

Abstract
Benzodiazepines (BZDs) have been used extensively for more than 40 years because of their high therapeutic index and low toxicity. Although BZDs are understood to act primarily as allosteric modulators of GABAA receptors, the mechanism of modulation is not well understood.
The applicability of an allosteric model with two binding sites for γ-aminobutyric acid (GABA) and one for a BZD-like modulator was investigated.
This model predicts that BZDs should enhance the efficacy of partial agonists.
Consistent with this prediction, diazepam increased the efficacy of the GABAA receptor partial agonist kojic amine in chick spinal cord neurons.
To further test the validity of the model, the effects of diazepam, flurazepam, and zolpidem were examined using wild-type and spontaneously active mutant α1(L263S)β3γ2 GABAA receptors expressed in HEK-293 cells.
In agreement with the predictions of the allosteric model, all three modulators acted as direct agonists for the spontaneously active receptors.
The results indicate that BZD-like modulators enhance the amplitude of the GABA response by stabilizing the open channel active state relative to the inactive state by less than 1 kcal, which is similar to the energy of stabilization conferred by a single hydrogen bond.
Keywords: Two state model, diazepam, zolpidem, flumazenil, kojic amine, partial agonist
Introduction
Since the discovery that benzodiazepines (BZDs) produce their pharmacological effects by allosterically modulating GABAA receptors, numerous other GABAA receptor modulators have been identified, including barbiturates, steroids, and certain divalent and trivalent metal cations (Choi et al., 1977; Leeb-Lundberg et al., 1980; Chan & Farb, 1985; Majewska et al., 1986; 1988; 1990; Majewska & Schwartz, 1987; Kleckner & Dingledine, 1988; Macdonald et al., 1989; Turner et al., 1989; Draguhn et al., 1990; Roca et al., 1990; Smart & Constanti, 1990; Wu et al., 1990; Celentano et al., 1991; Ma & Narahashi, 1993). N-methyl-D-aspartate (NMDA) receptors have been found to be modulated by glycine (Johnson & Ascher, 1987), steroids (Wu et al., 1991; Park-Chung et al., 1996), arachidonic acid (Miller et al., 1992), and polyamines (Sprosen & Woodruff, 1990). It is likely that the capacity for allosteric modulation is a general characteristic of neurotransmitter receptors. The physiological significance of allosteric modulation to nervous system function remains unclear, but there is evidence that at least some endogenous modulators, such as neurosteroids and glycine (Wood, 1995), may play significant regulatory roles.
From a pharmacological and therapeutic perspective, drugs that act as allosteric modulators can offer significant advantages over classical agonists and antagonists. For example, the BZDs exhibit large therapeutic indexes, probably because they enhance the action of endogenous γ-aminobutyric acid (GABA) without activating the receptor directly. The low toxicity of the BZDs has contributed to their usefulness in a wide variety of clinical contexts, ranging from anxiety to epilepsy.
Allosteric modulation of neurotransmitter receptors remains poorly understood. A two-state allosteric model of receptor function (Karlin, 1967), based upon the allosteric transition model of Monod et al. (1965), has been found to provide a good description of agonist-mediated activation of acetylcholine (Jackson, 1989) and GABAA receptors (Chang & Weiss, 1999). In addition, the modulatory and direct activating effects of the general anesthetic etomidate on GABAA receptors have been shown to be consistent with an allosteric model (Rusch et al., 2004).
It is notable that despite decades of synthesis of BZD derivatives, no BZD modulators have been reported with the capacity to directly activate wild-type GABAA receptors. In contrast, direct activation is commonly observed with steroid and barbiturate modulators, as well as general anesthetics such as propofol and etomidate (Rusch et al., 2004). It has been suggested, based on single-channel results, that BZDs influence only the binding of GABA, and are therefore incapable of acting as agonists (Rogers et al., 1994). Alternatively, the inability of BZDs to directly activate the GABAA receptor could indicate that activation-associated changes in the BZD recognition site are small compared with the steroid and barbiturate-binding sites.
In the present study, we now examine whether an expanded version of the two-state allosteric model can also account for modulation of the GABAA receptor by diazepam (DZ) and related ligands that act via the BZD recognition site.
To test the applicability of the allosteric model, we examined the effects of BZD modulators on the response of neuronal GABAA receptors to kojic amine, a partial agonist at the GABAA receptor. The efficacy of kojic amine was enhanced in the presence of DZ as predicted by the allosteric model. We also examined the effects of BZD-like modulators on a mutant (α1L263Sβ3γ2) GABAA receptor that exhibits a high basal level of activity in the absence of GABA (Chang & Weiss, 1999). DZ, flurazepam (FZ), and zolpidem directly activated the mutant GABAA receptors, and the extent of direct activation was consistent with their modulatory effects on wild-type GABAA receptors. The results indicate that enhancement of GABAA receptor activation by BZDs primarily reflects stabilization of the active state of the receptor relative to the inactive state.
Methods
Chemicals
Drugs were purchased from Sigma (St Louis, MO, U.S.A.). DZ, zolpidem, flumazenil, and picrotoxin were prepared as stock solutions in DMSO; the final concentration of DMSO in all experiments was 0.5%, which was also present in the wash buffer.
Electrophysiology (neurons)
Primary cultures of embryonic chick spinal cord neurons were prepared as previously described (Park-Chung et al., 1999). Electrophysiological studies on primary embryonic chick spinal cord neurons were carried out after 2–4 weeks in culture. The whole-cell-patch-clamp method was used to record from neurons. The resting potential of most neurons was ca. −50 mV, and cells with a resting potential higher than −35 mV were rejected. All agonists and modulators were applied using a seven-barrel pressure application system, with a tip diameter of 100 μm per barrel. The drug application pipette was positioned ca. 50 μm from the cell body, and drug solution was applied via positive pressure from nitrogen gas. This method effectively replaces the buffer in the vicinity of the target neuron with the applied solution, with <10% dilution by the bath solution. The effective solution exchange time (90% exchange), determined by application of 140 mM KCl to neurons, was ca. 200 ms.
To study the effect of a modulator on the agonist response, the modulator was applied to the target neuron for 10 s, then agonist plus modulator was applied for 10 s. With high concentrations of agonist, the period of agonist exposure was reduced to 1–2 s to minimize agonist-dependent rundown (Gyenes et al., 1988).
Some neurons exhibited large GABA-induced currents, raising the concern that dose–effect curves could be distorted by voltage clamp mistracking due to series resistance error. To limit current amplitudes, solutions containing symmetrical 25 mM Cl− were used. Extracellular solution contained (mM) 133 Na-gluconate, 17 NaCl, 4 KCl, 1 CaCl2, 1 MgCl2, 10 HEPES, with the pH adjusted to 7.2 using NaOH. Intracellular solution contained (in mM) 120 K-gluconate, 20 KCl, 3 NaCl, 1 MgCl2, 11 EGTA, 10 HEPES, and 3.8 mM Mg-ATP, with the pH adjusted to 7.2 using KOH. In some experiments, the electrode was coated with Sylgard (Dow Corning) near the tip to reduce pipet capacitance to allow greater series resistance compensation. Series resistance compensation was 20–60% with coated electrodes and 10–20% with uncoated electrodes. Data were accepted only from cells for which the calculated series resistance error was less than 15 mV.
Transient expression of recombinant GABAA receptors
Human embryonic kidney cells (HEK-293, American Type Culture Collection, Rockville, MD, U.S.A.) were cultured in untreated 25 cm2 flasks (Costar) in Dulbecco's modified Eagle's medium (D-MEM) (Invitrogen, Carlsbad, CA, U.S.A.) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA, U.S.A.) and 1% MEM Non-Essential Amino Acids Solution (Invitrogen, Carlsbad, CA, U.S.A.). The cells were transiently transfected with α1 (wild-type or mutant)+γ2s GABAA receptor subunits (the HEK-293 cells express an endogenous β3 subunit) using Fugene 6 (Roche, Indianapolis, IN, U.S.A.) at a 3 : 1 Fugene : DNA ratio. All cells were cotransfected with the CD8-α expression plasmid πH3-CD8 (Jurman et al., 1994), which was a gift from Dr Brian Seed (Harvard Medical School, Boston, MA, U.S.A.). Cells were cultured for 48–72 h following transfection at 37°C and 5% CO2. At 12 h prior to recording, the transfected cells were briefly (0.05%, 1 min, 23°C) trypsinized (Invitrogen, Carlsbad, CA, U.S.A.) to detach them from the flask, centrifuged, and resuspended in culture media. Magnetic polystyrene microspheres coated with anti-CD8 antibody (Dynal, Great Neck, NY, U.S.A.) were added to the cells in suspension and incubated for 30 min while gently shaking, after which the beaded (transfected) cells were magnetically isolated. The bead-purified cells were plated onto 35 mm dishes (Corning) for electrophysiology.
Electrophysiology (HEK-293)
Patch pipettes were fabricated from borosilicate glass (A-M Systems, Inc.). with a vertical pipette puller (David Kopf Instruments) and filled with intracellular solution containing (in mM): 140 CsCl, 1 MgCl2, 11 EGTA, and 10 HEPES (pH adjusted to 7.3), supplemented with 4 mM Mg2+-ATP (Gyenes et al., 1994). Prior to each experiment, all drugs were freshly dissolved in extracellular solution (in mM): 135 NaCl, 5.4 KCl, 1.8 CaCl2, 1 MgCl2, and 5 HEPES (pH adjusted to 7.2). Small, beaded cells were selected for recording using phase-contrast microscopy, the whole-cell configuration was obtained, and the cell was lifted off the dish surface and placed into the flow of solution. Experiments were performed at room temperature using standard whole-cell patch-clamp techniques. Pipette and cell capacitance and series resistance were compensated in the patch-clamp amplifier (Dagan 3900). Current was filtered at 2 kHz by a four-pole Bessel, lowpass filter and digitized at 5–10 kHz using custom software running in the Labview environment (National Instruments, Austin, TX, U.S.A.).
Rapid solution exchange was carried out using a piezo-electric driven theta-tube apparatus based upon a design by Dr Stuart Forman (Harvard Medical School, Boston, MA, U.S.A.). Flow velocity was 8.2 cm s−1. Solution exchange, measured as the time for the 10–90% response to a 140 mM change in K+concentration with an open tip pipette, was typically less than 1 ms. The recording chamber was continuously perfused with bath solution to prevent drugs from accumulating. Current measurements were taken at the response peak.
The standard recording protocol employed 0.01–1 s applications of GABA or other drugs separated by a 60 s wash period to prevent accumulation of desensitization. A 100 μM GABA standard was applied after every 2–4 agonist applications. At least two different cells from at least two different transfections were studied for each experiment.
Site-directed mutagenesis
cDNAs for the rat α1, β2, and γ2s GABAA receptor subunits in the pRc/CMV vector were obtained from Dr Phil Skolnick (Eli Lilly). The α1 subunit L263S mutation (Chang & Weiss, 1999) was introduced by the site-specific mutagenesis by overlap extension method using custom-made primers (Oligos Etc. Inc., Wilsonville, OR, U.S.A.), which flanked the mutation site. The primer pair upstream of the mutation site was 5′-gaa gtt gtc tat gag tgg and 5′-gac gac cgt ttc gac cat gac aac c and downstream of the mutation site 5′-ggt tgt cat ggt cga aac ggt cgt c and 5′-ccc aat aga cta agt taa ag. The mismatched base pairs are indicated in bold. Dideoxynucleotide sequencing confirmed that the mutation and its orientation were correct, and that no stray mutations occurred (Harvard sequencing facility, Harvard Medical School, Boston, MA, U.S.A.).
Data analysis
Baseline, measured as the average current before drug application, was subtracted from all measured currents. The leak-subtracted currents were then normalized by linear regression to the flanking 100 μM GABA standard measurements to correct for receptor rundown and cell-to-cell differences. EC50 and Emax were estimated by least-squares nonlinear regression using the Hill equation: I=Emax ([agonist]n/(EC50n+[agonist]n).
Fits to the allosteric model were carried out by least-squares nonlinear regression, using the equilibrium state equation for the model depicted in Figure 1 (see Appendix A for derivation)
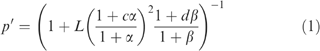 |
where p′ is the equilibrium fraction of receptors in the active R′ state, α is the normalized agonist concentration ratio [A]K′A−1, β is the normalized modulator concentration [B]K′B−1, c=K′AKA−1, d=K′BKB−1, and K′ and K are, respectively, the dissociation constants for binding of the subscripted ligand to the active and inactive states (Karlin, 1967). Global fits of the allosteric model to normalized, unpooled current measurements were carried out using either Excel (Microsoft, Redmond, WA, U.S.A.), MATLAB (The Mathworks, Northhampton, MA, U.S.A.), or Prism (Graphpad Software) by minimizing the total sum of the squared deviations of the individual data points from the calculated values. Confidence limits (95%) for a fitted parameter were determined by refitting the equation to the data while holding a single parameter constant at successively increasing or decreasing values, and determining the minimum positive or negative change in the value of that parameter that resulted in a significant deterioration of the fit (P<0.05), as evaluated by F-test using the extra sum of squares method (Munson & Rodbard, 1980). Pooled data are presented as mean±s.e.m. and statistical significance was determined at the P<0.05 level. Estimation of EC50 and maximum response from experimental data was carried out either with Excel (Microsoft, Redmond, WA, U.S.A.), MATLAB (The Mathworks, Northhampton, MA, U.S.A.) or Prism (Graphpad Software) by nonlinear regression using the Hill equation.
Figure 1.

Two-state model of allosteric modulation. (a) Extended two site model with two identical sites for an agonist A and one site for a heterologous modulator B. R denotes the inactive conformation and R′ the active conformation. Binding sites for A are treated as identical; factors of 1/2 and 2 are stoichiometric. Desensitization is not modeled. (b) Simulated concentration–response–surface for interaction of a full agonist (A) with a positive modulator (B). Parameter values are L=3000, c=0.002, d=0.3. (c) Simulated concentration–response–surface for interaction of a partial agonist with a positive modulator. Parameter values are L=3000, c=0.025, d=0.3.
Real-time PCR
Total RNA was extracted from HEK-293 cells with the RNeasy® midi kit (Qiagen). Primers (Oligos Etc) and TaqMan probes (Applied Biosystems) were designed to amplify segments of the human GABAA receptor β2 and β3 subunit cDNAs encoding amino acids located in the cytoplasmic loop between transmembrane domains M3 and M4, via Primer Express software (Applied Biosystems). Primers and probes sequences: β2 forward primer: 5′- GACCCCAGAAGCACAATGCTA-3′, β2 reverse primer: 5′- GCCTGGGCAACCCAGC-3′, β2 probe: 5′- CCTATGATGCCTCCAGCATCCAGTATCG-3′; β3 Forward primer: 5′- CAGTACAGGAAACAGAGCATGCC-3′, β3 Reverse primer: 5′- TCTTGTGCGGGAGGCTTC-3′, β3 probe: 5′- CGAGAAGGGCATGGGCGATTCC-3′.
Ribosomal RNA probe and primers were purchased from Applied Biosystems. Quantitative one-step real-time PCR was performed with an ABI Prism Applied Biosystems 7900HT Sequence Detection System using a QuantiTect™ Probe RT-PCR kit (Qiagen). Reactions were performed in triplicate in a total volume of 50 μl containing 5–200 ng of total RNA, QuantiTect RT mix, QuantiTect Probe RT-PCR master mix, 250 nM receptor probe, 900 nM receptor primers, 200 nM rRNA probe and 50 nM rRNA primers. Two aliquots of 20 μl per reaction were loaded in a 384-well plate. Incubation conditions were 48°C for 30 min, 95°C for 10 min followed by 50 cycles of 95°C for 15 s, and 60°C for 1 min.
Western blot analysis
HEK-293 cells were washed with 20 mM TBS (20 mM Tris-HCl buffer, 137 mM NaCl, pH 7.6), scraped with a rubber policeman and centrifuged at low speed. The pellet was resuspended in lysis buffer (20 mM TBS, 1% Nonidet P-40, 10% glycerol, 2 mM phenylmethylsulfonyl fluoride, 10 μg ml−1 aprotinin, 10 μg ml−1 pepstatin, 2 mM EDTA, 0.2 mM EGTA, 5 mM DTT, and 1 μg ml−1 leupeptin) and mixed with constant agitation for 25 min at 4°C. This suspension was centrifuged at low speed, the pellet discarded, and the protein concentration of the supernatant was measured. Aliquots of 40 μg protein were loaded on a 10% SDS-polyacrylamide gel. After electrophoresis, the samples were electroblotted onto nitrocellulose filters (Amersham). Blots were blocked for 2 h at 25°C in 20 mM TBS containing 5% nonfat dry milk and 0.05% Tween-20 (TBS-MLK-T). The blots were then incubated overnight at 4°C with 2 μg ml−1 anti-GABAA receptor β2/3 subunit antibody (mouse monoclonal IgG clone 62-361, Upstate biotechnology) in TBS-MLK-T. After washing with TBS-T, nitrocellulose filters were incubated for 1.5 h at room temperature with goat anti-mouse horseradish peroxidase-conjugated antibody (Vector, 1 : 3000 dilution in TBS-MLK-T) to reveal the presence of β2/3 protein, followed by enhanced chemiluminiscence (ECL) with Hyperfilm ECL (Amersham).
Results
Allosteric model of receptor activation
The two-state allosteric model for receptor activation as proposed by Karlin (1967) and Thron (1973) was based on the allosteric enzyme model of Monod et al. (1965). According to this model, a receptor at rest is regarded as being in equilibrium between an inactive state R and an active state R′, with proportions defined by an isomerization equilibrium constant L=([R]/[R′])eq. The equilibrium normally favors the inactive state (L≫1) so that basal activity of the receptor in the absence of ligand is low. A ligand that binds with greater affinity to the active state than to the inactive state (i.e. K′A<KA) will act as an agonist, shifting the conformational equilibrium to favor the active state (Colquhoun, 1978).
Allosteric modulation
The allosteric model can readily be expanded to accommodate additional binding sites. Since current evidence favors two GABA-binding sites, we have used a slightly more elaborate version of the two-state model (Figure 1) with two identical binding sites for agonist A and one site for a heterologous modulator B.
This model makes two key predictions regarding agonist-modulator interactions:
Allosteric modulators influence the efficacy of partial agonists. If an agonist A has high efficacy (K′A≪KA), then a positive modulator binding at the B site will produce a finite, parallel, leftward shift in the concentration–response curve for A, such as has been described for BZD modulation of GABAA receptor activation by GABA (Choi et al., 1981). In contrast, if A is a partial agonist, with insufficient efficacy to fully activate the receptor, then the effect of a positive modulator B will be to increase the maximum response to A. Therefore, the efficacy of A determines whether the modulatory effect of B manifests primarily as enhancement of the potency (Figure 1b) or the efficacy of A (Figure 1c).
Allosteric modulators have agonist activity. The distinction between an agonist and a modulator in this model is somewhat arbitrary. Ligands that exhibit greater affinity for the active state than for the inactive state will produce some degree of activation, whether binding is at the A sites or the B site. If L≫1, as expected for the wild-type receptor, a compound that has only slightly greater affinity for the active state will produce negligible direct activation. However, there is a strong synergistic interaction between the two sites so that such a ligand binding at the B site can act as a positive modulator, substantially enhancing the activation produced by an agonist binding at the A sites (Figure 1b and c).
Kojic amine is a partial agonist in chick spinal cord neurons
To select a suitable partial agonist to evaluate the effect of BZDs on partial agonist efficacy, we examined responses of spinal cord neurons in culture to high concentrations (1–10 mM) of 3-amino-4-propanesulfonic acid (3APS), imidazole-4-acetic acid (IAA), isoguvacine, 4,5,6,7-tetrahydroisoxazol[5,5-c]pyridin-3-ol (THIP), piperidine-4-sulfonic acid (P4S), isonipecotic acid, thiomuscimol, taurine, and kojic amine. As an internal standard, the response of each neuron to 100 μM GABA, a concentration that in these neurons typically elicits 60–70% of a maximum GABA response, was also measured.
Although P4S has low efficacy on some GABAA receptors (Ebert et al., 1994), it elicited a response nearly as large as that produced by the 100 μM GABA standard, suggesting that its efficacy on chick spinal cord neurons approaches that of GABA. 3APS, IAA, thiomuscimol, isonipecotic acid, and isoguvacine also elicited responses that approached or exceeded the 100 μM GABA standard (Figure 2a–f). Currents induced by isoguvacine or THIP exhibited complex kinetics characterized by a rapidly fading current, followed by a ‘rebound' current when the agonist was washed off (Figure 2f and g), suggesting that these agonists may have inhibitory activity at a separate site.
Figure 2.

Identification of taurine and kojic amine as possible GABAA receptor partial agonists. Sample traces are shown illustrating responses of chick spinal cord neurons to (a) 100 μM 3APS; (b) 100 μM P4S; (c) 100 μM IAA; (d) 100 μM thiomuscimol; (e) 100 μM isonipecotic acid; (f) 100 μM isoguvacine; (g) 100 μM THIP; (h) 10–100 mM taurine; (i) 100 μM–10 mM kojic amine. Responses are compared to the response of the same neuron to 100 μM GABA, used as an internal standard. Bar indicates period of agonist exposure.
Taurine and kojic amine elicited currents that were substantially less than the 100 μM GABA standard, without the kinetic complexities seen with THIP and isoguvacine (Figure 2h and i). The maximum taurine response was less than that for GABA, but the potency of taurine was very low, with concentrations of 60–100 mM required for saturation. Kojic amine also produced a maximum response that was less than the 100 μM GABA standard, but was more potent than taurine. The EC50 of kojic amine was 4.7 mM and the extrapolated maximum response to kojic amine was 59% of the 100 μM GABA response. The kojic amine-induced current was completely blocked by 100 μM gabazine, demonstrating that kojic amine-induced current is mediated by the GABAA receptor (Figure 3a).
Figure 3.

Surmountable antagonism of the GABA response by kojic amine. (a) Kojic amine-induced current is completely blocked by 100 μM gabazine. (b) Kojic amine (25 mM) reversibly inhibits the current induced by 100 μM GABA when applied midway during the GABA pulse. Inhibition by kojic amine is not observed when the concentration of GABA is increased to 3 mM. (c) Current induced by 100 μM GABA+25 mM kojic amine (middle trace) is less than that induced by 100 μM GABA alone (left trace). Trace on right shows recovery. (d) Simultaneous application of 3 mM GABA+25 mM kojic amine (middle) reveals little inhibition compared to 3 mM GABA alone (left and right traces).
To verify that kojic amine is a partial agonist at the GABA recognition site, we examined the effect of applying GABA and kojic amine in combination. As shown in Figure 3b and c, the response to 100 μM GABA plus 25 mM kojic amine is less than the response to 100 μM GABA alone, consistent with competition by a partial agonist. As shown in Figure 3b and d, increasing GABA from 100 μM to 3 mM surmounts the inhibitory effect of kojic amine. This result indicates that the inhibitory effect of kojic amine is due to a competitive interaction between GABA and kojic amine at the GABA recognition site, and not the result of a separate noncompetitive inhibitory activity of kojic amine, confirming that kojic amine is a partial agonist.
DZ enhances the efficacy of kojic amine
Concentration–response studies (Figure 4) show that DZ shifts the GABA concentration–response curve to the left, with no change in the maximum response, in agreement with previous reports (Choi et al., 1981; Hattori et al., 1986; Mehta & Ticku, 1989). In contrast, DZ significantly increases the maximum kojic amine induced current (Figure 4b, c).
Figure 4.

Modulation of GABA and kojic amine concentration–response curves by DZ. (a) DZ (1 μM) enhances the 10 mM kojic amine response. (b) Effect of DZ on maximal GABA and kojic amine responses. Bars show the percentage change in the 1 mM GABA response and 25 mM kojic amine response in the presence of 1 μM DZ. Number of cells tested is shown adjacent. *Indicates significant change (P⩽0.01, paired two-tail t-test). (c) Pooled concentration–response curves for GABA and kojic amine alone and in the presence of 1 μM DZ. Responses are normalized to the 100 μM GABA response of the same neuron (indicated by +). Smooth curves are calculated using Equation (1) with the parameters determined by simultaneous least-squares fitting of the entire data set to Equation (1). Fitted parameters are cGABA=0.002, cKojic=0.026, dDZ=0.29. As the experiments were carried out at a single saturating concentration of DZ, the results provide no information about KDZ, which was fixed at 50 nM. Since equally good fits were obtained over a large range of values of L, L was fixed and set equal to 3000. Error bars indicate s.e.m. Number of neurons tested is shown adjacent. *Indicates significant effect of DZ relative to paired GABA response of same neuron in the absence of DZ (P<0.03, paired t-test).
To determine whether the modulatory effects of DZ on the GABA and kojic amine dose–effect curves are quantitatively consistent with the two-state model, dose-response data for GABA and kojic amine in the presence and absence of DZ were simultaneously fitted to Equation (1). As shown in Figure 4c, the two-state model is able to fit the effects of DZ on both the GABA and kojic amine dose–response curve. Sensitivity analysis indicates that the data contain little information about the value of the resting isomerization equilibrium constant L, as fit residuals do not change significantly when L is varied over a wide range. In the absence of information about the value of L, it is also not possible to obtain a reliable estimate of the GABA affinity ratio cGABA=K′GABA/KGABA or the kojic amine affinity ratio ckojic=K′kojic/Kkojic, because the values of these parameters covary with L. However, the DZ affinity ratio dDZ=K′DZ/KDZ is virtually insensitive to the specific value of L, yielding an estimate of dDZ=0.29 (95% confidence limits 0.16–0.32).
Modulation of spontaneous activity of α1L263S GABAA receptors
Mutation of the conserved M2 lysine of one or more GABAA receptor subunits has been reported to result in receptors with a measurable level of spontaneous activity, consistent with a decrease in L (Chang & Weiss, 1999). Such spontaneously active receptors offer the opportunity to examine the effects of modulators in the absence of GABA. In particular, the allosteric model predicts that BZD-like positive modulators should enhance spontaneous activity in the absence of GABA. An additional advantage of this mutant is that it is possible to determine the value of L for the mutant experimentally from the level of spontaneous activity.
Wild-type or mutant (L263S) α1 subunits were transfected into HEK293 cells along with β2 and/or γ2 subunits. Solutions were applied to lifted cells using a piezoelectric rapid solution switching system to minimize desensitization. Untransfected HEK-293 cells had no measurable GABA-induced current (n=8), whereas cells transfected with wild-type α1+β2+γ2 subunits exhibited clear GABA responses. However, there was little difference in the amplitude of the GABA-induced current between cells transfected with α1+β2+γ2 subunits and cells transfected with only α1+γ2 subunits. Since a β subunit is critical for organization of the GABA-binding site, this observation implies endogenous expression of a β subunit in the HEK293 cells (Davies et al., 2000; Kirkness & Fraser, 1993). A second batch of HEK293 cells obtained from ATCC yielded similar results.
Western blot analysis of total protein from untransfected HEK293 cells indicated the presence of an endogenous β2/β3 subunit (Figure 5). Real-time PCR analysis showed that the HEK293 cells have both β2 and β3 mRNAs, but that β3 mRNA is approximately 250 × more abundant than β2 mRNA (the efficiency of the β2 and β3 PCR reactions was approximately equal). Thus, the endogenous β subunit is most likely β3. For subsequent experiments, cells were transfected only with α1 and γ2, relying upon the endogenous β subunit. GABA responses were resistant to 10 μM Zn2+(5±2% inhibition, n=5), indicating efficient incorporation of γ subunits. In contrast, GABA-induced currents of HEK293 cells transfected with α1 alone were inhibited 71±9% (n=8) by 10 μM Zn, consistent with the absence of a γ subunit (Draguhn et al., 1990; Hosie et al., 2003).
Figure 5.

Untransfected HEK293 cells contain endogenous GABAA receptor β subunit protein. Results of a Western blot analysis of total protein from untransfected HEK293 cells, HEK293 cells transfected with GABAA receptor α1 and γ2 subunits, and rat cerebral cortical cultured neurons.
Expression of α1L263S in combination with γ2 and (endogenous) β3 subunits results in enhanced potency of GABA; the GABA EC50 for the α1L263S mutant receptor is 210 nM, a 96-fold increase in potency over wild-type. The increased potency of GABA is characteristic of the α1L263S mutation, and is consistent with a decreased value of L (Chang & Weiss, 1999). Cells expressing α1L263Sβ3γ2 subunits exhibited a large holding current in the absence of GABA that was blocked by 1 mM picrotoxin, indicating a high level of basal GABAA receptor activation. Figure 6 shows an example of a maximal (100 μM) GABA response and the 1 mM picrotoxin ‘response' (reduction of the holding current by picrotoxin) from the same cell. Assuming that the maximal GABA response reflects full receptor activation, the isomerization equilibrium constant of the α1L263S mutant receptor is given by the ratio of GABA-dependent current to the picrotoxin-sensitive current, yielding Lmut=1.01±0.07 (n=23), indicating that about half of the available receptors are active in the absence of GABA.
Figure 6.

Mutant receptors exhibit picrotoxin-sensitive holding current. Application of 1 mM picrotoxin reversibly inhibited the holding current of HEK293 cells expressing α1L263Sβ3γ2 subunits. Response to 100 μM GABA is of similar magnitude but opposite direction, indicating that about half of the GABAA receptor channels are spontaneously active. Inset: Same data shown with an expanded time base.
In the absence of GABA, DZ directly activated the α1L263S mutant receptor (Figure 7a) with an EC50 of 30 nM and a maximal current of 48±5% (n=10) of the response to 100 μM GABA. In contrast, no measurable response was observed from the wild-type GABAA receptor when exposed to DZ concentrations as high as 10 μM. The high potency of GABA at the α1L263S mutant receptor raises the concern that the apparent direct activation by BZDs could instead reflect potentiation of trace amounts of contaminating GABA present in the medium. To test this possibility, we examined the effect of the competitive inhibitor gabazine. In the absence of GABA or DZ, gabazine alone produced a small (13±3%) decrease in the spontaneous picrotoxin-sensitive current, consistent with a previous report that gabazine has weak inverse agonist activity at the GABA recognition site (Chang & Weiss, 1999). As shown in Figure 8, gabazine completely blocked activation of the α1L263S mutant receptor by 30 nM GABA (which produces a current similar to that evoked by 1 μM DZ), while inhibiting the response to 1 μM DZ by only 12±4%, which was not significantly different (P=0.98, unpaired two-tail t-test, n=6) from the gabazine-induced inhibition of the picrotoxin-sensitive spontaneous current. Therefore, activation of the α1L263S mutant receptor by DZ is not due to traces of GABA.
Figure 7.

Diazepam modulation of GABA-induced current of wild-type receptor and spontaneous current of mutant receptor is consistent with an allosteric model. (a) Response of α1L263S receptor to 1 μM DZ alone. (b) Concentration–response dependence of activation of the wild-type and α1L263S mutant receptors by GABA and diazepam. Response of wild-type receptor to GABA alone and to GABA in the presence of 1 μM DZ is shown, along with the response of α1L263S mutant GABAA receptors to DZ alone and GABA alone. Error bars indicate standard error (n⩾5). Smooth curves are calculated from Equation (1) based upon simultaneous fits of the entire data set to Equation (1). Parameters are given in Table 1. (c) Dose–response surfaces for GABA and DZ acting at wild-type or α1L263S mutant GABAA receptors. Surfaces were calculated from Equation (1) using the parameters in Table 1.
Figure 8.

DZ-induced current is resistant to gabazine. Gabazine (GZ; 10 μM) completely blocked the response of HEK293 cells expressing αL263Sβ3γ2 subunits to 0.03 μM GABA (G), but failed to block the response to 1 μM DZ alone. Gabazine alone slightly inhibited the spontaneous current of the mutant receptor. Responses are scaled to full range of activation (i.e. the sum of the picrotoxin-sensitive current and the maximal GABA current), with the abscissa set at the level of the spontaneous (picrotoxin-sensitive) current. Bars show mean normalized current±s.e.m (n=6). *Significantly different from GABA alone (P<0.03, unpaired two-tail t-test). †Significantly different from DZ alone (P<0.03, unpaired two-tail t-test). #Significant decrease in holding current (P<0.001 one sample two-tail t-test).
To determine whether the allosteric model can quantitatively account for the effects of DZ, GABA concentration–response data on wild-type and α1L263S mutant receptors in the presence and absence of DZ and concentration–response data for DZ alone on the α1L263S mutant were fitted simultaneously with Equation (1). As shown in Figure 7b, the allosteric model was able to fit both the direct activation of the α1L263S mutant receptor by DZ and the modulatory effect of DZ at the wild-type receptor, yielding an estimate of dDZ=0.35 (95% confidence limits 0.24–0.48), which is similar to the value of 0.29 determined for DZ potentiation of kojic amine efficacy with chick spinal cord neurons. The fit also yielded an estimate for the wild-type α1β3γ2 isomerization equilibrium constant of Lwt=3304. Sensitivity analysis indicates that this value is fairly well defined by the data set, with 95% confidence limits of 1452–5398. The GABA potency ratio was c=K′GABA/KGABA=0.0027 (95% confidence limits 0.001–0.004).
Direct activation of the α1L263S mutant receptor was also observed with the BZD flurazepam (FZ) (Figure 9a). Direct activation of the α1L263S mutant receptor by FZ was inhibited by flumazenil (P=0.0006, unpaired two-tail t-test, n=4), indicating that direct activation by flurazepam is mediated by the BZD recognition site. Flumazenil alone weakly but significantly activated the α1L263S mutant receptor (P<0.003, two-tail one-sample t-test, n=8), in agreement with a previous report that flumazenil is a very low-efficacy positive modulator (Chan & Farb, 1985). Flurazepam and GABA concentration–response data were generally well fit by the allosteric model, except that the model consistently overestimated maximal direct activation of the α1L263S mutant receptor by FZ. Notably, the current elicited by 100 μM FZ was consistently lower than that elicited by 10 μM FZ, suggesting that high concentrations of FZ may exert a secondary inhibitory effect. For this reason, data at the highest concentration (100 μM) of FZ was excluded from the fit. Fitted parameters for Lwt, KGABA, and K′GABA were similar to those obtained with DZ, with dFZ=0.18 (Table 1).
Figure 9.

Modulation by FZ and zolpidem of GABA-induced current of wild-type receptor and spontaneous current of mutant receptor and is consistent with an allosteric model. (a) Response of wild-type receptor to GABA alone and to GABA in the presence of 1 μM FZ is shown, along with the response of α1L263S mutant GABAA receptors to FZ alone and GABA alone. Error bars indicate standard error (n⩾4). Smooth curves are calculated based upon simultaneous fits of the entire dataset (except for the highest concentration of FZ alone, data point in parentheses) to Equation (1). Parameters are given in Table 1. (b) FZ-induced activation is inhibited by the BZD antagonist flumazenil (flum). *Significantly less than FZ alone (P=0.0006); #Significantly greater than 0 (P<0.003). (c) Response of wild-type receptor to GABA alone and to GABA in the presence of 1 μM zolpidem (Zol) is shown, along with the response of α1L263S mutant GABAA receptors to zolpidem alone and GABA alone. Smooth curves are calculated based upon simultaneous fits of the entire dataset to Equation (1). Parameters are given in Table 1. Error bars indicate standard error (n⩾5). Results for GABA alone are the same as in Figure 7, and are repeated for comparison.
Table 1.
Fitted model parameters
| Model parameter | Diazepam | Flurazepam | Zolpidem |
|---|---|---|---|
| K′GABA | 1.14 × 10−7 | 1.22 × 10−7 | 2.78 × 10−7 |
| KGABA | 4.22 × 10−5 | 2.90 × 10−5 | 7.94 × 10−5 |
| c | 0.0027 | 0.0042 | 0.0035 |
| K′MOD | 1.73 × 10−8 | 2.88 × 10−6 | 1.82 × 10−7 |
| KMOD | 5.01 × 10−8 | 1.61 × 10−5 | 7.55 × 10−7 |
| d | 0.3453 | 0.1789 | 0.2412 |
| Lwt | 3304 | 2260 | 2764 |
| Lmut | 1.01a | 1.01a | 1.01a |
Parameters were estimated by simultaneous nonlinear least-squares regression of dose–response data for GABA alone (wild-type and mutant receptors), the indicated modulator alone (mutant receptor), and GABA in the presence of the indicated modulator (wild-type receptor). Lmut was calculated from the ratio of the GABA-dependent current to the picrotoxin-sensitive current, and was held constant in the fit.
Value fixed based upon experimental measurements of spontaneous current and maximum GABA-induced current.
We also examined the effects of zolpidem, an imidazopyridine that is believed to act through the BZD recognition site. Like DZ and FZ, zolpidem directly activated the α1L263S receptor (Figure 9c). Global fitting yielded dZol=0.24.
Discussion
Two models have been proposed to account for the modulatory effects of BZDs on the GABAA receptor. The idea that GABAA receptor modulation could be understood in terms of an allosteric model was first suggested by Gee (1988). In this model, the modulatory effect of BZDs is primarily an allosteric gating effect due to stabilization of the active state of the receptor relative to the inactive state. Alternatively, it has been proposed based upon single-channel kinetic studies that BZDs act by increasing the association rate constant for binding of GABA to one of the two agonist recognition sites of the GABAA receptor (Rogers et al., 1994).
Although both the allosteric gating model and the binding model are consistent with the known effects of BZDs on the wild-type GABA concentration–response curve, the two models make different predictions as to the effects of BZD-site modulators on GABAA receptor partial agonist efficacy and the effects of modulators on α1L263S mutant receptors in which there is a high level of basal activity. The allosteric gating model predicts that positive modulators should increase the efficacy of partial agonists. This is a strong test of allosteric gating models; if partial agonist efficacy is not affected by modulators, it would eliminate this model as well as any other gating model in which modulators act by stabilizing the open-channel conformation of the receptor. In contrast, the binding model predicts that these modulators will not affect partial agonist efficacy, because at saturation all agonist-binding sites are fully occupied, and the rate of agonist binding is therefore not limiting.
We tested the efficacy of a number of GABA analogs on primary chick spinal cord neurons. Kojic amine was selected for additional study because it induced a response that was clearly smaller than the maximum GABA response, with greater potency than taurine. The present results show that DZ increases the maximum response to the partial agonist kojic amine in chick spinal cord neurons, in agreement with the two-state model.
It has been reported by Kristiansen & Lambert (1996) that midazolam enhances the response of hippocampal neurons to 1 mM 5-(4-piperidyl)isoxazol-3-ol (4-PIOL), a weak partial agonist of the GABAA receptor. As 1 mM 4-PIOL has previously been reported to be a saturating concentration (Kristiansen et al., 1991), this result suggests that enhancement of partial agonist efficacy by BZDs is not unique to kojic amine or DZ. Kristiansen and Lambert also point out that this result is inconsistent with the idea that potentiation is solely due to enhancement of agonist binding affinity.
The allosteric gating model and the binding kinetics model also differ in their predictions as to the effects of BZDs in the absence of GABA. The binding kinetics model predicts that BZDs should not directly activate the GABAA receptor in the absence of agonist. The allosteric gating model predicts that BZDs alone should be able to activate the receptor, but that the extent of direct activation will be negligible for wild-type receptors with L≫1. However, measurable direct activation should be evident with a receptor for which L≈1. To test this prediction, we used a previously described GABAA receptor mutation in which L is reduced without affecting the GABA recognition site, resulting in a measurable basal current in the absence of GABA (Chang & Weiss, 1999). The extent of direct activation of α1L263S mutant receptors by DZ, FZ, and zolpidem is consistent with the magnitude of the shift of the wild-type receptor GABA concentration–response curve. Direct activation of spontaneously active α1β3γ2L(L245S) by DZ (Bianchi & Macdonald, 2001), and direct activation of spontaneously active α2(S270W)β1γ2S receptors by FZ (Findlay et al., 2000) have been reported previously. The present results show that the effect of BZDs on receptor gating is sufficient to account for enhancement of the GABA response of wild-type GABAA receptors.
The results indicate that the shift in the GABA concentration response curve by BZDs and the resulting enhancement of GABA response at low GABA concentrations can be attributed to stabilization of the active state relative to the inactive state, rather than acceleration of GABA binding. However, the results do not rule out BZD-induced alterations in GABA binding kinetics, which may contribute to the ability of BZDs to shape the kinetics of GABA-mediated synaptic responses (Vicini et al., 1986).
Single-channel studies indicate that GABAA receptor activation is kinetically complex, and characterized by multiple open and closed states (Newland et al., 1991; Rogers et al., 1994). For our analysis, we have used a simplified model consisting of a single open state and a single closed state, so it is unclear whether all open states or only certain ones exhibit elevated affinity for BZD-like positive modulators.
Enhancement of the GABA response and direct activation of α1β2γ2L receptors by the general anesthetic etomidate has also been shown to be consistent with a two-state allosteric model (Rusch et al., 2004), suggesting that this model of allosteric modulation may be a generally applicable for GABA receptor modulators.
HEK293 cells expressing mutant rat α1L263S subunits in conjunction with rat γ2 and an endogenous human β3 subunit exhibited approximately 50% spontaneous GABAA receptor activation, which is somewhat greater than the 18% spontaneous activity reported for α1(L263S)β2γ2 receptors expressed in Xenopus oocytes (Chang & Weiss, 1999). The difference could be due to the use of the endogenous HEK293 β3 subunit in our study, or to the different characteristics of the HEK293 and oocyte membranes.
Although modulation of the GABA and kojic amine dose–response curves by DZ in chick spinal cord neurons is consistent with the two-state model, the model parameters were underdetermined by the data, and it was not possible to obtain reliable estimates of the parameters L and c=K′GABA/KGABA (Equation 1). With α1(L263S)β3γ2 receptors, however, it was possible to measure Lmut directly, reducing the number of free parameters and permitting determination of Lwt and c. Simultaneous fitting of the control GABA concentration–response data for wild-type and α1(L263S) mutant receptors together with the wild-type GABA response data and the response of the α1(L263S) mutant to DZ alone yielded Lwt=3304. In comparison, Chang & Weiss (1999) estimated a wild-type isomerization equilibrium constant of Lwt=100,744 based upon analysis of GABA responses of wild-type and mutant α1β2γ2 receptors expressed in Xenopus oocytes. The difference could be due either to the different expression systems or to the different receptor subunit composition. The fit also yielded dissociation constants of K′GABA=0.11 μM for binding of GABA to the active state and KGABA=42 μM for binding of GABA to the inactive state. This compares well with the values of 0.12 and 79 μM determined by Chang & Weiss (1999). Based upon these results, the probability of channel opening for the wild-type receptor is (Lwt+1)−1=0.0003 in the absence of GABA and (Lwtc2+1)−1=0.98 at saturating GABA.
For DZ, the dissociation constants for binding are K′DZ=17 nM for the active state and KDZ=50 nM for the inactive state, yielding a potency ratio dDZ=K′DZ/KDZ=0.35. For the wild-type receptor, this yields a probability of channel opening of (LwtdDZ+1)−1=0.0009 for the wild-type receptor, consistent with the absence of detectable activation by DZ alone.
Activation of ligand-gated ion channels is presumed to involve a global change in receptor structure, which will likely shift the positions of many functional groups that could be involved in ligand binding. According to the two-state model, even a small preference in ligand binding to one state vs the other can result in significant modulatory effects. The effect of a ligand on the conformational equilibrium is dependent on the difference in the free energy of agonist binding to the active and inactive states, ΔΔGbinding=RT ln(d). ΔΔGbinding offers a useful method of ranking modulatory efficacy. Based on the present results, flurazepam has the greatest efficacy at α1β3γ2 receptors, followed by zolpidem and DZ (Figure 10). Notably, the value of dDZ=0.35 estimated for DZ corresponds to a ΔΔGbinding of 0.6 kcal per mol between the active and inactive states (Figure 10), which is comparable to the energy of a single hydrogen bond (Fersht, 1987). Thus, the modulatory effect of DZ could be explained if receptor activation allows the formation of one additional hydrogen bond between DZ and the receptor.
Figure 10.

Energetics of GABAA receptor modulation. Difference in energy of binding to the active and inactive states was calculated as ΔΔGbinding=RT ln(d), using the values in Table 1. Dashed line indicates the ΔΔGbinding required to activate 50% of wild-type or α1L263S mutant receptors.
By comparison, ΔΔGbinding for binding of a single GABA molecule is 3.4 kcal per mol, enabling two GABA molecules to produce nearly complete activation. The BZD-binding site is structurally homologous to the GABA recognition site, but is located at the αγ interface rather than at an αβ interface. However, the fact that no BZD-site ligand has been identified with the capacity to directly activate wild-type GABAA receptors suggests that activation-associated changes of the BZD-binding site are slight compared to the GABA recognition site.
Acknowledgments
We thank Dr Maria Gravielle for expert assistance with the Western blot and real-time PCR experiments. Financial support was provided by NINDS R01NS035700.
Abbreviations
- BZD
benzodiazepine
- DZ
diazepam
- FZ
flurazepam
- GABA
γ-aminobutyric acid
Appendix A
Assumptions
The receptor can exist in two conformational states, R and R′, with R′ designated as the active state.
Ligand affinity for a particular site is determined solely by the state of the receptor. Ligand Lj binds to state R with dissociation constant Kj and to state R′ with dissociation constant K′j.
Each ligand binds to a single site.
Derivation of state equationThe fraction of receptors in the active conformational state R′ is given by
where the square brackets denote concentration. If there are n ligands binding to n distinct sites, then for each conformational state there are 2n distinct binding states, since each ligand can be either bound or not bound to its respective site. Let Ljℜ represent an arbitrary binding state in which ligand Lj is bound to state R at site j (possibly along with other ligands bound at other sites), and let ℜ represent the binding state that is identical in terms of ligand binding, except that Lj is not bound (i.e. site j is empty). These are related at equilibrium by the equation
where the brackets denote concentration (strictly speaking, activity). The total concentration of receptors is thus given by
Likewise, for each binding state of R′,
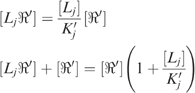 |
Binding to site 1 can thus be factored out as follows:
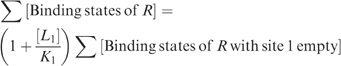 |
Similarly,
Factoring out binding to each successive site in this manner yields
Likewise, for R′,
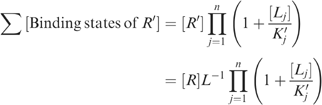 |
where [R] represents the concentration of receptors in state R with all sites empty, and L=([R][R′]−1)eq is the allosteric equilibrium constant for the unliganded receptor.
Substitution into Equation (2) yields
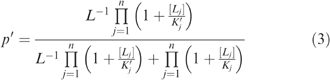 |
To model the GABAA receptor, sites 1 and 2 are assumed to be identical recognition sites for an agonist (A), and a third site is assumed to be a recognition site for a BZD-like modulator (B). For notational convenience, the following substitutions are made: α=[A]K′A−1, β=[B]K′B−1, c=K′AKA−1, d=K′BKB−1. Equation (3) then becomes
In the absence of agonist or modulator, this reduces to p′(α=0,β=0)=(1+L)−1, which is the basal activity in the absence of agonist, and approaches 0 for L≫1. The maximum response in the presence of a saturating concentration of A alone is given by
Similarly, the maximum response in the presence of a saturating concentration of B alone is
The direct effect of B alone will be negligible if L≫d−1, but will be substantial when L and d−1 are of similar magnitude. The maximum response in the presence of saturating concentrations of both A and B is
If c and d are both less than 1 (as must be the case for agonists and positive modulators), then p′(α=∞,β=∞)>p′(α=∞,β=0); that is a modulator will increase the maximum response to an agonist. With a high-efficacy agonist for which Lc2≪1, the magnitude of this effect is small (Figure 1b), but for a lower-efficacy modulator such that Lc2≈1, the enhancement of the maximum response can be substantial (Figure 1c).
References
- BIANCHI M.T., MACDONALD R.L. Agonist trapping by GABAA receptor channels. J. Neurosci. 2001;21:9083–9091. doi: 10.1523/JNEUROSCI.21-23-09083.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CELENTANO J.J., GIBBS T.T., FARB D.H. Negative modulation of the GABA response by extracellular zinc. Mol. Pharmacol. 1991;40:766–773. [PubMed] [Google Scholar]
- CHAN C.Y., FARB D.H. Modulation of neurotransmitter action: control of the γ-aminobutyric acid response through the benzodiazepine receptor. J. Neurosci. 1985;5:2365–2373. doi: 10.1523/JNEUROSCI.05-09-02365.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHANG Y., WEISS D.S. Allosteric activation mechanism of the α1β2γ2 γ-aminobutyric acid type A receptor revealed by mutation of the conserved M2 leucine. Biophys. J. 1999;77:2542–2551. doi: 10.1016/s0006-3495(99)77089-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOI D.W., FARB D.H., FISCHBACH G.D. Chlordiazepoxide selectively augments GABA action in spinal cord cell cultures. Nat. (London) 1977;269:342–344. doi: 10.1038/269342a0. [DOI] [PubMed] [Google Scholar]
- CHOI D.W., FARB D.H., FISCHBACH G.D. Chlordiazepoxide selectively potentiates GABA conductance of spinal cord and sensory neurons in cell culture. J. Neurophysiol. 1981;45:621–631. doi: 10.1152/jn.1981.45.4.621. [DOI] [PubMed] [Google Scholar]
- COLQUHOUN D.The relation between classical and cooperative models for drug action Drug Receptors 1978Baltimore: University Park Press; 149–182.ed. Rang, H.P. pp [Google Scholar]
- DAVIES P.A., HOFFMANN E.B., CARLISLE H.J., TYNDALE R.F., HALES T.G. The influence of an endogenous β3 subunit on recombinant GABAA receptor assembly and pharmacology in WSS-1 cells and transiently transfected HEK293 cells. Neuropharmacology. 2000;39:611–620. doi: 10.1016/s0028-3908(99)00163-x. [DOI] [PubMed] [Google Scholar]
- DRAGUHN A., VERDOORN T.A., EWERT M., SEEBURG P.H., SAKMANN B. Functional and molecular distinction between recombinant rat GABAA receptor subtypes by Zn2+ Neuron. 1990;5:781–788. doi: 10.1016/0896-6273(90)90337-f. [DOI] [PubMed] [Google Scholar]
- EBERT B., WAFFORD K.A., WHITING P.J., KROGSGAARD-LARSEN P., KEMP J.A. Molecular pharmacology of γ-aminobutyric acid type A receptor agonists and partial agonists in oocytes injected with different α, β, and γ receptor subunit combinations. Mol. Pharmacol. 1994;46:957–963. [PubMed] [Google Scholar]
- FERSHT A.R. The hydrogen bond in molecular recognition. Trends Biochem. Sci. 1987;12:301–304. [Google Scholar]
- FINDLAY G.S., UENO S., HARRISON N.L., HARRIS R.A. Allosteric modulation in spontaneously active mutant gamma-aminobutyric acid(A) receptors. Neurosci. Lett. 2000;293:155–158. doi: 10.1016/s0304-3940(00)01503-2. [DOI] [PubMed] [Google Scholar]
- GEE K.W. Steroid modulation of the GABA/benzodiazepine receptor-linked chloride ionophore. Mol. Neurobiol. 1988;2:291–317. doi: 10.1007/BF02935636. [DOI] [PubMed] [Google Scholar]
- GYENES M., FARRANT M., FARB D.H. ‘Run-down' of γ-aminobutyric acidA receptor function during whole-cell recording: a possible role for phosphorylation. Mol. Pharmacol. 1988;34:719–723. [PubMed] [Google Scholar]
- GYENES M., WANG Q., GIBBS T.T., FARB D.H. Phosphorylation factors control neurotransmitter and neuromodulator action at the γ-aminobutyric acid type A receptor. Mol. Pharmacol. 1994;46:542–549. [PubMed] [Google Scholar]
- HATTORI K., OOMURA Y., AKAIKE N. Diazepam action on γ-aminobutyric acid-activated chloride currents in internally perfused frog sensory neurons. Cell. Mol. Neurobiol. 1986;6:307–322. doi: 10.1007/BF00711116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOSIE A.M., DUNNE E.L., HARVEY R.J., SMART T.G. Zinc-mediated inhibition of GABAA receptors: discrete binding sites underlie subtype specificity. Nat. Neurosci. 2003;6:362–369. doi: 10.1038/nn1030. [DOI] [PubMed] [Google Scholar]
- JACKSON M.B. Perfection of a synaptic receptor: kinetics and energetics of the acetylcholine receptor. Proc. Natl. Acad. Sci. U.S.A. 1989;86:2199–2203. doi: 10.1073/pnas.86.7.2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- JOHNSON J.W., ASCHER P. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nat. (London) 1987;325:529–531. doi: 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- JURMAN M.E., BOLAND L.M., LIU Y., YELLEN G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques. 1994;17:876–881. [PubMed] [Google Scholar]
- KARLIN A. On the application of ‘a plausible model' of allosteric proteins to the receptor for acetylcholine. J. Theor. Biol. 1967;16:306–320. doi: 10.1016/0022-5193(67)90011-2. [DOI] [PubMed] [Google Scholar]
- KIRKNESS E.F., FRASER C.M. A strong promoter element is located between alternative exons of a gene encoding the human γ-aminobutyric acid-type A receptor β3 subunit (GABRB3) J. Biol. Chem. 1993;268:4420–4428. [PubMed] [Google Scholar]
- KLECKNER N.W., DINGLEDINE R. Requirement for glycine in activation of NMDA receptors expressed in Xenopus oocytes. Science (Washington, DC) 1988;241:835–837. doi: 10.1126/science.2841759. [DOI] [PubMed] [Google Scholar]
- KRISTIANSEN U., LAMBERT J.D. Benzodiazepine and barbiturate ligands modulate responses of cultured hippocampal neurones to the GABAA receptor partial agonist, 4-PIOL. Neuropharmacology. 1996;35:1181–1191. doi: 10.1016/s0028-3908(96)00070-6. [DOI] [PubMed] [Google Scholar]
- KRISTIANSEN U., LAMBERT J.D.C., FALCH E., KROGSGAARD-LARSEN P. Electrophysiological studies of the GABAA receptor ligand, 4-PIOL, on cultured hippocampal neurones. Br. J. Pharmacol. 1991;104:85–90. doi: 10.1111/j.1476-5381.1991.tb12389.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LEEB-LUNDBERG F., SNOWMAN A.M., OLSEN R.W. Barbiturate receptors are coupled to benzodiazepine receptors. Proc. Natl. Acad. Sci. U.S.A. 1980;77:7468–7472. doi: 10.1073/pnas.77.12.7468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MA J.Y., NARAHASHI T. Differential modulation of GABAA receptor-channel complex by polyvalent cations in rat dorsal root ganglion neurons. Brain Res. 1993;607:222–232. doi: 10.1016/0006-8993(93)91510-y. [DOI] [PubMed] [Google Scholar]
- MACDONALD R.L., ROGERS C.J., TWYMAN R.E. Barbiturate regulation of kinetic properties of the GABAA receptor channel of mouse spinal neurones. J. Physiol. (London) 1989;417:483–500. doi: 10.1113/jphysiol.1989.sp017814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MAJEWSKA M.D., DEMIRGÖREN S., SPIVAK C.E., LONDON E.D. The neurosteroid dehydroepiandrosterone sulfate is an allosteric antagonist of the GABAA receptor. Brain Res. 1990;526:143–146. doi: 10.1016/0006-8993(90)90261-9. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., HARRISON N.L., SCHWARTZ R.D., BARKER J.L., PAUL S.M. Steroid hormone metabolites are barbiturate-like modulators of the GABA response. Science (Washington, DC) 1986;232:1004–1007. doi: 10.1126/science.2422758. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., MIENVILLE J.-M., VICINI S. Neurosteroid pregnenolone sulfate antagonizes electrophysiological responses to GABA in neurons. Neurosci. Lett. 1988;90:279–284. doi: 10.1016/0304-3940(88)90202-9. [DOI] [PubMed] [Google Scholar]
- MAJEWSKA M.D., SCHWARTZ R.D. Pregnenolone-sulfate: an endogenous antagonist of the γ-aminobutyric acid receptor complex in brain. Brain Res. 1987;404:355–360. doi: 10.1016/0006-8993(87)91394-1. [DOI] [PubMed] [Google Scholar]
- MEHTA A.K., TICKU M.K. Benzodiazepine and beta-carboline interactions with GABAA receptor-gated chloride channels in mammalian cultured spinal cord neurons. J. Pharmacol. Exp. Therap. 1989;249:418–423. [PubMed] [Google Scholar]
- MILLER B., SARANTIS M., TRAYNELIS S.F., ATTWELL D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- MONOD J., WYMAN J., CHANGEUX J.-P. On the nature of allosteric transitions: a plausible model. J. Mol. Biol. 1965;12:88–118. doi: 10.1016/s0022-2836(65)80285-6. [DOI] [PubMed] [Google Scholar]
- MUNSON P.J., RODBARD D. LIGAND: a versatile computerized approach for characterization of ligand-binding systems. Anal. Biochem. 1980;107:220–239. doi: 10.1016/0003-2697(80)90515-1. [DOI] [PubMed] [Google Scholar]
- NEWLAND C.F., COLQUHOUN D., CULL-CANDY S.G. Single channels activated by high concentrations of GABA in superior cervical ganglion neurones of the rat. J. Physiol. (London) 1991;432:203–233. doi: 10.1113/jphysiol.1991.sp018382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PARK-CHUNG M., MALAYEV A., PURDY R.H., GIBBS T.T., FARB D.H. Sulfated and unsulfated steroids modulate γ-aminobutyric acidA receptor function through distinct sites. Brain Res. 1999;830:72–87. doi: 10.1016/s0006-8993(99)01381-5. [DOI] [PubMed] [Google Scholar]
- PARK-CHUNG M., WU F.-S., PURDY R.H., GIBBS T.T., FARB D.H. Distinct sites for positive and negative modulation of NMDA receptors by sulfated steroids. Soc. Neurosci. Abst. 1996;22:1280. [Google Scholar]
- ROCA D.J., FRIEDMAN L., SCHILLER G.D., ROZENBERG I., GIBBS T.T., FARB D.H. γ-Aminobutyric acidA receptor regulation in culture: altered allosteric interactions following prolonged exposure to benzodiazepines, barbiturates, and methylxanthines. Mol. Pharmacol. 1990;37:710–719. [PubMed] [Google Scholar]
- ROGERS C.J., TWYMAN R.E., MACDONALD R.L. Benzodiazepine and β-carboline regulation of single GABAA receptor channels of mouse spinal neurons in culture. J. Physiol. (London) 1994;475:1. doi: 10.1113/jphysiol.1994.sp020050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUSCH D., ZHONG H., FORMAN S.A. Gating allosterism at a single class of etomidate sites on αβ2γ2L GABAA receptors accounts for both direct activation and agonist modulation. J. Biol. Chem. 2004;279:20982–20992. doi: 10.1074/jbc.M400472200. [DOI] [PubMed] [Google Scholar]
- SMART T.G., CONSTANTI A. Differential effect of zinc on the vertebrate GABAA–receptor complex. Br. J. Pharmacol. 1990;99:643–654. doi: 10.1111/j.1476-5381.1990.tb12984.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SPROSEN T.S., WOODRUFF G.N. Polyamines potentiate NMDA induced whole-cell currents in cultured striatal neurons. Eur. J. Pharmacol. 1990;179:477–478. doi: 10.1016/0014-2999(90)90193-a. [DOI] [PubMed] [Google Scholar]
- THRON C.D. On the analysis of pharmacological experiments in terms of an allosteric receptor model. Mol. Pharmacol. 1973;9:1–9. [PubMed] [Google Scholar]
- TURNER D.M., RANSOM R.W., YANG J.S.-J., OLSEN R.W. Steroid anesthetics and naturally occurring analogs modulate the γ-aminobutyric acid receptor complex at a site distinct from barbiturates. J. Pharmacol. Exp. Ther. 1989;248:960–966. [PubMed] [Google Scholar]
- VICINI S., ALHO H., COSTA E., MIENVILLE J.-M., SANTI M.R., VACCARINO F.M. Modulation of γ-aminobutyric acid-mediated inhibitory synaptic currents in dissociated cortical cell cultures. Proc. Natl. Acad. Sci. U.S.A. 1986;83:9269–9273. doi: 10.1073/pnas.83.23.9269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WOOD P.L. The co-agonist concept: is the NMDA-associated glycine receptor saturated in vivo. Life Sci. 1995;57:301–310. doi: 10.1016/0024-3205(95)00288-h. [DOI] [PubMed] [Google Scholar]
- WU F.-S., GIBBS T.T., FARB D.H. Inverse modulation of γ-aminobutyric acid- and glycine-induced currents by progesterone. Mol. Pharmacol. 1990;37:597–602. [PubMed] [Google Scholar]
- WU F.-S., GIBBS T.T., FARB D.H. Pregnenolone sulfate: a positive allosteric modulator at the NMDA receptor. Mol. Pharmacol. 1991;40:333–336. [PubMed] [Google Scholar]