-NAME or L-NIL, respectively. In control animals, the administration of L-NAME, L-NIL or D-NAME did not alter gastrointestinal transit.
Table 1.
Gastrointestinal transit (%) in CD-1 mice treated with saline (SS) or croton oil (CO) and co-treated with vehicle, L-NAME, L-NIL or D-NAME
| Treatment | Saline (SS) | Croton oil (CO) |
|---|---|---|
| Vehicle | 48.6±1.5 | 75.2±2.3* |
| L-NAME | 54.8±2.2 | 52.8±3.3** |
| L-NIL | 48.1±1.3 | 55.2±4.8*** |
| D-NAME | 46.1±2.4 | 70.2±2.3* |
Results are expressed as mean values±s.e.m. for six to eight animals per group. For each group, *compares CO with SS, P<0.001; **compares L-NAME-CO with vehicle-CO and D-NAME-CO-treated animals, P<0.001 and, ***compares L-NIL-CO with vehicle-CO and D-NAME-CO-treated animals, P<0.03 (Tukey's test).
Antitransit effects of morphine
The involvement of nitric oxide in the enhanced antitransit effects of morphine observed in CD-1 mice with intestinal inflammation was evaluated by measuring their antitransit effects in controls and animals with intestinal inflammation treated with L-NAME or L-NIL. In control animals, the subcutaneous administration of morphine produced a dose-related inhibition of gastrointestinal transit, which was significantly shifted to the left in croton oil-treated mice (Figure 2). The dose–response curves to morphine after cotreatment with L-NAME (Figure 2a) or L-NIL (Figure 2b) were unaltered in controls and did not shift to the left in croton oil-treated animals. Thus, treatment with NOS inhibitors prevented the shift to the left of the dose response to morphine induced by croton oil.
Figure 2.

Inhibition of gastrointestinal transit induced by the subcutaneous administration of morphine in controls (SS) and croton oil-(CO) treated animals, cotreated with L-NAME (a) or L-NIL (b). Each point represents the mean of six to eight animals; vertical lines show s.e.m. *P<0.01, indicates significant differences in the dose–response curve of morphine in croton oil-treated animals when compared to the other groups (Tukey's test).
Table 2 shows the ED50 mean values of morphine on the inhibition of gastrointestinal transit in saline- and croton oil-treated CD-1 mice co-treated with vehicle, L-NAME, L-NIL or D-NAME. Results show that the administration of croton oil significantly enhanced (9.5-fold) the antitransit effect of morphine when compared to saline-treated animals (P<0.001; Tukey's test). Similar effects were observed in D-NAME-treated mice, where inflammation (CO) increases 8.9 times the antitransit effects of morphine as compared to their respective control group (P<0.001; Tukey's test). Treatment with NOS inhibitors did not alter the potency of morphine in control animals, but completely abolished the enhanced potency of morphine observed during inflammation (P<0.001; Tukey's test). The ED50 value of morphine in mice treated with croton oil and L-NAME animals was significantly higher than that obtained in all of the other groups (P<0.01; Tukey's test), except the group of mice treated with croton oil and the specific iNOS inhibitor, L-NIL.
Table 2.
ED50 mean values (mg kg−1) of morphine on the inhibition of gastrointestinal transit in CD-1 mice, treated with saline (SS) or croton oil (CO), and cotreated with vehicle, L-NAME, L-NIL or D-NAME
| Treatment | Saline (SS) | Croton oil (CO) |
|---|---|---|
| Vehicle | 1.43±0.11 | 0.15±0.03* |
| L-NAME | 1.74±0.13 | 2.57±0.15** |
| L-NIL | 1.73±0.13 | 2.06±0.20*** |
| D-NAME | 1.60±0.07 | 0.18±0.04* |
Results are expressed as mean values±s.e.m. for six to eight animals per group. For each group *compares CO with SS, P<0.001, **compares L-NAME-CO with vehicle-CO- and D-NAME-CO-treated animals, P<0.001 and, ***compares L-NIL-CO with vehicle-CO- and D-NAME-CO-treated animals, P<0.001 (Tukey's test).
In addition, the antitransit effects produced by a fixed dose of morphine (1 mg kg−1) were evaluated in WT and iNOS-deficient mice (Figure 3). Two-way ANOVA indicated a significant genotype (P<0.001) and croton oil treatment effect (P<0.012), and also their interaction (P<0.05). A significant increase in the inhibitory effects of morphine was observed in WT but not in iNOS knockout mice after croton oil treatment (P<0.001, Tukey's test).
Figure 3.

Inhibition of gastrointestinal transit induced by the administration of a fixed dose of morphine (1 mg kg−1) in WT and iNOS−/− mice treated with saline (SS) or croton oil (CO). Each column represents the mean of six to eight animals; vertical lines show s.e.m., *compares CO with SS, P<0.001 and #compares iNOS-CO with WT-CO group, P<0.05 (Tukey's test).
MPO activity
The severity of intestinal inflammation induced by croton oil in CD-1 mice treated with or without nitric oxide inhibitors was assessed by measurement of MPO activity (Table 3). The results showed a 3.5-fold increase in the MPO activity in the jejunum of croton oil-treated animals when compared to controls (saline-treated mice; P<0.001, Tukey's test). The enhanced jejunal MPO activity induced by croton oil was significantly inhibited by treatment with L-NAME or L-NIL (P<0.001, Tukey's test), although their administration did not modify the basal MPO activity in saline treated animals. In addition, the MPO activity in croton oil-treated animals was significantly enhanced after D-NAME administration (3.3 times; P<0.001 Tukey's test). A significant correlation (r=0.70; P<0.001, Pearson's test) between the levels of intestinal inflammation and the morphine antitransit effects in animals treated with and without NOS inhibitors has also been demonstrated.
Table 3.
MPO activity (U g wet tissue−1) in the jejunum of CD-1 mice treated with saline or croton oil and cotreated with vehicle, L-NAME, L-NIL or D-NAME
| Treatment | Saline (SS) | Croton oil (CO) |
|---|---|---|
| Vehicle | 1.17±0.28 | 4.16±0.22* |
| L-NAME | 1.27±0.19 | 1.81±0.16** |
| L-NIL | 1.40±0.11 | 2.02±0.20*** |
| D-NAME | 1.19±0.16 | 3.93±0.29* |
Results are expressed as mean values±s.e.m. for six to eight animals per group. For each group *compares CO with SS, P<0.001; **compares L-NAME-CO with vehicle-CO- and D-NAME-CO-treated animals, P<0.001 and ***compares L-NIL-CO with vehicle-CO- and D-NAME-CO-treated animals, P<0.001 (Tukey's test).
Expression of iNOS mRNA levels in the gut
The mRNA levels of iNOS in the jejunum of WT mice with and without intestinal inflammation were determined by using relative real-time PCR with PGK-1 as an endogenous control. PGK-1 gene expression is unaffected by croton oil treatment.
Intestinal inflammation induces a 10-fold increase in the levels of iNOS messenger RNA (Figure 4a). The statistical analysis of the iNOS transcript expression revealed a significant increase in the iNOS mRNA levels in the gut of mice with intestinal inflammation (P<0.0001; Student's t-test). Each experiment was repeated in samples obtained from four different animals (control and inflamed) and similar results were obtained.
Figure 4.

(a) Is a graphical representation of the relative expression of iNOS mRNA in the jejunum samples of WT mice with (CO) and without intestinal inflammation (SS). Inflammation induced by croton oil enhances the iNOS mRNA levels. *P<0.0001 when croton oil-treated and saline-treated animals are compared (Student's t-test). (b) Represents the relative expression of MOR mRNA in the jejunum of WT and iNOS−/− mice treated with vehicle (SS) or croton oil (CO). Inflammation enhances the intestinal MOR mRNA levels in WT but not in iNOS−/− mice. For each genotype, *compares CO with SS, P<0.01 and #compares iNOS-CO with WT-CO group, P<0.05 (Tukey's test). Each column represents the mean±s.e.m. of four different samples from independent experiments.
Transcript expression of MOR in the gut
To evaluate if nitric oxide released during intestinal inflammation could modulate MOR gene expression, we tested their expression in the gut from mice lacking the iNOS gene. Figure 4b shows the mRNA relative expression of MOR from WT and iNOS−/− mice 5 days after the intragastric administration of saline or croton oil. Two-way ANOVA demonstrated a significant effect of genotype (P<0.033), croton oil treatment (P<0.004) and their interaction (P<0.046). Thus, although the basal levels of MOR mRNA were similar between WT and iNOS animals, intestinal inflammation only increased the expression of MOR in WT but not in iNOS−/− mice (P<0.01, Tukey's test).
Discussion
In this study, we showed that nitric oxide derived from the increased expression of iNOS is implicated in the upregulation of MOR gene transcription as well as in the increase of gastrointestinal transit observed during intestinal inflammation. Nitric oxide liberated during inflammation is also implicated in the increase of the antitransit effects of morphine during croton oil-induced inflammation. Thus, the absence of iNOS gene and treatment with NOS inhibitors, both prevent the increased gastrointestinal transit induced by croton oil, in a manner similar to the amelioration of the hypermotility induced by the iNOS inhibitors after Trichinells spiralis infection or lipopolysaccharide treatment (Torrents et al., 2003; Mathison et al., 2004). This effect could not be mimicked by the administration of the inactive stereoisomer, D-NAME. In addition, an increased expression of iNOS has been found in different models of intestinal inflammation and in inflammatory bowel disease (Evans et al., 2000; Yue et al, 2001). Our results support these findings showing that intestinal inflammation provoked by croton oil induces iNOS expression in the gut of CD-1 mice. Owing to the primary source of iNOS, mRNA and protein expression in the inflammed tissue is activated in resident immune cells and immune cells that traffic to sites of inflammation from the systemic circulation; then, these cell population could contribute to the increased iNOS gene expression reported in our study (Kalff et al., 2000). In summary, our results show that the increased gastrointestinal transit induced by croton oil appears to be mediated by nitric oxide derived from iNOS since: (1) the inducible form of NOS is increased during croton oil-induced inflammation, (2) the specific inhibitor of the enzyme normalizes transit and (3) gastrointestinal transit is not altered in iNOS−/− deficient mice treated with croton oil.
The role of nitric oxide on the enhanced intestinal effects of μ-opioids during inflammation was evaluated by measuring the antitransit effects of morphine (MOR agonist) in animals treated with NOS inhibitors. According to previous studies, the antitransit effects of morphine were significantly increased during intestinal inflammation (Puig & Pol, 1998). However, this increased potency of morphine (9.5 times) was significantly diminished in iNOS−/− mice and in WT mice treated with L-NAME or L-NIL. In control animals, the administration of NOS inhibitors did not alter the antitransit effects of morphine. Thus, nitric oxide appears to mediate the enhanced effects of μ-opioids during peripheral inflammation. The fact that treatment with a specific (L-NIL) or the nonspecific NOS inhibitor (L-NAME) produced similar effects suggests that nitric oxide synthesized by iNOS could be primarily responsible for the observed effects. This hypothesis is supported by the 10-fold increase in the iNOS mRNA levels in the gut of WT mice with intestinal inflammation. Other inflammatory agents such as indomethacin and TNBS, also increase iNOS mRNA levels (Evans et al., 2000; Yue et al, 2001). This is the first study to report that the nitric oxide pathway is involved in the antitransit effects of morphine during intestinal inflammation. Our results agree with data reported in other nociceptive models of peripheral inflammation in which the analgesic effects of μ-opioids were enhanced or diminished by the local administration of nitric oxide donors or NOS inhibitors, respectively (Nozaki-Taguchi & Yamamoto, 1998; Tasatargil & Sadan, 2004). Granados-Soto et al. (1997) also demonstrated that peripheral administration of methylene blue (a soluble guanylyl cyclase inhibitor) significantly attenuated the antinociceptive effects of morphine, supporting the view that the activated L-arginine/nitric oxide/cGMP pathway during inflammation is implicated in the antinociceptive effects produced by μ-opioids in this experimental condition.
In this work, intestinal inflammation induced by croton oil was confirmed by the measurement of MPO activity (3.5-fold increase) and according to other inflammatory models (Kolios et al., 2004), the administration of a non-(L-NAME) and a specific iNOS inhibitor (L-NIL), both attenuated the intestinal inflammation induced by croton oil. Since the increased potency of morphine in croton oil-treated animals was also diminished after L-NAME or L-NIL treatment, a significant correlation between the levels of intestinal inflammation and the morphine antitransit effects in animals treated with and without NOS inhibitors has been demonstrated.
We and other investigators have shown that intestinal inflammation enhances the transcription and expression of MOR in the gut, thus explaining the increased antitransit and also the anti-inflammatory effects induced by μ-opioids during intestinal inflammation (Pol et al., 2001; Philippe et al., 2003). Likewise, the analgesic effects of μ-opioids were also augmented in animals with peripheral inflammation related to an upregulation of peripheral MOR (mRNA and proteins) that occurs under inflammatory conditions (Zöllner et al., 2003; Puehler et al., 2004). In croton oil-treated animals, the augmented effect of morphine was demonstrated by an increase in the levels of MOR mRNA and proteins located in the myenteric plexus of animals, which mainly control intestinal motility (Pol et al., 2001). However, due to immune cells such as macrophages and lymphocytes constitutively express opioid receptors (Tomassini et al., 2003) and their number increases at the site of inflammation, we cannot exclude that these cell populations could contribute to the increased MOR gene expression reported in this study.
In addition, morphine as an endogenous signaling molecule is also involved in controlling gut motility via nitric oxide release (Stefano et al., 2004). Thus, the specific binding of morphine with MOR can stimulate the production of cNOS-derived nitric oxide, as well as other inflammatory mediators, from activated immune cells (Kowalski, 1998; Stefano et al., 2000). Nitric oxide synthesized by constitutive nitric oxide synthase (cNOS) is involved in the tonic inhibition of the intestine. Thus, the enhanced inhibitory effects of morphine on gastrointestinal transit during inflammation could come from two sources: (1) the increased expression of MOR and its inhibitory effects on the myenteric plexus and (2) the increased production of the inhibitory mediator nitric oxide from activated immune cells.
Recent studies have shown that some inflammatory mediators (e.g. IL-4, TNFα) increase the MOR gene transcription in different types of cells (Kraus et al., 2001; 2003). MOR gene transcription may also be positively controlled by other factors such as the activation of L-type calcium channels or retinoic acid (Chen & Loh, 2001; Jenab & Inturrisi, 2002) and various transcription factor-binding sites in the MOR gene have been described, whose activation could stimulate (Sp1, Sp3, mPy; Sox18) or repress (Oct-1) MOR gene transcription (Wei & Loh, 2002). However, the precise mechanisms implicated in the increased expression of MOR during intestinal inflammation are not yet elucidated.
In this report, we have investigated the role of nitric oxide in the enhanced expression of MOR during inflammation by using mice lacking the inducible NOS gene. Results showed that although the basal levels of MOR mRNA were similar in WT and iNOS−/− animals, intestinal inflammation only increased the expression of MOR in WT but not in iNOS−/− mice. These results suggest that nitric oxide derived from the increased expression of iNOS is implicated in the upregulation of MOR gene transcription observed in the gut during peripheral inflammation. This fact is supported by the colocalization of MOR and NOS in this tissue (Ho et al., 2003) and because nitric oxide activates diverse signaling pathways to regulate gene expression (Hemish et al., 2003). The possible novel signaling pathway of nitric oxide on transcriptional regulation of MOR gene is under investigation in our laboratory.
In summary, our results showed that during intestinal inflammation, inhibition of the synthesis of nitric oxide suppresses the enhanced effects of morphine and the expression of MOR in the gut only increases in WT but not in iNOS−/− mice. In conclusion, nitric oxide is involved in the MOR inhibition of gastrointestinal transit and required for the increased expression of MOR during inflammatory processes.
Acknowledgments
We thank Sergi Leánez for his excellent technical assistance. Ted M. Dawson is the Leonard and Madlyn Professor of Neurodegenerative Diseases. This work was supported by grants from FIS (00/0658) and CICYT (SAF2003-02578) Madrid, Spain. Part of these results have been presented as communication to the 31th Annual Meeting Society for Neuroscience, held in Orlando, U.S.A., November 2002 and to the International Symposium on Nitric Oxide-Cyclic GMP Signal Transduction in Brain, held in Valencia, Spain, November 2003.
Abbreviations
- ANOVA
analysis of variance
- CT
threshold cycle
- D-NAME
NG-nitro-D-arginine-methyl ester hydrochloride
- IL-6
interleukin-6
- iNOS
inducible nitric oxide synthase
- L-NAME
NG-nitro-L-arginine-methyl ester hydrochloride
- L-NIL
L-N6-(1-iminoethyl)-lysine hydrochloride
- MOR
μ-opioid receptor
- MPO
myeloperoxidase
- PCR
polymerase chain reaction
- PGK-1
phosphoglycerate kinase-1
- RT
reverse transcriptase
- TNBS
trinitrobenzene sulfonic acid
- WT
wild type
References
- BAGNOL D., MANSOUR A., AKIL H., WATSON S.J. Cellular localization and distribution of the cloned μ and κ opioid receptors in rat gastrointestinal tract. Neuroscience. 1997;81:579–591. doi: 10.1016/s0306-4522(97)00227-3. [DOI] [PubMed] [Google Scholar]
- BECK P.L., XAVIER R., WONG J., EZEDI I., MASHIMO H., MIZOGUCHI A., MIZOGUCHI E., BHAN A.K., PODOLSKY D.K. Paradoxical roles of different nitric oxide synthase isoforms in colonic injury. Am. J. Physiol. Gastrointest. Liver Physiol. 2004;286:G137–G147. doi: 10.1152/ajpgi.00309.2003. [DOI] [PubMed] [Google Scholar]
- BIANCHI M., MAGGI R., PIMPINELLI F., RUBINO T., PAROLARO D., POLI V., CILIBERTO G., PANERAI A.E., SACERDOTE P. Presence of a reduced opioid response in interleukin-6 knockout mice. Eur. J. Neurosci. 1999;11:1501–1507. doi: 10.1046/j.1460-9568.1999.00563.x. [DOI] [PubMed] [Google Scholar]
- BÖRNER C., KRAUS J., SCHRÖDER H., AMMER H., HÖLLT V. Transcriptional regulation of the human μ-opioid receptor gene by interleukin-6. Mol. Pharmacol. 2004;66:1719–1726. doi: 10.1124/mol.104.003806. [DOI] [PubMed] [Google Scholar]
- CHEN H.C., LOH H.H. μ-Opioid receptor gene expression: the role of NCAM. Neuroscience. 2001;108:7–15. doi: 10.1016/s0306-4522(01)00397-9. [DOI] [PubMed] [Google Scholar]
- COGGESHALL R.E., ZHOU S., CARLTON S.M. Opioid receptors on peripheral sensory axons. Brain Res. 1997;764:126–132. doi: 10.1016/s0006-8993(97)00446-0. [DOI] [PubMed] [Google Scholar]
- DIKOPOULOS N., NÜSSLER A.K., LIPTAY S., BACHEM M., REINSHAGEN M., STIEGLER M., SCHMID R.M., ADLER G., WEIDENBACH H. Inhibition of nitric oxide synthesis by aminoguanidine increases intestinal damage in the acute phase of rat TNB colitis. Eur. J. Clin. Invest. 2001;31:234–239. doi: 10.1046/j.1365-2362.2001.00802.x. [DOI] [PubMed] [Google Scholar]
- ESPLUGUES J.V. No as a signaling molecule in the nervous system. Br. J. Pharmacol. 2002;135:1079–1095. doi: 10.1038/sj.bjp.0704569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EVANS S.M., LÁSZLÓ F., WHITTLE B.J.R. Site-specific lesion formation, inflammation and inducible nitric oxide synthase expression by indomethacin in the rat intestine. Eur. J. Pharmacol. 2000;388:281–285. doi: 10.1016/s0014-2999(99)00869-9. [DOI] [PubMed] [Google Scholar]
- GRANADOS-SOTO V., RUFINO M.O., GOMES-LOPES L.D., FERREIRA S.H. Evidence for the involvement of the nitric oxide-cGMP pathway in the antinociception of morphine in the formalin test. Eur. J. Pharmacol. 1997;340:177–180. doi: 10.1016/s0014-2999(97)01399-x. [DOI] [PubMed] [Google Scholar]
- GRISHAM M.B., PAVLICK K.P., LAROUX F.S., HOFFMAN J., BHARWANI S., WOLF R.E. Nitric oxide and chronic gut inflammation: controversies in inflammatory bowel disease. J. Invest. Med. 2002;50:272–283. doi: 10.2310/6650.2002.33281. [DOI] [PubMed] [Google Scholar]
- HEMISH J., NAKAYA N., MITTAL V., ENIKOLOPOV G. Nitric oxide activates diverse signaling pathways to regulate gene expression. J. Biol. Chem. 2003;278:42321–42329. doi: 10.1074/jbc.M308192200. [DOI] [PubMed] [Google Scholar]
- HO A., LIEVORE A., PATIERNO S., KOHLMEIER S.E., TONINI M., STERNINI C. Neurochemically distinct classes of myenteric neurons express the mu-opioid receptor in the guinea pig ileum. J. Comp. Neurol. 2003;458:404–411. doi: 10.1002/cne.10606. [DOI] [PubMed] [Google Scholar]
- HOKARI R., KATO S., MATSUZAKI K., KUROKI M., IWAI A., KAWAGUCHI A., NAGAO S., MIYAHARA T., ITOH K., SEKIZUKA E., NAGATA H., ISHII H., MIURA S. Reduced sensitivity of inducible nitric oxide synthase-deficient mice to chronic colitis. Free Radic. Biol. Med. 2001;31:153–163. doi: 10.1016/s0891-5849(01)00565-2. [DOI] [PubMed] [Google Scholar]
- JENAB S., INTURRISI C.E. Retinoic acid regulation of mu opioid receptor and c-fos mRNAs and AP-1 DNA binding in SH-SY5Y neuroblastoma cells. Mol. Brain Res. 2002;99:34–39. doi: 10.1016/s0169-328x(01)00343-6. [DOI] [PubMed] [Google Scholar]
- KALFF J., SCHRAUT W.H., BILLIAR T.R., SIMMONS R.L., BAUER A.J. Role of inducible nitric oxide synthase in postoperative intestinal smooth muscle dysfunction in rodents. Gastroenterology. 2000;118:316–327. doi: 10.1016/s0016-5085(00)70214-9. [DOI] [PubMed] [Google Scholar]
- KIMURA H., HOKARI R., MIURA S., SHIGEMATSU T., HIROKAWA M., AKIBA Y., KUROSE I., HIGUCHI H., FUJIMORI H., TSUZUKI Y., SERIZAWA H., ISHII H. Increased expression of an inducible isoform of nitric oxide synthase and the formation of peroxynitritite in colonic mucosa of patients with active ulcerative colitis. Gut. 1998;42:180–187. doi: 10.1136/gut.42.2.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOLIOS G., VALATAS V., WARD S.G. Nitric oxide in inflammatory bowel disease: a universal messenger in an unsolved puzzle. Immunology. 2004;113:427–437. doi: 10.1111/j.1365-2567.2004.01984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KOWALSKI J. Augmenting effect of opioids on nitrite production by stimulate murine macrophages. Neuropeptides. 1998;32:287–291. doi: 10.1016/s0143-4179(98)90050-6. [DOI] [PubMed] [Google Scholar]
- KRAUS J., BÖRNER C., GIANNINI E., HICKFANG K., BARUN H., MAYSER P., HOEHE M.R., AMBROSCH A., KÖNING W., HÖLLT V. Regulation of μ-opioid receptor gene transcription by interleukin-4 and influence of an allelic variation within a STAT6 transcription factor binding site. J. Biol. Chem. 2001;276:43901–43908. doi: 10.1074/jbc.M107543200. [DOI] [PubMed] [Google Scholar]
- KRAUS J., BÖRNER C., GIANNINI E., HÖLLT V. The role of nuclear factor κB in tumor necrosis factor-regulated transcription of the human μ-opioid receptor gene. Mol. Pharmacol. 2003;64:876–884. doi: 10.1124/mol.64.4.876. [DOI] [PubMed] [Google Scholar]
- KRAWISZ J.E., SHARON P., STENSON W.F. Quantitative assay for acute intestinal inflammation based on myeloperoxidase activity. Assessment of inflammation in rat and hamster models. Gastroenterology. 1984;87:1344–1350. [PubMed] [Google Scholar]
- KRIEGLSTEIN C.F., CERWINKA W.H., LAROUX F.S., SALTER J.W., RUSSELL J.M., SCHUERMANN G., GRISHAM M.B., ROSS C.R., GRANGER D.N. Regulation of murine intestinal inflammation by reactive metabolites of oxygen and nitrogen: divergent roles of superoxide and nitric oxide. J. Exp. Med. 2001;5:1207–1218. doi: 10.1084/jem.194.9.1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LIVAK K.J., SCHMITTGEN T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−(ΔΔCT) method. Methods. 2001;25:402–408. doi: 10.1006/meth.2001.1262. [DOI] [PubMed] [Google Scholar]
- MANSOUR A., FOX C.A., AKIL H., WATSON S.J. Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 1995;18:22–29. doi: 10.1016/0166-2236(95)93946-u. [DOI] [PubMed] [Google Scholar]
- MATHISON R., HO W., PITTMAN Q.J., DAVISON J.S., SHARKEY K.A. Effects of cannabinoid receptor-2 activation on accelerated gastrointestinal transit in lipopolysaccharide-treated rats. Br. J. Pharmacol. 2004;142:1247–1254. doi: 10.1038/sj.bjp.0705889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCAFFERTY D.M., MIAMPAMBA M., SIHOTA E., SHARKEY K.A., KUBES P. Role of inducible nitric oxide synthase in trinitrobenzene sulphonic acid induced colitis in mice. Gut. 1999;45:864–873. doi: 10.1136/gut.45.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MCCAFFERTY D.M., MUDGETT J.S., SWAIN M.G., KUBES P. Inducible nitric oxide synthase plays a critical role in resolving intestinal inflammation. Gastroenterology. 1997;112:1022–1027. doi: 10.1053/gast.1997.v112.pm9041266. [DOI] [PubMed] [Google Scholar]
- NOZAKI-TAGUCHI N., YAMAMOTO T. The interaction of FK409, a novel nitric oxide releaser, and peripherally administered morphine during experimental inflammation. Anesth. Analg. 1998;86:367–373. doi: 10.1097/00000539-199802000-00028. [DOI] [PubMed] [Google Scholar]
- PHILIPPE D., DUBUQUOY L., GROUX H., BRUN V., VAN CHUOÏ-MARIOT M.T., GAVERIAUX-RUFF C., COLOMBEL J.F., KIEFFER B.L., DESREUMAUX P. Anti-inflammatory properties of the μ-opioid receptor support its use in the treatment of colon inflammation. J. Clin. Invest. 2003;111:1329–1338. doi: 10.1172/JCI16750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POL O., ALAMEDA F., PUIG M.M. Inflammation enhances μ-opioid receptor transcription and expression in mice intestine. Mol. Pharmacol. 2001;60:894–899. doi: 10.1124/mol.60.5.894. [DOI] [PubMed] [Google Scholar]
- POL O., PUIG M.M. Expression of opioid receptors during peripheral inflammation. Curr. Top. Med. Chem. 2004;4:51–61. doi: 10.2174/1568026043451519. [DOI] [PubMed] [Google Scholar]
- POL O., VALLE L.L., SÁNCHEZ-BLÁZQUEZ P., GARZÓN J., PUIG M.M. Antibodies and antisense oligodeoxynucleotides to μ-opioid receptor selectively block the effects of μ-opioid agonists on intestinal transit and permeability in mice. Br. J. Pharmacol. 1999;127:397–404. doi: 10.1038/sj.bjp.0702570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PUEHLER W., ZÖLLNER C., BRACK A., SHAQURA M.A., KRAUSE H., SCHÄFER M., STEIN C. Rapid upregulation of μ opioid receptor mRNA in dorsal root ganglia in response to peripheral inflammation depends on neuronal conduction. Neuroscience. 2004;129:473–479. doi: 10.1016/j.neuroscience.2004.06.086. [DOI] [PubMed] [Google Scholar]
- PUIG M.M., POL O. Peripheral effects of opioids in a model of chronic intestinal inflammation in mice. J. Pharmacol. Exp. Ther. 1998;287:1068–1075. [PubMed] [Google Scholar]
- STEFANO G.B., GOUMON Y., BILFINGER T.V., WELTERS I.D., CADET P. Basal nitric oxide limits immune, nervous and cardiovascular excitation: human endothelia express a mu opiate receptor. Prog. Neurobiol. 2000;60:513–530. doi: 10.1016/s0301-0082(99)00038-6. [DOI] [PubMed] [Google Scholar]
- STEFANO G.B., ZHU W., CADET P., BILFINGER T.V., MANTIONE K. Morphine enhances nitric oxide release in the mammalian gastrointestinal tract via the μ3 opiate receptor subtype: a hormonal role for endogenous morphine. J. Physiol. Pharmacol. 2004;55:279–288. [PubMed] [Google Scholar]
- STERNINI C. Receptors and transmission in the brain–gut axis: potential for novel therapies. III. Mu-opioid receptors in the enteric nervous system. Am. J. Physiol. Gastrointest. Liver Physiol. 2001;281:G8–15. doi: 10.1152/ajpgi.2001.281.1.G8. [DOI] [PubMed] [Google Scholar]
- TALLARIDA R.J., MURRAY R.B. Manual of Pharmacological Calculations with Computer Programs. New York: Springer-Verlag; 1986. [Google Scholar]
- TASATARGIL A., SADAN G. Reduction in [D-Ala2, NMePhe4, Gly-ol5]Enkephalin-induced peripheral antinociception in diabetic rats: the role of the L-arginine/nitric oxide/cyclic guanosine monophosphate pathway. Anesth. Analg. 2004;98:185–192. doi: 10.1213/01.ANE.0000093250.59364.EB. [DOI] [PubMed] [Google Scholar]
- TOMASSINI N., RENAUD F.L., ROY S., LOH H.H. Mu and delta receptors mediate morphine effects on phagocytosis by murine peritoneal macrophages. J. Neuroimmunol. 2003;136:9–16. doi: 10.1016/s0165-5728(02)00463-0. [DOI] [PubMed] [Google Scholar]
- TORRENTS D., PRATS N., VERGARA P. Inducible nitric oxide synthase inhibitors ameliorate hypermotility observed after T. spiralis in the rat. Dig. Dis. Sci. 2003;48:1035–1049. doi: 10.1023/a:1023796108391. [DOI] [PubMed] [Google Scholar]
- VALLE L.L., PUIG M.M., POL O. Effects of μ-opioid receptor agonists on intestinal secretion and permeability during acute intestinal inflammation in mice. Eur. J. Pharmacol. 2000;389:235–242. doi: 10.1016/s0014-2999(99)00871-7. [DOI] [PubMed] [Google Scholar]
- VIDAL E.L., PATEL N.A., DEWU G., FIALA M., CHANG S.L. Interleukin-1 induces the expression of μ opioid receptors in endothelial cells. Immunopharmacology. 1998;38:261–266. doi: 10.1016/s0162-3109(97)00085-4. [DOI] [PubMed] [Google Scholar]
- WEI L., LOH H.H. Regulation of opioid expression. Curr. Opin. Pharmacol. 2002;2:69–75. doi: 10.1016/s1471-4892(01)00123-0. [DOI] [PubMed] [Google Scholar]
- YUE G., LAI P., YIN K., SUN F.F., NAGELE R.G., LIU N., LIU X., LINASK K.K., WANG C., LIN K.T., WONG P.Y.K. Colon epithelial cell death in 2,4,6 trinitrobenzenesulfonic acid-induce colitis is associated with increased inducible nitric-oxide synthase expression and peroxynitrite production. J. Pharmacol. Exp. Ther. 2001;297:915–925. [PubMed] [Google Scholar]
- ZÖLLNER C., SHAQURA M.A., BOPAIAH C.P., MOUSA S., STEIN C., SCHÄFER M. Painful inflammation-induced increase in μ-opioid receptor binding and G-protein coupling in primary afferent neurons. Mol. Pharmacol. 2003;64:202–210. doi: 10.1124/mol.64.2.202. [DOI] [PubMed] [Google Scholar]