

Abstract
Bile acid synthesis and pool size increases in diabetes, while insulin inhibits bile acid synthesis. The objective of this study is to elucidate the mechanism of insulin regulation of cholesterol 7α-hydroxylase gene expression in human hepatocytes. Real-time PCR assays showed that physiological concentrations of insulin rapidly stimulated CYP7A1 mRNA expression in primary human hepatocytes, but inhibited CYP7A1 expression after extended treatment. The insulin-regulated forkhead box O1 (FoxO1) and steroid regulatory element-binding protein-1c (SREBP-1c) strongly inhibited hepatocyte nuclear factor 4α and peroxisome proliferator-activated receptor γ coactivator-1α transactivation of the CYP7A1 gene.FoxO1 binds to an insulin response element in the rat CYP7A1 promoter, which is not present in the human CYP7A1 gene. Insulin rapidly phosphorylates and inactivates FoxO1, while insulin induces nuclear SREBP-1c expression in human primary hepatocytes. Chromatin immunoprecipitation assay shows that insulin reduced FoxO1 and PGC-1α, but increased SREBP-1c recruitment to CYP7A1 chromatin. We conclude that insulin has dual effects on human CYP7A1 gene transcription: physiological concentrations of insulin rapidly inhibit FoxO1 activity leading to stimulation of the human CYP7A1 gene, whereas prolonged insulin treatment induces SREBP-1c, which inhibits human CYP7A1 gene transcription. Insulin may play a major role in the regulation of bile acid synthesis and dyslipidemia in diabetes.
The liver plays a central role in lipid metabolism and maintaining whole body lipid homeostasis, which is dysregulated in metabolic syndrome (Syndrome X), obesity and diabetes (1). Bile acid synthesis in the liver is the predominant pathway for cholesterol catabolism and is regulated by cholesterol 7α-hydroxylase (CYP7A1) (2). Bile acids are physiological agents that facilitate biliary cholesterol excretion, intestinal absorption of nutrients, and disposal of toxic metabolites. Bile acids are also signaling molecules that activate bile acid receptors to regulate bile acid synthesis and glucose metabolism (2). In vivo studies show that bile acid pool and excretion increase in diabetic human patients (3,4) and in experimental diabetic animals (5,6). Insulin treatment restores bile acid pool and synthesis to the normal levels. It has been reported that physiological concentrations of insulin (1.4 to 14 nM) inhibit bile acid synthesis by down regulation of CYP7A1 (7–9), sterol 12α -hydroxylase (CYP8B1) (10), and sterol 27-hydroxylase (CYP27A1) gene transcription (9). We have previously reported that insulin plays a dominant role in the inhibition of CYP7A1 gene transcription (7,8). How insulin regulates human CYP7A1 and what factors mediate the insulin effects remain unknown.
Insulin is known to induce over 100 genes but inhibit only a few hepatic genes involved in glucose metabolism (11). The consensus sequence of negative insulin response element (IRE), T(G/A)TTT(T/G)(G/T), has been identified in promoters of phosphoenolpyruvate carboxykinase (PEPCK), glucose 6-phosphatase (G6Pase), insulin-like growth factor binding protein-1 (IGFBP-1), tyrosine aminotransferase (TAT) and apolipoprotein CIII (ApoCIII) genes for a long time (12). These IREs are known to bind hepatocyte nuclear factor 3α (HNF3α or FoxA1), but HNF3α does not mediate insulin action (12). Recently forkhead box O1 (FoxO1), a mammalian homolog of Caenorhabditis elegans DAF-16 and a winged/helix forkhead transcription factor, has been identified as an insulin-regulated transcription factor that plays a critical role in mediating insulin inhibition of the PEPCK, G6Pase, IGFBP-1 and ApoCIII genes (13–17). Akt/protein kinase B, a downstream serine kinase of the insulin-signaling pathway, phosphorylates FoxO1 (18). The phosphorylated-FoxO1 is excluded from the nucleus and degraded by the ubiquitin proteosome pathway, which results in inhibition of FoxO1 target genes (19,20). FoxO1 exerts a positive effect by binding to its target gene promoters (15). FoxO1 also regulates genes as a DNA-binding independent co-activator or co-repressor of many transcription factors (21). Dysregulation of FoxO1 expression and function leads to hyperglycemia and hypertriglyceridemia in the mouse models of Type I and Type II diabetes (17).
Insulin stimulates the activity of sterol regulatory element-binding proteins (SREBPs), which are key regulators of lipid homeostasis and insulin actions (22). The predominant SREBP isoform, SREBP-1c in liver and adipose tissues preferentially induces genes in fatty acid and triglyceride synthesis (23). A SREBP precursor (125 kDa) forms a complex with SREBP cleavage activating protein (SCAP) in the endoplasmic reticulum. Insulin inducing gene-1 and -2a (Insig) anchor the SREBP/SCAP complex to the endoplasmic reticulum membrane (24). When cellular sterol levels are low, Insigs release the SCAP/SREBP complex and allow SCAP to escort SREBPs to the Golgi apparatus. Two sterol-regulated proteases act sequentially to release the N-terminal basic-helix-loop-helix leucine zipper domain. The mature SREBP (68 kDa) enters the nucleus, binds to the sterol regulatory element (SRE) and induces target gene transcription. Insulin increases SREBP-1c mRNA expression via induction of liver orphan receptor α (LXRα) (25,26). Insulin also promotes SREBP-1c cleavage and increases insulin-stimulated lipogenesis by inhibition of Insig-2a expression in livers (11,24). Studies have shown that insulin-dependent proteolytic cleavage is crucial and sufficient for SREBP-1c activation, whereas LXRα induces SREBP-1c, but not SREBP-2 gene transcription (27).
In this study, we investigated the insulin regulation of CYP7A1 gene transcription in human hepatocytes. Results indicate that short-term treatment of physiological concentrations of insulin rapidly induces while prolonged treatment of insulin represses human CYP7A1 gene expression. This dual effect of insulin action may be mediated by two insulin sensitive transcription factors, FoxO1 and SREBP-1c. This study provides new insights into how bile acid synthesis is regulated by insulin under normal physiology and how dysregulation of bile acid homeostasis may contribute to glucose and lipid abnormalities in diabetes.
EXPERIMENTAL PROCEDURES
Cell Culture
The human hepatoblastoma cell line, HepG2, was purchased from American Type Culture Collection (Manassas, VA). The cells were cultured in Dulbecco’s modified Eagle medium and F-12 (Sigma, St. Louis, MO) supplemented with 100U/ml penicillin G/streptomycin sulfate (Mediatech, Herndon, VA) and 10% (v/v) heat inactivated fetal bovine serum (Irvine Scientific, Santa Ana, CA). Primary human hepatocytes: HH1247 (3 y, M), HH1248 (42 y, F), HH1249 (59 y, M), HH1251 (29 y, M), HH1274 (55 y, F), HH1281 (16 y, M), HH1286 (50 y, M), and HH1308 (64 y, M) were isolated from human donors and were obtained through the Liver Tissue Procurement and Distribution System of National Institutes of Health (S. Strom, University of Pittsburgh, Pittsburgh, PA). Cells were maintained in Hepatocyte Maintenance Medium supplemented with 10−7 M of insulin and dexamethasone (Cambrex, NJ).
Reporters and expression plasmids
Rat and human CYP7A1/luciferase (Luc) reporter were constructed as previously described (Crestani, 1995; Wang, 1996). Expression plasmid for human peroxisome proliferator-activated receptor γ coactivator-1α (P G C - 1 α) (pcDNA3/HA-PGC-1α) was obtained from A. Kralli (The Scripps Research Institute, La Jolla, CA). Expression plasmid for pCMV-rHNF3α was kindly provided by Robert H. Costa (University of Illinois at Chicago, IL). pCMV5-FoxO1 and pBluescript-FoxO1 were kindly provided by D. Accili (Columbia University, NY). SREBP-1c expression plasmid (pCMV-SREBP-1c-436) was purchased from ATCC. pCMX-hLXRα and pCMX-RXRα were provided by D. Mangelsdorf (UT Southwestern) and R. Evans (The Salk Institute for Biological Studies, La Jolla, CA), respectively. The construction of the expression plasmid for HNF4α (pCMV-HNF4α) was previously described (8). The β-galactosidase expression plasmid (pCMV-β-gal) and the mammalian expression vector pcDNA3 were obtained from Clontech (Palo Alto, CA). The reporter vector (pGL3-Basic) was purchased from Promega (Madison, WI). The PEPCK reporter plasmid, p2000Luc was obtained from R. Hanson (Case Western Reserve University). The synthetic reporter 5XUAS-TK-Luc, which contains 5 copies of Gal4 binding site UAS located upstream of the thymidine kinase promoter and luciferase gne, was provided by A. Takeshita (Toranomon Hospital, Tokyo, Japan) (28). Gal4-HNF4α fusion plasmid pBx-HNF4-LBD was obtained from I. Talianidis (Institute of Molecular Biology and Biotechnology Foundation for Research and Technology, Hellas, Herakleion Crete, Greece).
Transient transfection assay
HepG2 cells were grown to approximately 80% confluence in 24-well tissue culture plates. Luciferase reporters and expression plasmids were transfected using LipofectAMINE 2000 reagent (Life Technologies, Inc., Gaithersbrug, MD) following manufacturer’s instructions. The pcDNA3 empty vector was added to normalize the amounts of DNA transfected in each assay. Luciferase and β-galactosidase activities were assayed and expressed in relative luciferase units as described previously (28).
RNA isolation and quantitative real-time PCR
Primary human hepatocytes were cultured in serum-free and insulin-free media for 24 h and treated with insulin (Sigma, St. Louis, MO) as indicated. RNA isolation, reverse transcription reactions and real time PCR were performed as described previously(29). All TaqMan Probes: CYP7A1 (HS00167982), FoxO1 (HS00231106), SREBP-1 (HS00231674), HNF4α (HS00230853) and UBC (HS00824723) were ordered from “Gene Expession Assays” (Applied Biosystems Inc., Foster City, CA). PEPCK probe was custom made from “Custom Gene Expression Assays” (Applied Biosystems, Inc.). Ubiquitin C (UBC) was used as an internal control for all PCR amplification reactions. Relative mRNA expression was quantified using the comparative Ct (ΔCt) method and expressed as 2 −ΔΔCt. Each assay was done in triplicate and expressed as mean ± standard deviation.
Electrophoretic mobility shift assay (EMSA)
FoxO1 and FoxA1 were synthesized in vitro using the transcription/translation (TNT) system programmed with the FoxO1 and FoxA1 expression plasmids according to the manufacturer’s instruction (Promega, Madison, WI). [α32P] dCTP (3000 Ci/mol) was obtained from Perkin Elmer Life Science Products (Boston, MA). Synthetic oligonucleotides of complimentary strands were labeled with [α-32P] dCTP and incubated with in vitro translated proteins (5 μl) as described previously (28). Gels were dried and autoradiographed using a Phosphor Imager 445Si (Molecular Dynamics, Sunnyvale, CA). Sequences of the probes (tagged with GATC at 5’ end for labeling) used in the experiments are listed in Supplemental Data. Oligonucleotides were synthesized by MWG Biotech (High Point, NC).
Site directed mutagenesis
A PCR-based QuickChange Site-directed Mutagenesis kit (Stratagene, La Jolla, CA) was used for mutation of the reporter constructs: mutant rIRE2 sequence was introduced into rat p-344/luc plasmid, and HNF4α binding site mutation was introduced into ph-1887/Luc plasmid as described previously (28).
Chromatin immunoprecipitation (ChIP) assay
Human primary hepatocytes were obtained in T75 tissue culture flasks. HepG2 cells were over-expressed with HA-PGC-1α and cultured in serum-free and insulin-free media for 24 h. ChIP assays were performed as described previously (28). Antibodies against HNF4α (sc-8987), SREBP-1 (sc-366), and HA-tag (sc-805, Lot#K0303) were purchased from Santa Cruz Biotechnology (Santa Cruz, CA), and FoxO1 (FKHR, #9462) from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies were used to immuno-precipitate chromatin. Non-immuno IgG (Santa Cruz) was used as negative controls for IP. A 391-bp fragment containing the HNF4α binding site was amplified by PCR and analyzed on a 1.5% agarose gel.
Isolation of cell nuclei
Approximately 107 cells were washed with cold 1X PBS for three times. Cells were collected by centrifugation at 4 °C and re-suspended in 1 ml Buffer A (10 mM Tris-Cl, pH 7.5; 10 mM NaCl; 3 mM MgCl2) containing protease inhibitors (Sigma, St. Louis, MO) and incubated on ice for 10 min. 100 μl of 10% NP40 was then added and cells were incubated for another 10 min on ice. Cells were lysed by passing through a 22-gauge needle 20 times. Cell lysates were then centrifuged at 3000 rpm at 4 °C for 10 min to precipitate the nuclei. Nuclei were then washed 3 times in ice cold buffer A and lysed in SDS lysis buffer for immunoblotting analysis.
Immunoblotting analysis
Primary human hepatocytes were treated with 10 nM insulin as described above. Total cell lysates or nuclei fractions were analyzed by SDS-polyacrylamide gel electrophoresis. An antibody against Phospho-FoxO1 (Ser-256) (#9461) (Cell Signaling Technology, Inc.), and antibodies against FoxO1 (FKHR, sc-9462), HNF4α (sc-8987) and Actin (sc-1615) (Santa Cruz Biotechnology) were used for western blotting and detected by ECL Western blotting detection kit (Amersham Biosciences, UK).
RESULTS
Effect of Insulin on CYP7A1 and transcription factor mRNA expression in human primary hepatocytes
We first studied the insulin effect on CYP7A1 mRNA expression in human primary hepatoctyes using quantitative real time PCR (Q-PCR). Fig. 1A shows that increasing doses of insulin (0.1 nM to 100 nM) rapidly induced CYP7A1 mRNA levels in 2 h, with a maximum induction of ~22-fold at the physiological concentration of 10 nM in this donor liver. Higher concentrations of insulin had less stimulatory effect on CYP7A1 mRNA expression. In contrast, insulin treatment for 6 h dose-dependently inhibited CYP7A1 mRNA by ~ 40% at 100 nM (Fig.1B). As a positive control, insulin strongly reduced PEPCK mRNA expression in primary hepatocytes at both 2 and 6 h (Fig. 1A&B). To further study the insulin effect on human CYP7A1 mRNA expression, we treated primary hepatocytes with insulin (10 nM) for a period of time from 2 to 24 h. Q-PCR analysis of primary hepatocytes from five donors showed that insulin induced CYP7A1 mRNA expression levels at 2 h by an average of ~9-fold, and inhibited CYP7A1 mRNA at 6 h and 24 h by ~36% and ~39%, respectively (Table 1, Supplemental data). As a positive controls, insulin strongly inhibited PEPCK mRNA in a time-dependent manner.
Fig. 1.
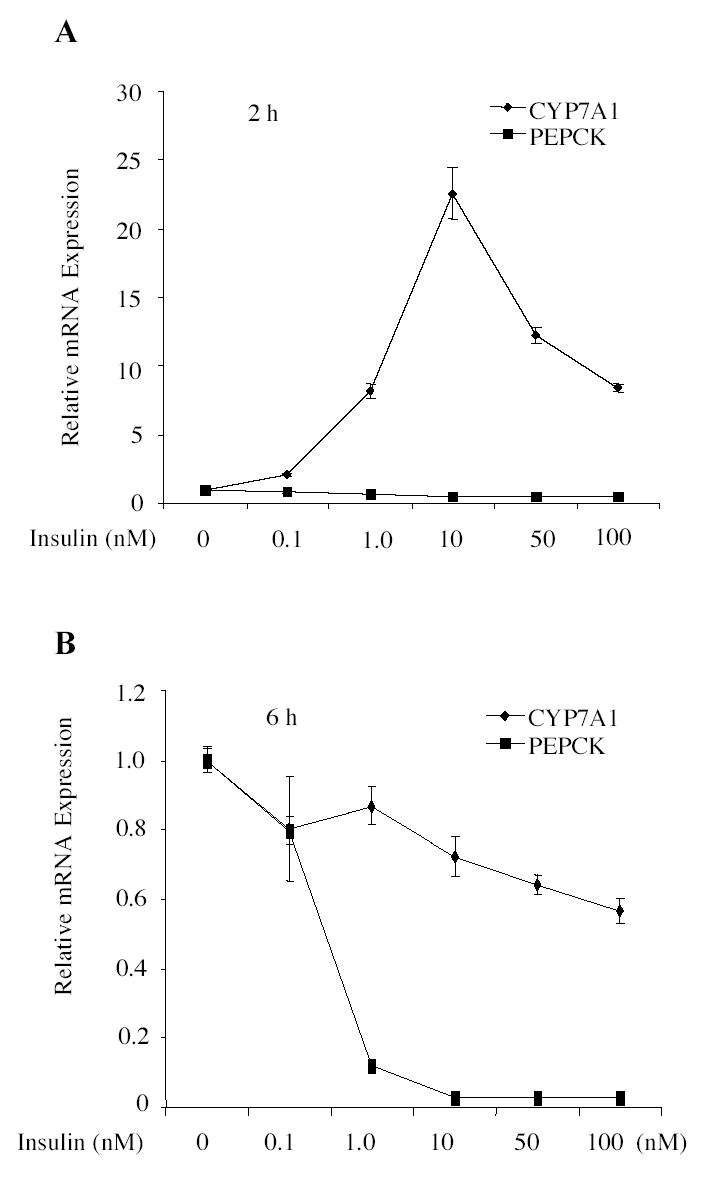
Real time PCR assays of insulin effect on CYP7A1 and PEPCK mRNA expressions in human primary hepatocytes. Primary human hepatocytes (HH 1251) were treated with increasing concentrations of insulin for 2 h (A) or 6 h (B). CYP7A1 or PEPCK mRNA expression was assayed in triplicates and expressed as mean ± standard deviation.
Table 1.
Q-PCR analysis of insulin Effects on CYP7A1, PEPCK, HNF4α, SREBP-1c and FoxO1 mRNA expressions in primary human hepatocytes
| Gene | Hepatocytes | 0h | 2h | 6h | 24h |
|---|---|---|---|---|---|
| CYP7A1 | HH1247 | 1±0.197 | 6.27±0.713 | 0.6±0.1 | 0.59±0.15 |
| HH1248 | 1±0.026 | 2.73±0.173 | 0.92±0.047 | 0.75±0.046 | |
| HH1249 | 1±0.049 | 2.15±0.062 | 0.22±0.016 | 0.49±0.013 | |
| HH1251 | 1±0.125 | 22.5±1.79 | 0.72±0.058 | 0.47±0.012 | |
| HH1274 | 1±0.066 | 11.58±0.95 | 0.73±0.047 | 0.77±0.024 | |
| MEAN | 1 | 9.05±8.41* | 0.64±0.26* | 0.61±0.14* | |
| PEPCK | HH1247 | 1±0.057 | 0.32±0.014 | 0.046±0.01 | 0.2±0.012 |
| HH1248 | 1±0.03 | 0.55±0.035 | 0.11±0.015 | 0.26±0.016 | |
| HH1249 | 1±0.09 | 0.12±0.005 | 0.009±0.0003 | 0.047±0.003 | |
| HH1251 | 1±0.047 | 0.52±0.031 | 0.028±0.002 | 0.045±0.013 | |
| HH1274 | 1±0.054 | 0.49±0.02 | 0.034±0.001 | 0.134±0.007 | |
| MEAN | 1 | 0.4±0.18* | 0.045±0.04* | 0.137±0.094* | |
| HNF4α | HH1247 | 1±0.064 | 1.63±0.164 | 1.04±0.053 | 1.03±0.056 |
| HH1248 | 1±0.056 | 1.24±0.054 | 0.83±0.085 | 0.98±0.068 | |
| HH1249 | 1±0.095 | 1.5±0.117 | 0.58±0.042 | 0.77±0.064 | |
| HH1251 | 1±0.059 | 1.17±0.097 | 0.67±0.032 | 0.98±0.004 | |
| HH1274 | 1±0.069 | 0.99±0.036 | 0.7±0.08 | 0.8±0.024 | |
| MEAN | 1 | 1.31±0.26* | 0.76±0.18* | 0.91±0.12 | |
| SREBP-1 | HH1247 | 1±0.057 | 2.25±0.079 | 1.76±0.06 | 1.73±0.082 |
| HH1248 | 1±0.032 | 1.8±0.073 | 1.68±0.176 | 1.53±0.104 | |
| HH1249 | 1±0.1 | 3.12±0.163 | 1.57±0.05 | 1.49±0.047 | |
| HH1251 | 1±0.046 | 2.44±0.12 | 2.28±0.246 | 2.17±0.012 | |
| HH1274 | 1±0.069 | 2.27±0.113 | 2.05±0.077 | 1.83±0.221 | |
| MEAN | 1 | 2.38±0.48* | 1.87±0.29* | 1.75±0.27* | |
| FoxO1 | HH1247 | 1±0.052 | 1.07±0.05 | 1.08±0.067 | 1.06±0.052 |
| HH1248 | 1±0.043 | 1.0±0.034 | 0.79±0.08 | 0.91±0.066 | |
| HH1249 | 1±0.083 | 1.28±0.079 | 0.64±0.046 | 1.22±0.074 | |
| HH1251 | 1±0.045 | 1.04±0.055 | 0.76±0.035 | 0.89±0.026 | |
| HH1274 | 1±0.055 | 0.82±0.046 | 0.94±0.033 | 1.36±0.008 | |
| MEAN | 1 | 1.04±0.16 | 0.84±0.17 | 1.09±0.20 |
In each hepatocyte preparation, assays were done in triplicates and expressed as mean± standard deviation. Mean values of five donors were pooled together and expressed as mean ± S.D. of the mean. Statistical analysis was performed using Paired t-test.
P< 0.05, insulin vs. control at 0h. Ct values at 0h in hepatocytes HH1247, HH1248, HH1249, HH1251 and HH1274, respectively, are: CYP7A1: 35.2, 34.9, 28, 35.8, 33.1; PEPCK: 24.7, 29.3, 22.9, 26.9, 25.1; HNF4α: 24.9, 26.3, 23.9, 25.2, 25.9; SREBP1: 27, 28, 28, 31, 30.1; Foxo1: 27, 26.9, 27.6, 26.4, 25.8.
We also measured the mRNA expression levels of HNF4α, the key regulator of human CYP7A1 gene transcription, and SREBP-1c and FoxO1, two transcription factors that are known to mediate the effects of insulin (Table 1, Supplemental data). Insulin significantly induced HNF4α mRNA levels at 2 h by ~ 30% but inhibited it at 6 h and 24 h. On the other hand, insulin induced SREBP-1c by about 2-fold, consistent with a previous report (30). Insulin had no effect on FoxO1 mRNA expression, consistent with previous studies that insulin regulates FoxO1 mainly at post-translational levels (15,18,19).
Identification of the insulin response elements (IREs) in the CYP7A1 gene
FoxO1 is known to bind to the consensus insulin response elements (IRE), T(G/A)TTT(T/G)(G/T) in the promoters of G6Pase, PEPCK, IGFBP-1 and TAT, and mediates the inhibitory effect of insulin on these genes (31). Analysis of the nucleotide sequences in the CYP7A1 promoter identified four putative IREs in the rat gene and two putative IREs in the human gene (Fig. 2A). Only the IRE located in the bile acid response element-I (−81 to −75) is conserved in rat and human CYP7A1 genes. We then performed EMSA to test if FoxO1 bound to these IREs in the CYP7A1 gene. Fig. 2B shows that FoxO1 bound strongly to the rat IRE2 (rIRE2), but did not bind to rIRE1, rIRE3, rIRE4, human IRE1 (hIRE1) and hIRE2 despite the similarity in nucleotide sequences (Fig. 2A). FoxO1 binding to rIRE2 was competed out by a large excess of un-labeled rIRE2 probe and a G6Pase probe that contained three FoxO1 binding sites (Fig 2C) (16). This rIRE2 also bound FoxA1 (HNF3α) as expected. A single mutation of G to T in the rIRE2 abolished Foxo1 binding (Fig. 2C). To test the effect of FoxO1 on rat CYP7A1 gene transcription, reporter assays were performed in HepG2 cells with a rat CYP7A1 promoter/luciferase construct. Co-transfection of FoxO1 expression plasmid stimulated rat CYP7A1 reporter activity by ~ 4-fold (Fig. 2D, left panel). When the single mutation that abolished FoxO1 binding was introduced into the rat CYP7A1 reporter, the basal reporter activity was reduced by more than 80%, and co-transfection of FoxO1 failed to stimulate the mutant reporter activity (Fig.2D, left panel). This mutant reporter did respond to HNF4α and LXRα because it contains the HNF4α and LXRα binding sites as in the wild type plasmid (Fig. 2D, right panel).
Fig. 2.
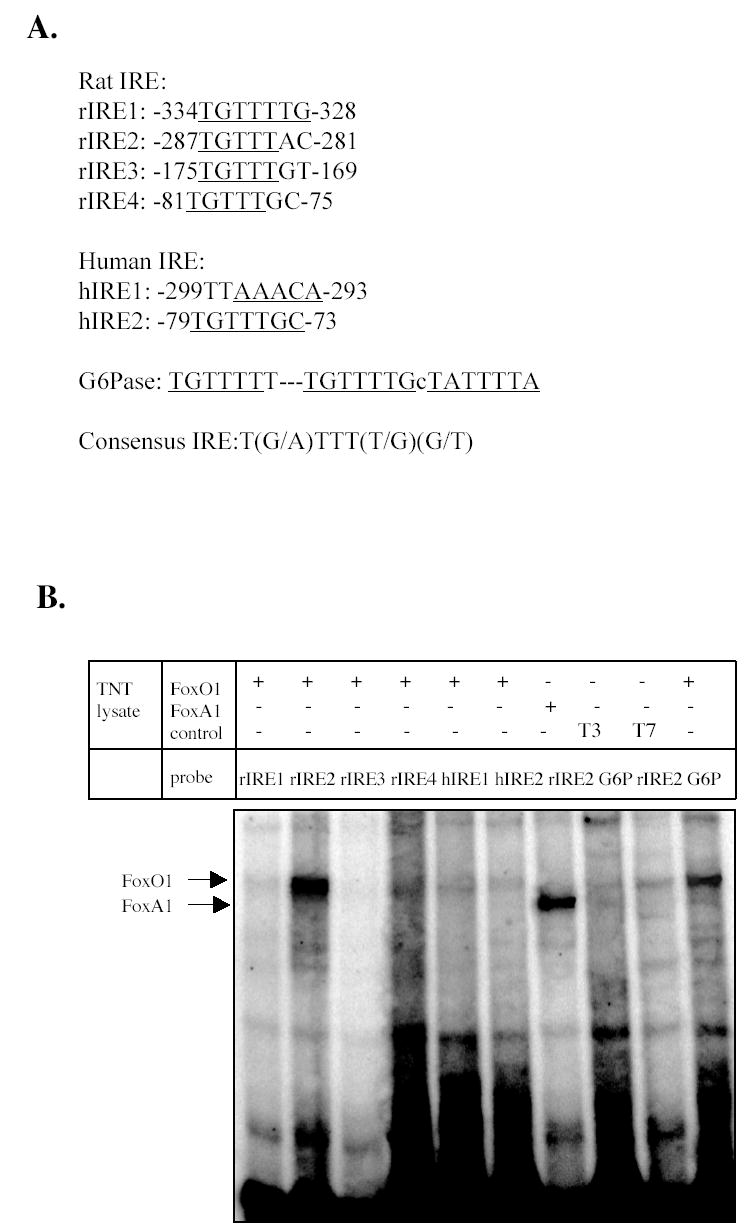

EMSA of FoxO1 and FoxA1 interaction with the putative rat and human IREs. A. Putative IREs in rat and human CYP7A1 promoter. B. EMSA of in vitro synthesized FoxO1 and FoxA1 binding to rat and human IRE probes. A G6Pase IRE probe was used as a positive control. Un-programmed TNT lysates (T3 and T7) were used as negative controls for non-specific bindings. Sequences of IRE probes used are described in Supplemental data. C. EMSA of in vitro synthesized FoxO1 and FoxA1 with 32P-labeled rat CYP7A1 IRE2 and mutant IRE2 probes. Competition assays were done with 100-fold excess of unlabeled probes. Mutation in rIRE2 probes is shown. D. Effects of FoxO1 on rat CYP7A1 reporter activities. WT: wild type rat CYP7A1 reporter (p-344/luc). IRE2 mutant: rat CYP7A1 reporter (p-344/luc) containing single nucleotide mutation in IRE2 sequence. Reporter (0.2 μg) and indicated expression plasmids (0.1 μg) were transfected into HepG2 cells. Transient transfection and luciferase assays were performed as described in Material and Methods. Mutation in rat IRE2 is shown. Statistical analysis was performed by student’s t-test, “*”, significant, P< 0.05.
FoxO1 inhibited human CYP7A1 reporter activity by blocking HNF4α trans-activation activity
The effect of FoxO1 on human CYP7A1 reporter activity was studied next using transfection assays. In contrast to the stimulatory effect on the rat CYP7A1 reporter, co-transfection of FoxO1 inhibited human CYP7A1 reporter activities of ph-1887/Luc, ph-371/Luc and ph-150/Luc by ~70 % (Fig.3A). Further deletion of the sequence downstream of –150 abolished the inhibitory effect of FoxO1. Thus, the inhibitory effect of FoxO1 required the region from –150 to −135, which was previously mapped as the bile acid response element-II that contained an HNF4α binding site (32). It has been reported that FoxO1 interacts with the DNA-binding domain of HNF4α and inhibits HNF4α trans-activation activity and its DNA binding (21). To test if the HNF4α binding site was involved in FoxO1 regulation of the human CYP7A1 gene, the HNF4α binding site in the ph-1887/Luc reporter was mutated. Fig. 3B shows that the HNF4α binding site mutant reporter (mHNF4α-ph-1887/luc) had a much lower basal activity and FoxO1 did not inhibit the mutant reporter activity. These results suggest that FoxO1 might inhibit human CYP7A1 gene by inhibiting HNF4α trans-activation activity. Mammalian one-hybrid assay was then used to study the effect of FoxO1 on H N F 4 α trans-activation of a Gal4 (5xUAS)/TK/Luc reporter. Fig. 3C shows that Gal4-HNF4α activation of the GAL4 reporter activity was drastically increased by PGC-1α, and FoxO1 dose-dependently inhibited reporter activity (Fig. 3C). These results suggest that in addition to interfering with HNF4α binding to DNA, FoxO1 might also block HNF4α and PGC-1α interaction. The effect of FoxO1 on HNF4α and PGC-1α co-activation of human CYP7A1 reporter activity (Fig 3D) was also studied. HNF4α or PGC-1α stimulated human CYP7A1 reporter activity by less than 100%, likely because of the abundant HNF4α expression in HepG2 cells. FoxO1 strongly inhibited the human CYP7A1 reporter activity stimulated by HNF4α and/or PGC-1α. Taken together, these results suggest that FoxO1 could act as a DNA-binding independent co-repressor of HNF4α and interfere with HNF4α and PGC-1α co-activation of CYP7A1 gene transcription.
Fig. 3.
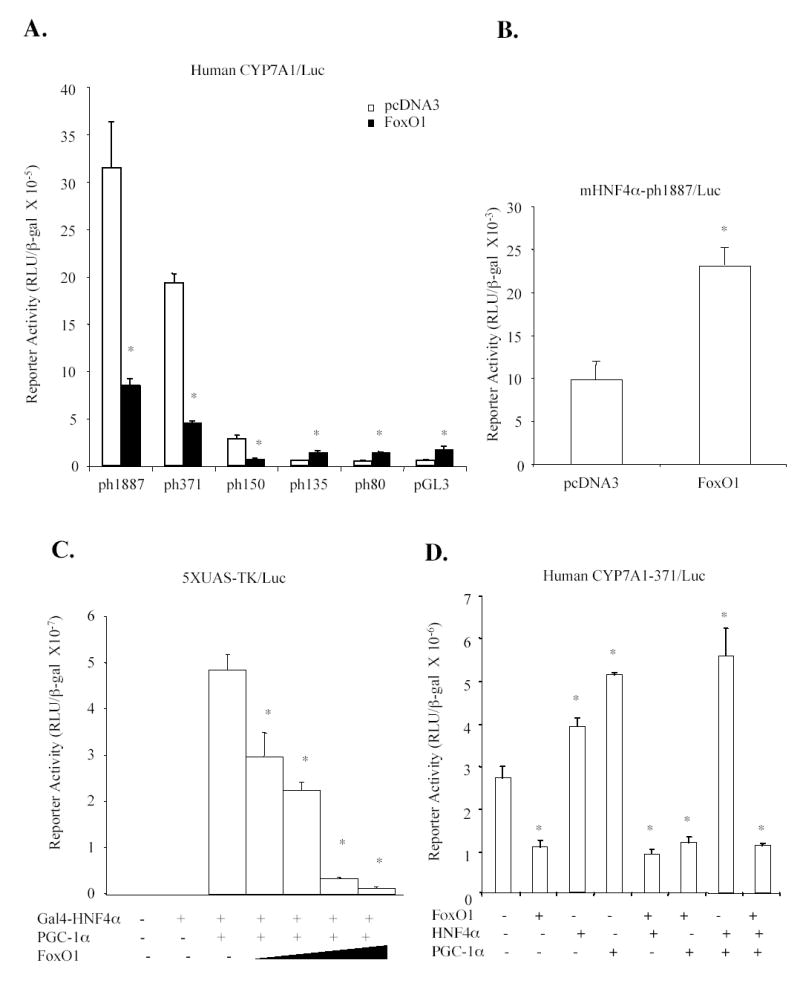
Effect of FoxO1 on human CYP7A1/Luc reporter activity. A. CYP7A1 reporter constructs (0.2 μg) were co-transfected with expression plasmids (0.1 μg) into HepG2 cells and luciferase activity was analyzed 40 h after transfection. B. HNF4α site mutant reporter (mHNF4-ph1887-Luc) was transfected in HepG2 cells. pcDNA3 empty vector or FoxO1 expression plasmid was co-transfected as indicated. C. Mammalian one-hybrid assay. The Gal4/luciferase reporter 5xUAS/TK/Luc (0.2μg) was co-transfected with Gal4 empty vector (−) (0.1μg), Gal4-HNF4α (0.1μg), PGC-1α (0.1μg) and increasing amounts of FoxO1expression plasmid (0.05-0.2μg) as indicated. D. A human CYP7A1 reporter, ph-371-Luc (0.2μg) was co-transfected with FoxO1, HNF4α, and/or PGC-1α (0.1 μg) as indicated into HepG2 cells. Each assay was performed in triplicates and statistical analysis was performed using student’s t-test, “*”, significant, P< 0.05.
SREBP-1c inhibited human CYP7A1 gene transcription by blocking HNF4α recruitment of PGC-1α
To test if SREBP-1c was involved in mediating insulin effect on human CYP7A1, the effect of SREBP-1c on human CYP7A1 reporters was studied by transfection assays in HepG2 cells. Co-transfection of the nuclear form of SREBP-1c strongly inhibited the human CYP7A1 reporter (ph-1887/Luc) by more than 80% (Fig 4A). Deletion analyses of the human CYP7A1 promoter localized a region from –150 to –135 that mediated the inhibitory effect of SREBP-1c (Fig 4A). We then tested the effect of SREBP-1c on the HNF4α binding site mutant reporter (mHNF4α-ph-1887/luc). As shown in Fig. 4B, mutation of the HNF4α binding site markedly reduced the basal reporter activity and SREBP-1c failed to inhibit the mutant reporter activity. These results suggest that SREBP-1c might inhibit human CYP7A1 by inhibiting HNF4α trans-activation of CYP7A1. SREBP-1c has been shown to interact with HNF4α-LBD and block its recruitment of PGC-1α to the PEPCK gene (33). Thus, mammalian one-hybrid assay was used to study the effect of SREBP-1c on PGC-1α and Gal4-HNF4α co-activation of a GAL4 5X UAS/TK/Luc reporter. Fig. 4C shows that SREBP-1c dose-dependently inhibited Gal4-HNF4α-LBD and PGC-1α co-activation of the reporter activity. Furthermore, SREBP-1c strongly inhibited the basal activity of a human CYP7A1 reporter activity and the activity stimulated by HNF4α and PGC-1α (Fig 4D). Thus, SREBP-1c could act as a DNA-binding independent co-repressor of HNF4α and interfere with HNF4α and PGC-1α co-activation of CYP7A1 gene transcription.
Fig. 4.
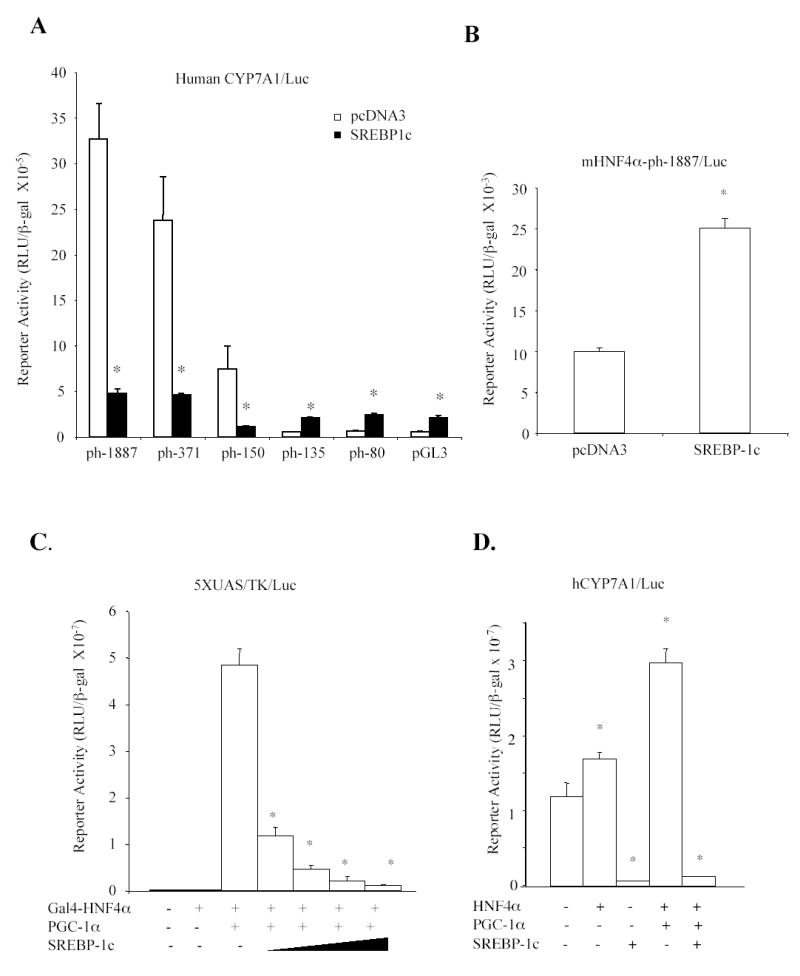
Effect of SREBP-1c on human CYP7A1 reporter activity. A. Human CYP7A1 reporter constructs (0.2μg) were co-transfected with pcDNA3 empty vector or SREBP-1c expression plasmids (0.1 μg) into HepG2 cells and luciferase activities were analyzed 40 hr after transfection. B. HNF4α site mutant reporter (mHNF4-ph1887/Luc) (0.2μg) was co-transfected with 0.1μg pcDNA3 or SREBP-1c expression plasmid into HepG2 cells. C. Mammalian one-hybrid assay. The Gal4/luciferase reporter, 5xUAS/TK/Luc (0.2μg) was co-transfected with Gal4 empty vector (−)(0.1μg), Gal4-HNF4α (0.1μg), PGC-1α (0.1μg) and increasing amounts of SREBP-1c expression plasmid (0.05-0.2μg) as indicated. D. Human CYP7A1 reporter construct, ph-371-Luc (0.2 μg) was co-transfected with 0.1 μg each of HNF4α and PGC-1α and/or SREBP-1c expression plasmid as indicated. Each assay was performed in triplicates and statistical analysis was performed using student’s t-test, “*”, significant, P< 0.05.
Western blotting analysis of insulin effects on FoxO1, SREBP-1c and HNF4α protein expressions in primary human hepatocytes
To study effect of insulin on phosphorylation of FoxO1 and expression of SREBP-1c protein in primary human hepatocytes, immunoblotting analysis of FoxO1 and SREBP protein expression was studied. Fig. 5A shows that without insulin treatment, FoxO1 was not phosphorylated and was mainly located in the nucleus. Insulin treatment for 30 min led to phosphorylation of FoxO1 and its rapid nuclear exclusion and degradation. Immunoblotting analysis did not detect SREBP-1c precursor (125 kDa) expression in hepatocytes (data not shown). Insulin treatment increased the amount of mature SREBP-1c (68 kDa) in cell lysates in a time-dependent manner (Fig. 5B). Insulin treatment did not significantly affect HNF4α protein levels in hepatocytes (Fig. 5C). The time courses of insulin inactivation of FoxO1 and induction of SREBP-1c in hepatocytes correlate well with Q-PCR results on CYP7A1 expression and support our hypothesis that insulin inactivates FoxO1 to rapidly induce CYP7A1 gene expression and that prolonged insulin treatment induces SREBP-1c to inhibit CYP7A1 gene expression.
Fig. 5.
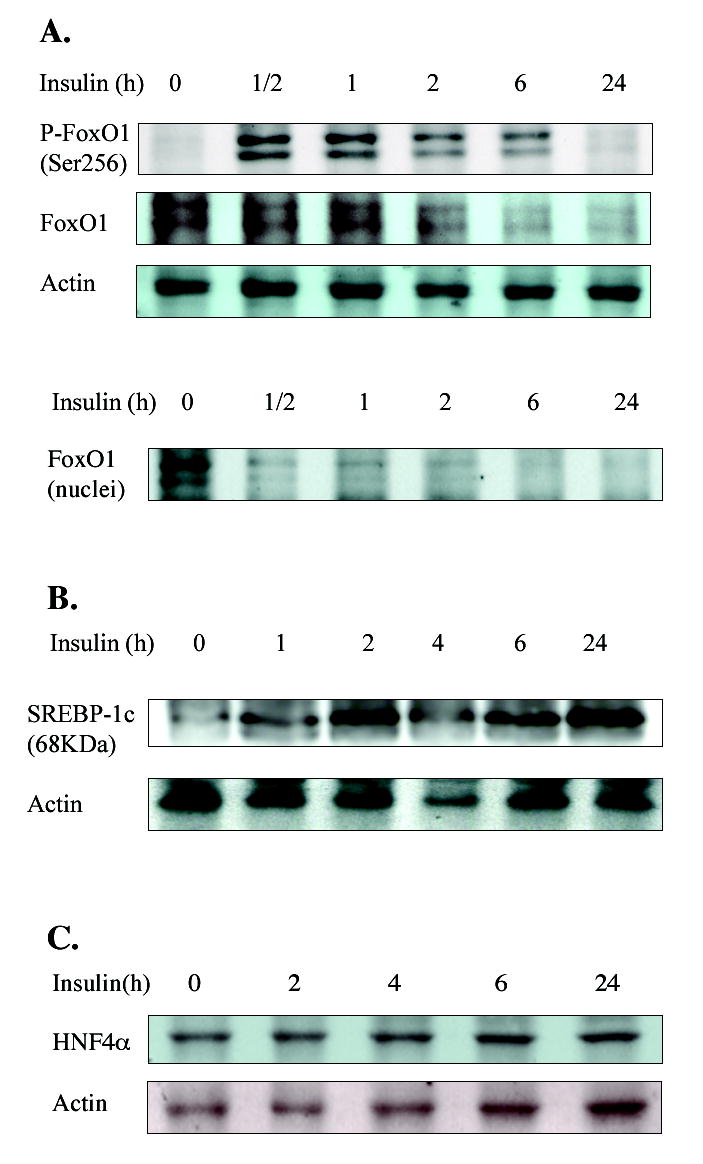
Western blotting analysis of FoxO1, SREBP-1c and HNF4α in primary human hepatocytes treated with insulin. Primary human hepatocytes were treated with 10 nM insulin for the time indicated. Total cell lysates or nuclei fractions from donor HH1286, HH1281 and HH1249 were used for detection of FoxO1 (A), SREBP-1c (B) and HNF4α (C) by antibodies, respectively.
ChIP assay of HNF4α binding and recruitment of SREBP-1c, FoxO1 and PGC-1α to the human CYP7A1 chromatin
To further confirm the roles of FoxO1 and SREBP-1c in mediating insulin regulation of human CYP7A1 gene transcription, ChIP assays were performed using primary human hepatocytes treated with 10 nM insulin for a period of time from 1 to 24 h. An antibody against FoxO1, SREBP-1c, HNF4α or PGC-1α was used to immunoprecipitate chromatin complexes for ChIP assays using primers designed to amplify a 391bp fragment of human CYP7A1 promoter region that included an HNF4α binding site. In the absence of insulin, ChIP assays detected FoxO1 and HNF4α in the CYP7A1 chromatin (Fig. 6A). Since FoxO1 did not bind to the human CYP7A1 promoter, it must have been recruited to the CYP7A1 chromatin by HNF4α. Insulin treatment rapidly abolished FoxO1 association with the CYP7A1 chromatin, consistent with FoxO1 inactivation and nuclear exclusion by insulin. In contrast, in the absence of insulin, SREBP-1c was not recruited to the CYP7A1 chromatin. Insulin treatment time-dependently increased SREBP-1c but decreased PGC-1α recruitment to CYP7A1 chromatin. These results are consistent with the induction of nuclear SREBP-1c by insulin in hepatocytes (Fig. 5B). Interestingly, insulin treatment for 1 h increased HNF4α and PGC-1α binding to CYP7A1 chromatin. Since FoxO1 is known to interact with HNF4α and inhibit its DNA binding activity (21), decreased FoxO1 protein in the nucleus allows HNF4α to bind to CYP7A1 chromatin. Taken together, our ChIP assay results suggest that insulin rapidly decreases FoxO1 in the nucleus, which leads to increased HNF4α and PGC-1α binding and transactivation of the CYP7A1 gene. Insulin induces SREBP-1c, which is recruited to CYP7A1 chromatin to block HNF4α recruitment of PGC-1α and results in inhibiting CYP7A1 gene transcription.
Fig. 6.
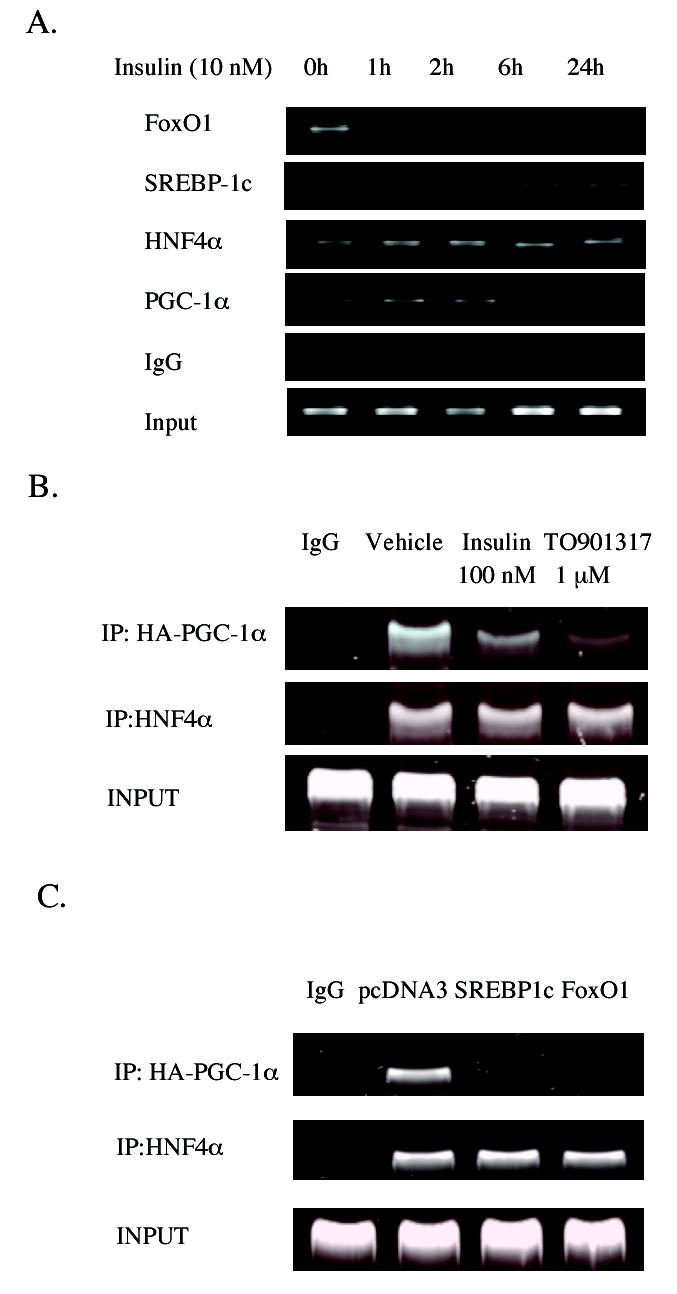
Chromatin immunoprecipitation assays of insulin effect on FoxO1, SREBP-1c and HNF4α recruitment to CYP7A1 chromatin. A. Human primary hepatocytes (HH1308) were treated with 10 nM insulin for the time indicated. Cells were harvested for ChIP assay as described under Materials and Methods. An antibody against HNF4α, SREBP-1 or FoxO1 was used to immunoprecipitate chromatins for PCR amplification and analysis. Ten % of cell lysate was used as input. Non-immune IgG was used as negative controls. B. ChIP assays of the effect of insulin and T0901317 on HNF4α recruitment of PGC-1α to CYP7A1 chromatin. HepG2 cells in 100 mm dish were transfected with HA-tagged PGC-1α (10 μg). Cells were then treated with insulin (100 nM) or T0901317 (1 μM) for 24 hr. Chromatins were precipitated with an antibody against HA tag or HNF4α for ChIP assays. Ten % of cell lysate was used as input. Non-immune IgG was used as negative controls. C. ChIP assays of HepG2 cells over-expressing SREBP-1c or FoxO1. HepG2 cells in 100 mm dish were transfected with 10 μg of HA-tagged PGC-1α, pcDNA3 empty vector, FoxO1 or SREBP-1c expression plasmid was over-expressed as indicated. Cells were harvested for ChIP assay as described under Experimental procedure. An antibody against HA-tag or HNF4α was used to immunoprecipitate chromatins for PCR amplification and analysis. Ten % of cell lysate was used as input. Non-immune IgG was used as negative controls.
ChIP assays were also performed using HepG2 cells overexpressing HA-PGC-1α to study insulin effect on HNF4α recruitment of PGC-1α to CYP7A1 chromatin. Fig. 6B shows that insulin (100 nM) treatment for 20 h reduced PGC-1α recruitment to CYP7A1 chromatin. T0901317, a potent LXRα agonist, also strongly reduced PGC-1α recruitment. These data are consistent with a previous report that insulin and LXRα induce SREBP-1c, which blocks PGC-1α interaction with HNF4α (33). Furthermore, we over-expressed SREBP-1c or FoxO1 in HepG2 cells to test their effect on PGC-1α recruitment to CYP7A1 chromatin. Fig. 6C shows that over-expression of SREBP-1c or FoxO1 abolished PGC-1α recruitment to CYP7A1 chromatin.
DISCUSSION
In the current study, we investigated the insulin regulation of human CYP7A1 gene transcription in human hepatocytes. Q-PCR analyses have revealed that physiological concentrations of insulin strongly and rapidly induce CYP7A1 mRNA expression. Interestingly, prolonged insulin treatment results in inhibition of human CYP7A1 mRNA expression. Results indicate that the dual effect of insulin action on human CYP7A1 is likely mediated through two insulin-regulated transcription factors, FoxO1 and SREBP-1c. FoxO1 bind to the IRE in the rat CYP7A1 gene promoter and stimulates rat CYP7A1 reporter activity. However, this IRE is not conserved in the human CYP7A1 promoter. Our results suggest that FoxO1 functions as a co-repressor that inhibits the human CYP7A1 gene by interfering with the HNF4α and PGC-1α co-activation of the gene. ChIP assay reveals that FoxO1 is present in CYP7A1 chromatin without insulin treatment and FoxO1 may interact with HNF4α to block its recruitment of PGC-1α to CYP7A1 chromatin (Fig. 6A). Upon insulin treatment, FoxO1 is released and PGC-1α is recruited by HNF4α to transiently stimulate CYP7A1 mRNA expression in human hepatocytes. Prolonged insulin treatment induces the mature form of SREBP-1c, which is recruited to CYP7A1 chromatin to inhibit HNF4α and PGC-1α trans-activation of the human CYP7A1 gene. This study suggests that CYP7A1 is a target gene of both FoxO1 and SREBP-1c, which play critical roles in regulating bile acid synthesis in response to insulin levels in hepatocytes.
Bile acids are known to activate a nuclear receptor Farnesoid X receptor (FXR), which induces a negative nuclear receptor small heterodimer partner (SHP) to inhibit nuclear receptor-regulated gene transcription (34). Several recent reports have shown that bile acids play critical roles in regulation of glucose and lipid metabolism (35–37). During the postprandial state, serum insulin levels increase transiently to stimulate glucose metabolism. According to our study, insulin may rapidly induce CYP7A1 and bile acid synthesis to facilitate nutrient absorption in the intestine during the postprandial state. Bile acids inhibit the PEPCK gene and prevent hyperglycemia (38). During post-absorptive state and starvation, glucagon levels increase to inhibit CYP7A1 and bile acid synthesis, while stimulating gluconeogenesis to prevent hypoglycemia (39).
In Type II diabetes, FoxO1 nuclear exclusion is impaired due to insulin resistance (17). Hyperinsulinemia may inhibit CYP7A1 and bile acid synthesis by inducing SREBP-1c, which stimulates lipogenesis and contributes to hypertriglyceridemia. Inhibition of bile acid synthesis stimulates gluconeogenesis and lipogenesis. In Type I diabetes, lack of insulin activates FoxO1, which stimulates gluconeogenesis, but inhibits bile acid synthesis and also causes hyperglycemia and dyslipidemia. All these metabolic changes are caused by insulin and may contribute to the development of diabetic dyslipidemia and cardiovascular diseases.
It should be noted that we have demonstrated distinct species differences in FoxO1 and SREBP-1c regulation of rat and human CYP7A1 gene transcription. FoxO1 induces rat CYP7A1 by binding to an IRE (TGTTTAC) in the rat gene promoter, but FoxO1 does not bind and inhibit the human CYP7A1 gene. Analysis of homologous CYP7A1 genes reveals that this rat IRE sequence is also conserved in mouse and hamster genes. In the mouse CYP7A1 gene, this IRE is located in the same region and is likely a FoxO1 binding site. A recent report shows a two-fold higher expression of CYP7A1 mRNA in FoxO1 transgenic mice suggesting induction of mouse CYP7A1 by FoxO1 (40). In the hamster CYP7A1 gene, this IRE is located upstream of a putative IRE that binds HNF3α and HNF4α (41). SREBP-1c also differentially regulates the rat and human CYP7A1 genes. SREBP-1c binds to the E-box (CANNTG) and SRE motifs, and activates gene transcription (42). However, SREBP-1c also can function as a DNA-binding-independent repressor by interfering with HNF4α interaction with PGC-1α and inhibiting PEPCK gene transcription (33). This study shows that the same mechanism also inhibits the human CYP7A1 gene. In contrast, we found that SREBP-1c strongly stimulated rat CYP7A1 reporter activity (data not shown). Analysis of the rat CYP7A1 gene promoter sequence reveals three E-boxes and several GC-rich sequences located downstream of –344 of the rat CYP7A1 promoter. These E-box motifs are not conserved in the human CYP7A1 gene. Further study is needed to identify the SREBP-1c binding site in the rat CYP7A1 gene and the mechanism by which FoxO1 and SREBP-1c regulates the rat CYP7A1 gene. Previously we have reported that in contrast to the induction of CYP7A1 mRNA expression in mouse liver, LXRα does not stimulate human CYP7A1 gene transcription because the human CYP7A1 gene lacking a L X R α response element (43). Species differences in regulation of the CYP7A1 gene and lipid metabolism underscore the importance of studying human CYP7A1 gene regulation in human hepatocyte models.
In summary, we studied the mechanisms of insulin regulation of CYP7A1 gene transcription mediated by FoxO1 and SREBP-1c. Our results suggest that insulin has a dual effect on human CYP7A1 gene transcription: a rapid induction at physiological concentrations to control glucose and lipid metabolism and later inhibition to maintain glucose and lipid homeostasis. Understanding the complex mechanism of insulin regulation of bile acid synthesis and lipid homeostasis will lead to developing novel drug therapies for treating diabetic dyslipidemia.
Supplemental data
Sequences of probes used in EMSA
Lower case indicates mutation and core IRE sequences are underlined.
G6PaseIRE: AGGCTGTTTTTGTGTGCCTGTTTTGCTATTTTACGTAA
Rat CYP7A1 IREs:
rIRE1:-248GGGAAGCTTCTGCCTGTTTTGC
TTTGCGTGAT−187
rIRE2: −299GATGTGCTCATCTGTTTACTTCT
TTTTCTACACA−266
rIRE3: −180GATCCTCTCTGTTTGTTCTGGAG
CCTCTTCTGAGAT−150
rIRE4: −91GATCGGACAAATAGTGTTTGCTT
TGGTCACTCAGAT−60
Human CYP7A1 IRE:
hIRE1:−306GCCCATCTTAAACAGGTTTATTT
GTTCTTTTTA−274
hIRE2:−86GGCTAATTGTTTGCTTTGTCAACC
AAGCTCAAGTTAATG−48
Mutant
rIRE2: −299GATGTGCTCATCTtTTTACTTCT
TTTTCTACACA−266
Acknowledgments
The technical assistance of Bethany Spalding-Yoder and Sandra Del Signore is gratefully acknowledged. Primary human hepatocytes were supplied by the Liver Procurement and Distribution System funded by a contract from NIDDK.
Footnotes
The abbreviations used are: Apo CIII, apolipoprotein CIII; ChIP, chromatin immunoprecipitation; CYP7A1, cholesterol 7α-hydroxylase; FoxO1, forkhead box O1; G6Pase, glucose 6-phosphatase; HNF4α, hepatocyte nuclear factor 4α; IGFBP, insulin-like growth factor binding protein; IRE, insulin response element; PEPCK, phosphoenolpyruvate carboxykinase; PGC-1α, peroxisome proliferators-activated receptor γ- coactivator-1α; Q-PCR, quantitative real-time PCR; SRE, sterol response element; SREBP, sterol regulatory element binding protein.
This study was supported by National Institute of Diabetes and Digestive and Kidney Diseases Grants DK 58379 and DK44442 (JYLC).
References
- 1.Reaven G, Abbasi F, McLaughlin T. Recent Prog Horm Res. 2004;59:207–223. doi: 10.1210/rp.59.1.207. [DOI] [PubMed] [Google Scholar]
- 2.Chiang JYL. Am J Physiol Gastrointest Liver Physiol. 2003;284:G349–G356. doi: 10.1152/ajpgi.00417.2002. [DOI] [PubMed] [Google Scholar]
- 3.Bennion LJ, Grundy SM. N Engl J Med. 1977;296:1365–1371. doi: 10.1056/NEJM197706162962401. [DOI] [PubMed] [Google Scholar]
- 4.Andersen E, Karlaganis G, Sjovall J. Eur J Clin Inv. 1988;18:166–172. doi: 10.1111/j.1365-2362.1988.tb02408.x. [DOI] [PubMed] [Google Scholar]
- 5.Hassan AS, Ravi Subbiah MT, Thiebert P. Proc Soc Exp Biol Med. 1980;164:449–452. doi: 10.3181/00379727-164-40894. [DOI] [PubMed] [Google Scholar]
- 6.Villanueva GR, Herreros M, Perez-Barriocanal F, Bolanos JP, Bravo P, Marin JJ. J Lab Clin Med. 1990;115:441–448. [PubMed] [Google Scholar]
- 7.Wang DP, Stroup D, Marrapodi M, Crestani M, Galli G, Chiang JY. J Lipid Res. 1996;37:1831–1841. [PubMed] [Google Scholar]
- 8.Crestani M, Sadeghpour A, Stroup D, Galli G, Chiang JY. J Lipid Res. 1998;39:2192–2200. [PubMed] [Google Scholar]
- 9.Twisk J, Hoekman MF, Lehmann EM, Meijer P, Mager WH, Princen HM. Hepatology. 1995;21:501–510. [PubMed] [Google Scholar]
- 10.Ishida H, Yamashita C, Kuruta Y, Yoshida Y, Noshiro M. J Biochem (Tokyo) 2000;127:57–64. doi: 10.1093/oxfordjournals.jbchem.a022584. [DOI] [PubMed] [Google Scholar]
- 11.Foufelle F, Ferre P. Biochem J. 2002;366:377–391. doi: 10.1042/BJ20020430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.O'Brien RM, Granner DK. Physiol Rev. 1996;76:1109–1161. doi: 10.1152/physrev.1996.76.4.1109. [DOI] [PubMed] [Google Scholar]
- 13.Hall RK, Yamasaki T, Kucera T, Waltner-Law M, O'Brien R, Granner DK. J Biol Chem. 2000;275:30169–30175. doi: 10.1074/jbc.M004898200. [DOI] [PubMed] [Google Scholar]
- 14.Schmoll D, Walker KS, Alessi DR, Grempler R, Burchell A, Guo S, Walther R, Unterman TG. J Biol Chem. 2000;275:36324–36333. doi: 10.1074/jbc.M003616200. [DOI] [PubMed] [Google Scholar]
- 15.Puigserver P, Rhee J, Donovan J, Walkey CJ, Yoon JC, Oriente F, Kitamura Y, Altomonte J, Dong H, Accili D, Spiegelman BM. Nature. 2003;423:550–555. doi: 10.1038/nature01667. [DOI] [PubMed] [Google Scholar]
- 16.Nakae J, Kitamura T, Silver DL, Accili D. J Clin Invest. 2001;108:1359–1367. doi: 10.1172/JCI12876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Altomonte J, Cong L, Harbaran S, Richter A, Xu J, Meseck M, Dong HH. J Clin Invest. 2004;114:1493–1503. doi: 10.1172/JCI19992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nakae J, Park BC, Accili D. J Biol Chem. 1999;274:15982–15985. doi: 10.1074/jbc.274.23.15982. [DOI] [PubMed] [Google Scholar]
- 19.Zhang X, Gan L, Pan H, Guo S, He X, Olson ST, Mesecar A, Adam S, Unterman TG. J Biol Chem. 2002;277:45276–45284. doi: 10.1074/jbc.M208063200. [DOI] [PubMed] [Google Scholar]
- 20.Matsuzaki H, Daitoku H, Hatta M, Tanaka K, Fukamizu A. Proc Natl Acad Sci U S A. 2003;100:11285–11290. doi: 10.1073/pnas.1934283100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hirota K, Daitoku H, Matsuzaki H, Araya N, Yamagata K, Asada S, Sugaya T, Fukamizu A. J Biol Chem. 2003;278:13056–13060. doi: 10.1074/jbc.C200553200. [DOI] [PubMed] [Google Scholar]
- 22.Foretz M, Pacot C, Dugail I, Lemarchand P, Guichard C, Le Liepvre X, Berthelier-Lubrano C, Spiegelman B, Kim JB, Ferre P, Foufelle F. Mol Cell Biol. 1999;19:3760–3768. doi: 10.1128/mcb.19.5.3760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Horton JD, Goldstein JL, Brown MS. J Clin Invest. 2002;109:1125–1131. doi: 10.1172/JCI15593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Engelking LJ, Kuriyama H, Hammer RE, Horton JD, Brown MS, Goldstein JL, Liang G. J Clin Invest. 2004;113:1168–1175. doi: 10.1172/JCI20978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tobin KA, Ulven SM, Schuster GU, Steineger HH, Andresen SM, Gustafsson JA, Nebb HI. J Biol Chem. 2002;277:10691–10697. doi: 10.1074/jbc.M109771200. [DOI] [PubMed] [Google Scholar]
- 26.Repa JJ, Liang G, Ou J, Bashmakov Y, Lobaccaro JM, Shimomura I, Shan B, Brown MS, Goldstein JL, Mangelsdorf DJ. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hegarty BD, Bobard A, Hainault I, Ferre P, Bossard P, Foufelle F. Proc Natl Acad Sci U S A. 2005;102:791–796. doi: 10.1073/pnas.0405067102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li T, Chiang JY. Am J Physiol Gastrointest Liver Physiol. 2004;288:G74–G84. doi: 10.1152/ajpgi.00258.2004. [DOI] [PubMed] [Google Scholar]
- 29.Song KH, Li T, Chiang JY. J Biol Chem. 2006;281:10081–10088. doi: 10.1074/jbc.M513420200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cagen LM, Deng X, Wilcox HG, Park EA, Raghow R, Elam MB. Biochem J. 2004;385:207–216. doi: 10.1042/BJ20040162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.O'Brien RM, Noisin EL, Suwanichkul A, Yamasaki T, Lucas PC, Wang JC, Powell DR, Granner DK. Mol Cell Biol. 1995;15:1747–1758. doi: 10.1128/mcb.15.3.1747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Stroup D, Chiang JY. J Lipid Res. 2000;41:1–11. [PubMed] [Google Scholar]
- 33.Yamamoto T, Shimano H, Nakagawa Y, Ide T, Yahagi N, Matsuzaka T, Nakakuki M, Takahashi A, Suzuki H, Sone H, Toyoshima H, Sato R, Yamada N. J Biol Chem. 2004;279:12027–12035. doi: 10.1074/jbc.M310333200. [DOI] [PubMed] [Google Scholar]
- 34.Chiang JY. Endocr Rev. 2002;23:443–463. doi: 10.1210/er.2000-0035. [DOI] [PubMed] [Google Scholar]
- 35.Stayrook KR, Bramlett KS, Savkur RS, Ficorilli J, Cook T, Christe ME, Michael LF, Burris TP. Endocrinology. 2005;146:984–991. doi: 10.1210/en.2004-0965. [DOI] [PubMed] [Google Scholar]
- 36.Watanabe M, Houten SM, Wang L, Moschetta A, Mangelsdorf DJ, Heyman RA, Moore DD, Auwerx J. J Clin Invest. 2004;113:1408–1418. doi: 10.1172/JCI21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cariou B, Duran-Sandoval D, Kuipers F, Staels B. Endocrinology. 2005;146:981–983. doi: 10.1210/en.2004-1595. [DOI] [PubMed] [Google Scholar]
- 38.Yamagata K, Daitoku H, Shimamoto Y, Matsuzaki H, Hirota K, Ishida J, Fukamizu A. J Biol Chem. 2004;279:23158–23165. doi: 10.1074/jbc.M314322200. [DOI] [PubMed] [Google Scholar]
- 39.Song KH, Chiang JY. Hepatology. 2006;43:117–125. doi: 10.1002/hep.20919. [DOI] [PubMed] [Google Scholar]
- 40.Zhang W, Patil S, Chauhan B, Guo S, Powell DR, Le J, Klotsas A, Matika R, Xiao X, Franks R, Heidenreich KA, Sajan MP, Farese RV, Stolz DB, Tso P, Koo SH, Montminy M, Unterman TG. J Biol Chem. 2006;281:10105–10117. doi: 10.1074/jbc.M600272200. [DOI] [PubMed] [Google Scholar]
- 41.De Fabiani E, Crestani M, Marrapodi M, Pinelli A, Golfieri V, Galli G. Biochem J. 2000;347:147–154. doi: 10.1042/0264-6021:3470147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Osborne TF. J Biol Chem. 2000;275:32379–32382. doi: 10.1074/jbc.R000017200. [DOI] [PubMed] [Google Scholar]
- 43.Chiang JY, Kimmel R, Stroup D. Gene. 2001;262:257–265. doi: 10.1016/s0378-1119(00)00518-7. [DOI] [PubMed] [Google Scholar]