

Summary
The Endosomal Sorting Complex Required for Transport (ESCRT) complexes are central to receptor downregulation, lysosome biogenesis, and budding of HIV. The yeast ESCRT-I complex contains the Vps23, Vps28, and Vps37 proteins and its assembly is directed by the C-terminal steadiness box of Vps23, the N-terminal half of Vps28, and the C-terminal half of Vps37. The crystal structures of a Vps23:Vps28 core subcomplex and the Vps23:Vps28:Vps37 core were solved at 2.1 and 2.8 Å resolution. Each subunit contains a structurally similar pair of helices that form the core. The N-terminal domain of Vps28 has a hydrophobic binding site on its surface that is conformationally dynamic. The C-terminal domain of Vps28 binds the ESCRT-II complex. The structure shows how ESCRT-I is assembled by a compact core from which the Vps23 UEVdomain, the Vps28 C-domain, and other domains project to bind their partners.
Introduction
Targeted degradation is a fundamental mechanism of protein regulation and quality control (Hershko et al., 2000; Hochstrasser, 2000; Pickart, 2001). Many integral membrane proteins are targeted for degradation by their covalent modification with a single moiety of the 76-amino acid protein ubiquitin (Haglund et al., 2003; Hicke, 2001; Katzmann et al., 2002; Raiborg et al., 2003; Sigismund et al., 2004). Monoubiquitinated membrane proteins are recognized by components of transport machinery with specific monoubiquitin-binding domains and delivered to the lysosome for degradation (Haglund et al., 2003; Hicke, 2001; Katzmann et al., 2002; Raiborg et al., 2003; Sigismund et al., 2004). Monoubiquitinated transmembrane proteins en route to the lysosome are sorted through multivesicular bodies (MVBs), a subset of late endosomes with a multivesicular appearance and containing vesicles that invaginate into the interior of the endosome (Gorden et al., 1978; Haigler et al., 1979); reviewed by (Gruenberg and Stenmark, 2004). Fusion of the limiting membrane of the MVB with the lysosomal membrane results in delivery of lumenal MVB vesicles and their contents into the lysosome where the vesicles and the transmembrane proteins are degraded. Membrane proteins that are excluded from the inner MVB vesicles remain within the limiting membrane of the MVB. Studies in mammalian cells have revealed critical roles for MVBs in such seemingly distinct processes as growth factor receptor downregulation (Katzmann et al., 2002), antigen presentation, developmental signaling and retroviral budding (Morita and Sundquist, 2004).
Sorting of monoubiquitinated proteins into the MVB pathway is a highly regulated process and requires the coordinated functions of 17 class E Vps (Vacuolar Protein Sorting) proteins (Bowers and Stevens, 2005; Katzmann et al., 2001; Odorizzi et al., 1998). The functional loss of any individual class E Vps protein results in a malformed late MVB known as the “class E compartment”. The characterization of these proteins has resulted in the identification of three high molecular weight cytoplasmic protein complexes, referred to as ESCRT (Endosomal Sorting Complex Required for Transport) complexes I, II and III (Babst et al., 2002; Katzmann et al., 2001); reviewed by (Conibear, 2002; Hicke and Dunn, 2003; Katzmann et al., 2002). The hetero-oligomeric ESCRT complexes are sequentially recruited to the late endosomal membrane and drive the formation of MVBs. The ESCRT complexes and associated proteins have been found to play roles in retrovirus budding, a process analogous to MVB vesicle formation with the same topological requirement. A number of viruses, including human immunodeficiency virus (HIV), equine infectious anaemia virus (EIAV) and Ebola, require the ESCRT machinery for budding (Morita and Sundquist, 2004).
The ESCRT-I complex directly binds to monoubiquitin moieties of protein cargo through its UEV (ubiquitin E2 variant) domain of Vps23, a catalytically inactive variant of an ubiquitin conjugating enzyme (Katzmann et al., 2001). The UEV domain interacts with not only ubiquitin, but also P(S/T)XP peptide sequence of the upstream Vps27 complex (Bache et al., 2003; Bilodeau et al., 2003; Katzmann et al., 2003), and a number of other cellular proteins, including the mammalian counterpart of Bro1 (Strack et al., 2003; von Schwedler et al., 2003) and the ubiquitin ligase Tal (Amit et al., 2004). P(S/T)XP motifs have been found in the Gag proteins of many retroviruses such as HIV-1, HTLV-I and MPMV (Goila-Gaur et al., 2003; Martin-Serrano et al., 2003; Pornillos et al., 2003; Scarlata and Carter, 2003; Strack et al., 2003; von Schwedler et al., 2003).These viruses use this motif to hijack the MVB sorting machinery to bud from host cells.
Over the past three years, there has been considerable progress in the structural analysis of the ESCRT complexes (reviewed by (Hurley and Emr)). The structures of the ubiquitin binding UEV domains of yeast (Teo et al., 2004b) and human (Sundquist et al., 2004) Vps23 have been determined in complex with ubiquitin, and the structure of the human UEV domain was solved in complex with a peptide containing a retroviral PTAP motif (Pornillos et al., 2002). The structure of the ubiquitin binding NZF-2 domain of yeast ESCRT-II has also been determined in complex with ubiquitin (Alam et al., 2004). The structure of the yeast ESCRT-II core complex has been determined (Hierro et al., 2004; Teo et al., 2004a).
The structure of the ESCRT-I core, arguably the most significant remaining piece of the structural puzzle, has remained unsolved. Apart from the UEV domain of Vps23, the subunits of ESCRT-I do not contain any well-studied domains. In order to physically map the structure of the core, we expressed a recombinant form of the yeast ESCRT-I complex in which the UEV domain was removed from Vps23, but with all other subunits intact. This complex was subjected to limited proteolysis, the digests were fractionated by gel filtration, and the resulting fragments analyzed by protein sequencing and mass spectrometry. This information was used to identify the protease-resistant core responsible for the assembly of the ternary complex. In the process, a core subcomplex of Vps23 and Vps28 and a Vps28 C-terminal domain were also identified. The structures of the complexes were determined, revealing how ESCRT-I assembles. The Vps28 C-terminal domain was shown to bind to ESCRT-II in vitro, showing how ESCRT-I and II likely interact. We went on to show that the subunit interfaces identified in the crystal structure are essential for ESCRT-I assembly and function in vivo in yeast.
Results
Identification of the ESCRT-I core
This study commenced with the expression and characterization of a recombinant form of the yeast ESCRT-I complex, referred to here as EIΔUEV-A, in which the UEV domain of Vps23, residues 1–161, was deleted (Fig. 1A). This was the smallest truncation of the intact complex that could be expressed as a soluble recombinant complex. EIΔUEV-A yielded a predominant peak with an apparent MW of 190 kDa by gel filtration (Fig. 1B). Sedimentation velocity studies of the EIΔUEV-A complex indicated the presence of two species having corrected sedimentation coefficients of approximately 3.6 and 4.8 S (Fig. 1C). A weight average buoyant molecular mass of approximately 25 kDa was obtained from a fit to a single solute model. This corresponds to a molecular mass of 98 kDa and is a value just larger than that expected for the monomeric 1:1:1 ternary complex (Mcalc = 78,057.3 Da), suggesting that the monomeric complex is the major species. An analysis in terms of two single ideal solutes yielded improved data fits and indicated that both monomeric and dimeric states of the ternary complexes were present. Modeling the equilibrium data to a monomerdimer mixture yielded excellent fits (Figure 1D ) with a dissociation constant Kd of 110 – 160 μM.
Figure 1. Identification and characterization of the ESCRT-I core.

A. Domain structure of ESCRT-I and complexes used in this study. B. Gel filtration of the EIΔUEV-A construct. C. Sedimentation velocity analysis of the 190 kDa peak obtained by gel filtration of the EIΔUEV-A construct in (B), showing the presence of two species. D. Sedimentation equilibrium analysis in terms of a monomer-dimer equilibrium of the EIΔUEV-A construct in (B) for data collected at 10 (blue), 12 (green) and 16 (red) krpm. The solid lines show the best-fit global analysis. Best-fit residuals are shown above the plot. E. Analysis of a partial endoproteinase Glu-C digest of EIΔUEV-B by gel filtration and protein sequencing showing the identification of Vps28 core boundaries. Additional analyses of Glu-C and tryptic digests were used to determine the core boundaries of Vps23 and Vps37 (not shown). F. Sedimentation equilibrium analysis of the core construct showing a single species consistent with a 1:1:1 complex. Data were collected at 11 (blue), 14 (green) and 17 (red) krpm for loading A280 of 0.50 (left), 0.41 (center) and 0.71 (right). The solid lines show the best-fit global analysis in terms of a single ideal solute, with the corresponding residuals shown in the panels above the plot.
In order to determine if the ESCRT-I complex contains a stable core structure, a pvariant of the EIΔUEV complex with Vps23 truncated to residue 199 and all Cys mutated to Ala (EIΔUEV-B) was expressed and purified. These modifications were made to reduce a propensity to age-dependent aggregation that was noticed for the EIΔUEV-A construct. EIΔUEV-B was subjected to limited proteolysis with trypsin and endoproteinase Glu-C. Proteolytic digests of EIΔUEV-B were subjected to gel filtration chromatography to separate potential core complexes, subcomplexes, and domains, and the components were chemically sequenced (Fig. 1E) and in some cases subjected to MALDI-TOF mass spectrometry. A binary complex consisting of Vps23(322–385) and Vps28(8–125) (binary-A) was identified, along with an isolated domain consisting of Vps28(145–242) (Vps28 C-domain). The Vps28 C-domain was expressed as a recombinant protein, purified, and found to have a native molecular weight of 14 kDa on gel filtration, as compared to a calculated molecular weight of 11.6 kDa (data not shown). We concluded that this fragment of Vps28 was a monomeric domain that was not part of the core.
The binary-A complex was made as a recombinant form and characterized. For high resolution crystallographic analysis, two further truncations of Vps28 were made: binary-B, with Vps28(13–118), and binary-C, with Vps28(13–125). Sedimentation equilibrium experiments on the binary-C (Supp. Fig. 1A) and the binary-B complexes (Supp. Fig. 1B) are consistent with monodisperse single ideal species having molecular masses of 63,900 ± 2,900 and 61,900 ± 600 Da, respectively, showing that these complexes are stable trimers (Supp. Table 1). This finding is inconsistent with the apparent monomeric 1:1 complex obtained by crystallography in the presence of the detergent N,N-dimethyldecylamine oxide (DDAO), as described below. It was not possible to carry out sedimentation analysis in the presence of DDAO, due to flotation of the detergent. The most reasonable interpretation is that the trimer is a non-physiological artifact due to the exposure of hydrophobic surfaces in the absence of Vps37. We suggest that detergent covers these surfaces in the binary complex crystal and thereby stabilizes the monomeric form.
To determine what portion of Vps37 was involved in the core complex, the binary-A construct and full-length Vps37 were co-expressed and purified. This complex was poorly stable and underwent partial proteolysis during purification. The proteolysis products were fractionated on gel filtration and sequenced, revealing cleavage products beginning at Vps37 residues 130 and 134. The core and core-A constructs were generated on this basis (Fig. 1A). Another variant in which Vps23 began at residue 257 (core-B) was designed to determine if the predicted coiled-coil from residues 257–300 contributed to higher-order oligomerization. Sedimentation equilibrium experiments on core (Fig. 1F), core-B (Supp. Fig. 1C), and core-A (Supp. Fig 1D) showed that these species are monodisperse monomers. The experimentally determined buoyant molecular masses obtained from single ideal solute fits (Supp. Table 1) are consistent with the presence of monomeric species having a 1:1:1 stoichiometry of the Vps28, Vps23 and Vps37 peptides in all of these core complex variants.
Structure of the Vps23:Vps28 subcomplex
The structure of the Vps23:Vps28 subcomplex was determined by multiwavelength anomalous dispersion (MAD) from a selenomethionyl derivative of the binary-A complex at 3.35 Å resolution (Supp. Table 2). This structure was partially refined and used as a molecular replacement (MR) search model to phase crystals of the binary-B complex that diffracted to 2.1 Å resolution (Fig. 2A). The asymmetric unit contains two copies of the Vps23:Vps28 heterodimer, which are related by approximate two-fold symmetry. The structure of Vps23 is well-defined throughout, and consists almost entirely of two long α-helices (Fig. 2B, Fig. 3). The Vps28 core contains an extended N-terminal loop from residue 18–30 and five helices: α0–4 (Fig. 3). The two helices of Vps23 are antiparallel to each other in a hairpin arrangement, as are the first two helices of Vps28. The two pairs of helices are strikingly similar to each other. Not only are the two pairs of helices aligned at essentially identical angles to each other, but the loop connecting the two helices is nearly identical in conformation. Every one of the 56 Cα carbon atoms in the section α1-loop-α2 in Vps23 can be superimposed on its counterpart in Vps28 without any breaks, and with an r.m.s.d. of just 1.0 Å.
Figure 2. Structure of the Vps23:Vps28 subcomplex.
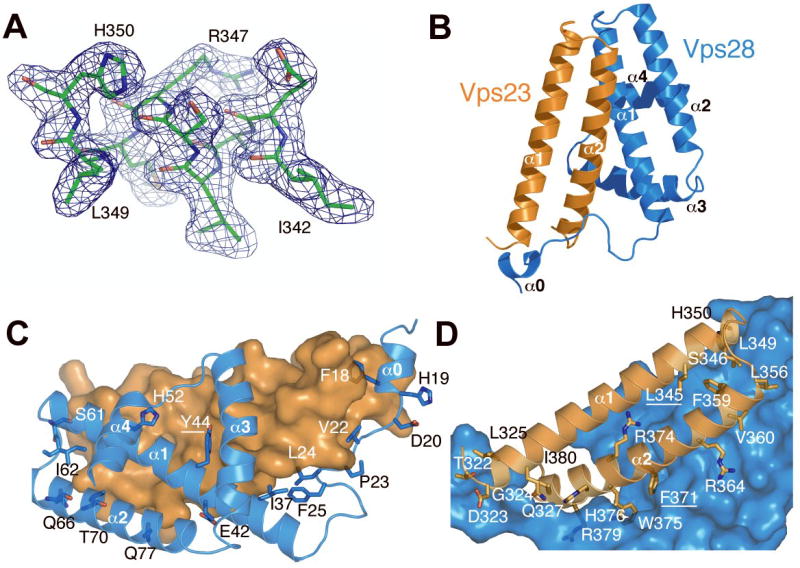
A. Composite 2Fo-Fc omit map contoured at 1.0 σshown with a portion of the refined model of Vps23. B. Ribbon model of the binary complex, Vps23 in orange, Vps28 in marine. C and D, the Vps23:Vps28 interfaces with Vps23 and Vps28 shown as a space filling surface model respectively.
Figure 3. Structure based sequence alignment.

A. Vps23 and its homologs. A structural alignment of Vps23 with Vps28 and Vps37 is shown immediately below. B. Vps28 and its homologs. C. Vps37 and its homologs. Colored circles indicate sites of interaction with other subunits, colored as in Fig. 2 and 4. Black triangles indicate mutations with no phenotype either alone or in any combination tested. Black squares indicate mutations with a class E phenotype taken alone or in combination with others. Black circles under the Vps28 alignment show residues involved in the hydrophobic hole in either the binary or ternary complexes, or both. Pink shading indicates complete conservation, cyan shading indicates >75% conservation.
The two subunits interact extensively over their entire length, burying a total of 2940 Å2 of solvent accessible surface area from the two proteins (Fig. 2C, D). Interactions between the two helical hairpins form the largest contact surface. The two hairpins are aligned roughly parallel to each other, and rotated about 30° with respect to each other. Their mutual interactions are centered at the bend in the hairpins, and continue along the length of Vps28:α1 and Vps23: α2. The Vps23:Phe-371 side-chain protrudes deep into a crevice in Vps28, where it is almost completely buried by interactions with Vps28: Ile-37, Leu-40, Tyr-44, and Ile-97. The N-terminal loop of Vps28 threads its way between the ends of the two Vps23 helices and proceeds to wrap around the C-terminal part of Vps23:α2. In particular, Vps28:Phe-18, and Val-22 bury themselves in the gap between the N- and C-terminal ends of Vps23 helices α1 and α2. Vps28:Leu-24 and Phe-25 form hydrophobic contacts with the Vps23:Leu-372 and Trp-375 at the C-terminus of Vps23:α2; and Vps28: Leu-14 interacts with the N-terminal part of Vps23:α1.
Relatively few polar interactions are observed as compared to the extensive hydrophobic interactions. With respect to the extended N-terminal portion of Vps28, the side-chain of Vps23:Gln-327 makes two hydrogen bonds to the main chain of Vps28:Val-22, stabilizing its unpartnered β-conformation. Vps23: His-376 also interacts with the Val-22 main-chain carbonyl. Vps23:Arg-379 stabilizes a turn in Vps28 by hydrogen bonding to carbonyls of Vps28:Pro-23 and Leu-24. Within the main helical interface, Vps23:Arg-364 is a linchpin for a hydrogen bonding network with Vps28:Asp-42, Gln-77, and Tyr-81.
The most prominent surface feature of the Vps23:Vps28 subcomplex is found on the back face of Vps28, distal to Vps23. All four helices of Vps28 together form a loose bundle with a large hydrophobic concavity in the center. The floor is formed by α1 residues Tyr-44, Val-47, and Ile-48, and the walls by Phe-78 and Leu-82 of α2; Phe-94 and Ile-97 of α3; Ile-106 of a α3- α4 loop; and Ile-112 and Leu-115 of α4. The pocket is largely empty. The tail of a DDAO molecule is bound near the bottom of the pocket adjacent to Tyr-44, Ile-97, and Ile-106, although the polar headgroup protrudes through an adjacent crevice and not through the central hole. The Vps23 face distal to Vps28 is rich in exposed hydrophobic residues. In the crystal, these faces interact with two ordered detergent molecules, which form bridges across a non-crystallographic axis of two fold symmetry.
Structure of the ESCRT-I core complex
The structure of the core complex was determined at 2.8 Å resolution by molecular replacement with the Vp23:Vps28 subcomplex (Fig. 4A; Supp. Table 2). Ternary complex crystals were grown in the presence of sulfate ions, but in the absence of detergent. There are two copies of a 1:1:1 Vps23:Vps28:Vps37 complex in the asymmetric unit. The two 1:1:1 complexes pack tightly against each other, with the C-terminal α3 and α4 helices of the Vps28 core packing against each other, against the Vps23 of the opposing complex, and with the opposing copy of Vps37 via a bound sulfate ion. The primary Vps23:Vps28 interface in the 1:1:1 complex is identical to that observed in the binary complex, so there is no ambiguity about which combination of subunits in the crystal corresponds to the 1:1:1 complex observed in solution.
Figure 4. Structure of the Vps23:Vps28:Vps37 core complex.

A. Density-modified map calculated by the prime-and-switch algorithm and contoured at 0.9 σ, overlaid on the refined structure of Vps37, which was not included in the phase calculation. B. Ribbon diagram of the core complex, colored in Fig. 2B and with Vps37 in green. C and D, the Vps23:Vps37 interface with Vps23 and Vps37 shown as a space filling surface model, respectively. E. Conformational change in the Vps28 subunit, with the conformation from the binary subcomplex colored marine and that from the core complex colored magenta. F. The hydrophobic hole in the distal side of the Vps28 subunit, with a crystal packing-related Vps28:Phe-100 from a shown in magenta as a marker. Hydrophobic residues on the surface of Vps28 are colored green.
The structure reveals that the core domain of Vps37 consists of an antiparallel helical hairpin closely similar to the core domains of Vps23 and Vps28 (Fig. 3, Fig. 4B), again despite a lack of detectable sequence similarity. 55 Cα atoms from Vps23 and Vps37 can be superimposed with an r.m.s.d. of 1.1 Å. The three subunits are arranged at nearly identical ~30° angles to each other, like three cards being held in a hand. Vps23 is the middle card in the hand, and there is no direct contact between Vps28 and Vps37. The exposed hydrophobic face of Vps23 observed in the binary subcomplex is completely buried in contacts with Vps37. The Vps23:Vps37 contacts are extensive, burying 1600 Å2 of solvent accessible surface area per subunit.
A series of hydrophobic residues centered on Leu-181, Phe-184, Ile-185, and Leu-189 of Vps37:α2 form the heart of the Vps23 binding surface of Vps37. These residues pack against a hydrophobic surface of Vps23 centered on α1 residues Leu-345 and Met-348, and α1-α2 loop residue Ile-354. These are the same exposed hydrophobic residues of Vps23 that were bound to an ordered detergent molecule in the binary complex. Vps23:Asp-340 makes two salt bridges across the interface, with Vps37:Arg-192 and His-196.
Conformational changes in Vps28
The α1/ α2 core of Vps28 preserves its binary complex conformation in the ternary complex, but beginning at the N-terminus of α3, the remainder of Vps28 undergoes a dramatic conformational change (Fig. 4E). Helix α3 pivots by ~20 °, and the last turn of the helix unravels in the ternary complex. Helix α4 occupies an almost completely different position in the ternary complex, with its C-terminus coinciding with the position observed in the binary complex. The N-terminus of α4 moves by 12 Å between the two complexes, and the helix shifts by 60° about an axis running through its C-terminus. Residues 118–125 are ordered and comprise a C-terminal loop in the ternary complex. These residues were present in the 3.35 Å SeMet crystal of the binary-A complex, and were observed to be disordered in that structure. The C-terminal loop in the ternary structure occupies the same position in space that α4 occupies in the binary structure. However, the direction of the chain is reversed such that the C-terminus of the Vps28 core region in the ternary structure coincides with the N-terminus of Vps28 helix α4 in the binary complex.
These large structural changes result in some repacking of the hydrophobic core of the Vps23:Vps28 portion of the ternary complex and a restructuring of the exposed hydrophobic hole on the distal face of Vps28. The most significant change is that Phe-100 points inward into the hydrophobic core in the binary complex, where it contacts Vps23:Ile-377, the aliphatic part of Vps23:Arg-374, and the ordered internal DDAO molecule. In the ternary complex, Phe-100 points outward and buries itself in the hydrophobic hole in the second ternary complex of the asymmetric unit (Fig. 4F). In place of the ordered DDAO molecule that occupies part of the Vps28 hydrophobic hole in the binary complex, the Phe-100 of the opposing complex fills in the hole, whose size is diminished in the ternary complex.
Phenotypic analysis of interfaces and Vps28 C-domain in sorting
To determine which subunit interfaces participate in the assembly of ESCRT-I, and which interactions are required for function, a series of mutations were engineered that blocked each of the subunit interfaces. Most interface mutants had normal phenotypes such as Vps23:A367P (Supp. Table 3), which was designed to destabilize Vps23 helix α2. Some of the mutants with normal phenotypes were tested for their ability to form normal complexes in vivo, and A367P (Fig. 5B) and all such mutants tested formed intact complexes (Supp. Table 3). We were able to engineer at least one mutant form of each participating subunit in each interface that disrupted the complex and led to an abnormal phenotype. To probe the Vps23:Vps28 interaction, the Vps23:F371D mutation was engineered to disrupt hydrophobic interactions between Vps23 and Leu 40, Tyr 44 of Vps28. Vps23 F371D has a class E phenotype (Fig. 5A), and forms a 1:1 Vps23:Vps37 subcomplex (Fig. 5B). The Vps28:L40D/Y44D double mutant disrupts the same hydrophobic cluster at Vps23:F371D. This mutant has a class E phenotype and forms a presumed Vps28 monomer (not detected by S300 gel filtration) and a 1:1 Vps23:Vps37 subcomplex (Fig. 5B). The size reduction of Vps23-containing complex in both mutant cells is similar to that observed in vps28Δ strains (Babst et al., 2000).
Figure 5. Sorting defects and complexex of ESCRT-I interface mutants.
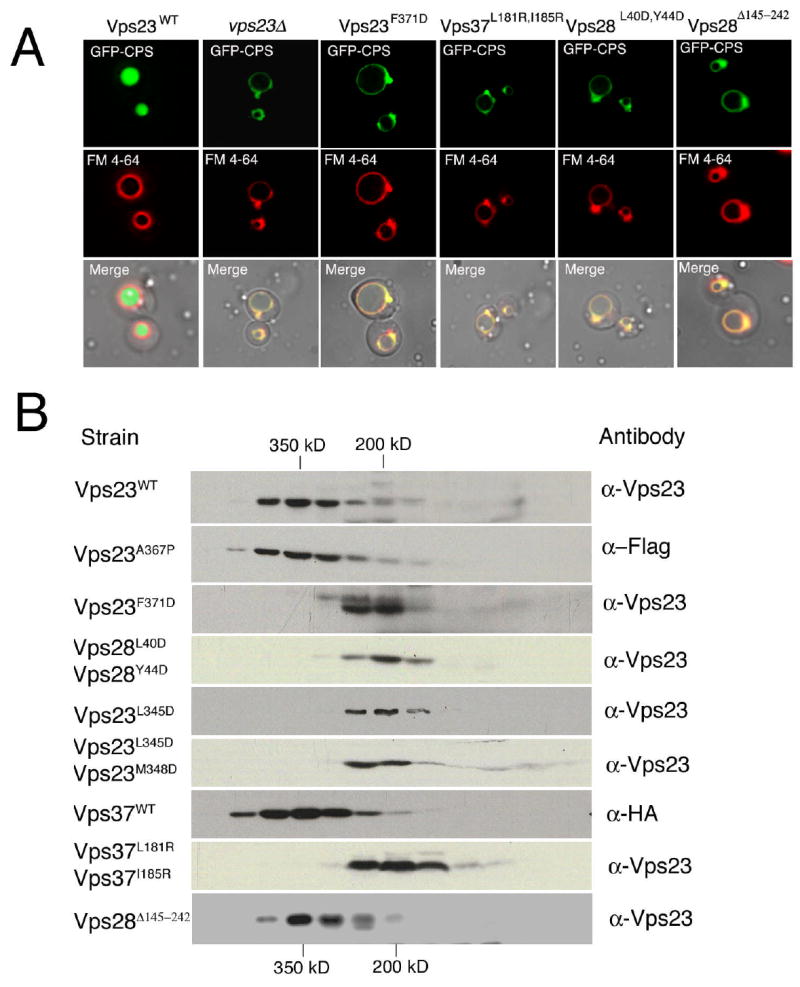
A. MVB cargo is visualized using a GFP-CPS fusion, and the limiting membrane of the vacuole is visualized with FM4–64. Normarski optics (bottom panels) and fluorescence localization of GFP-CPS (top panels) and FM 4–64 (middle panels) in vps23Δ cells expressing either wt Vps23, no Vps23 (vps23Δ) or Vps23F371D, in vps37Δ cells expressing Vps37L181R,I185R, in vps28Δ cells expressing Vps28L40D,Y44D or Vps28Δ145–242. B. Mutations at the molecular interface of ESCRT-I disrupt complex assembly in vivo as determined by gel filtration chromatography. Elution of molecular weight standards are indicated on the top and at the bottom. The column fractions were analyzed by western blot using antibodies specific for Vps23, HA and Flag epitopes.
To analyse interaction between Vps23 and Vps37, mutations were made in Vps23 and in the binding sites for Vps23 on Vps37. The Vps23:L345D/M348D double mutant disrupts hydrophobic interactions between Vps23 and Leu 181, Ile 185 of Vps37. Both Vps23:L345D and L345D/M348D have a class E phenotype, and form a 1:1 Vps23:Vps28 subcomplex. The Vps37:L181R/I185R double mutant disrupts the same hydrophobic interactions at Leu 345 and Met 348 of Vps23, leading to a class E phenotype and formation of a presumed Vps37 monomer (not detected by S300 gel filtration) and 1:1 Vps23:Vps28 subcomplexes. The size reduction of Vps23-containing complex is similar to that observed in vps37Δ strains (Katzmann et al., 2001). The behavior of these mutants shows that the Vps23:Vps28 and Vps23:Vps37 interfaces can be disrupted by mutations, that subcomplexes can exist in vivo when a given interface is disrupted, and that such subcomplexes are not functional in vivo. We find a one-to-one correspondence between those interface mutants that disrupt physical formation of the complex and that yield a class E phenotype.
As described below, we identified the Vps28 C-domain as the locus for binding to the ESCRT-II complex. Deletion of the Vps28 C-domain leads to a strong class E phenotype (Fig. 5a), consistent with an essential function for this domain. However, the ESCRT-I complex still assembles normally in vivo (Fig. 5b). In contrast to the interfacial mutants described above, the block in function is not due to disruption of complex formation. Rather, it is consistent with the loss of an essential interaction with a partner external to the ESCRT-I complex.
Protein:protein interactions
In order to determine which part of the ESCRT-I complex binds to the ESCRT-II complex, His6-tagged ESCRT-I core and Vps28 C-domain were immobilized on Co-Talon resin and assessed for the ability to bind the intact recombinant ESCRT-II complex. The Vps28 C-domain bound to the intact ESCRT-II complex. However, no interaction was observed with the ESCRT-I core, with the ESCRT-II core complex (Vps22:Vps25:Vps36 Δ1–393), or with a putative domain in Vps36 comprising residues 289–385 (Fig. 6A). This suggests that the Vps28 C-domain is the major locus for ESCRT-II binding by the ESCRT-I complex, and that Vps28 C-domain binding to ESCRT-II requires Vps36 residues 1–288. This region of Vps36 consists largely of a GLUE domain into which two NZF domains are inserted. The GLUE domain is conserved in human Vps36 (Slagsvold et al., 2005), but the NZF domains are not, so it seems likely that the Vps28 C-domain binds to the Vps36 GLUE domain.
Figure 6. Structural and functional organization of ESCRT-I.

A. The Vps28 C-domain binds to full length ESCRT-II complex as determined by a pull down assay. Lanes on the SDS-PAGE gel are labeled at top. Molecular weights at left are in kDa. B. Structures are shown for the UEV domain of human Vps23 in complex with ubiquitinated pro-CPS (red) (Sundquist et al., 2004) and the HIV-1 p6 PTAP-containing peptide (magenta) (Pornillos et al., 2002). The human domain is shown because the complexes with the PTAP peptide and ubiquitin are both available. The predicted coiled coil domain in the central part of Vps23 is omitted for simplicity.
Discussion
Structural analysis of the ESCRT-I complex allows us to understand the assembly and organization of this key trafficking complex (Fig. 6B). The core complex is a 1:1:1 heterotrimer. The UEV domain of Vps23 is monomeric in isolation and bound to ubiquitin and PTAP motif peptides (Pornillos et al., 2002; Sundquist et al., 2004; Teo et al., 2004b), and is unlikely to influence the oligomerization of the 1:1:1 heterotrimer. The fundamental unit of the full-length Vps23:Vps28:Vps37 complex is therefore a heterotrimer of 1:1:1 stoichiometry. The core of Vps23 identified here coincides with the region of human Vps23 identified as a steadiness box required for the stability of human ESCRT-I (Feng et al., 2000) and that interacts with human Vps28 (von Schwedler et al., 2003). Our finding that the C-terminus of Vps37 is part of the core agrees with the finding the corresponding part of the “ModR” motif of human Vps37A is critical for association with the rest of the human ESCRT-I complex (Bache et al., 2004).
The EIΔUEV-A core complex, the construct closest to intact ESCRT-I that we were able to characterize in recombinant form, has a lower native molecular weight than the naturally occurring ESCRT-I complex purified from yeast (Katzmann et al., 2001), which has a native molecular weight estimated in the 350 kDa range on the basis of gel filtration chromatography of complexes obtained from yeast. The UEV domain alone only accounts for 18.1 kDa per Vps23 subunit. The EIΔUEV-A complex shows a mixture of interconverting monomers and dimers species, while all other ternary complexes examined by sedimentation analysis were monomeric. Gel filtration results for EIΔUEV overstate the true native molecular weight, and the same could be true for the intact ESCRT-I complex. There is a time-dependent increase in the aggregation of this material. These factors may account in part of the difference between these results and the previous estimate of 350 kDa (Katzmann et al., 2001). We cannot exclude that there are factors that have yet to be identified that can promote oligomerization, of the fundamental 1:1:1 unit in vivo.
We found that ESCRT-I consists of a tightly associated core comprising one-third of the mass of the complex, with the remainder consisting of additional domains and their linkers projecting from the central core (Fig. 6B). This type of hub-and-spokes organization is now familiar in trafficking complexes. The best characterized of such complexes are the AP clathrin adaptor complexes. AP-1 and -2 consist of a core comprising just over two-thirds of the mass of the entire complex, and two large appendage domains linked to the core by long flexible linkers (Collins et al., 2002; Heldwein et al., 2004). The yeast ESCRT-II complex consists of a core comprising some two-thirds of the mass of the complex, with a series of additional domains linked to the N-terminus of the core portion of one subunit, Vps36 (Hierro et al., 2004; Teo et al., 2004a). The ESCRT-I complex is distinguished from complexes such as AP-2 and ESCRT-II in that relative to the total mass, the portion devoted to the core is about half of that found in these other complexes. However, the same basic core-and-appendages formula applies to all of these systems.
The three subunits of ESCRT-I have no detectable primary sequence similarity to each other, yet they associate with each other through two-helix core domains that are strikingly similar. This recapitulates a similar observation in ESCRT-II, that all three subunits of that complex, despite a complete lack of detectable sequence similarity, all contain core tandem winged-helix domains (Hierro et al., 2004; Teo et al., 2004a). Pseudosymmetric internal repeats are often seen in structures of single polypeptide chains. In such proteins it is usually speculated that these protein structures arose by the concatenation of duplicated genes, followed by their divergence over time. It is tempting to speculate that the multiple subunits of the ESCRT-I and -II complexes might have evolved from a common ancestor, one each for ESCRT-I and –II.
Much of the interest in the ESCRT pathway derives from its role in the budding of the HIV and other retroviruses. Most of the residues involved in subunit interfaces in the yeast complex are conserved in the human orthologs of ESCRT-I subunits. All but two of the eleven residues that are found in the hydrophobic hole on the surface of ESCRT-I are highly conserved from yeast to human (Fig. 3, 4F). This suggests that the human ESCRT-I complex will have a similar fundamental architecture and will possess a similar hydrophobic hole on its surface. Although we do not know the function of the hydrophobic hole, its conservation suggests it will have some functional importance. A major goal of research into the human ESCRT-I complex is to develop anti-HIV therapeutics targeted at this complex, as the human ESCRT-I complex has been shown to be essential for HIV budding (Demirov et al., 2002; Eastman et al., 2005; Garrus et al., 2001; Goila-Gaur et al., 2003; Martin-Serrano et al., 2001; Martin-Serrano et al., 2003; Scarlata and Carter, 2003; Strack et al., 2003; Stuchell et al., 2004; VerPlank et al., 2001; von Schwedler et al., 2003), as reviewed by (Freed, 2003; Morita and Sundquist, 2004). It is unlikely that a small molecule inhibitor could be developed to disrupt the assembly of the complex through competition with subunit interfaces, since these surfaces are so large. On the other hand, the hydrophobic hole presents a more attractive target. We have already found that it can bind an ordered detergent molecule, and that it can undergo conformational changes. A number of effective inhibitors of protein:protein complexes act not by blocking interfaces, but rather by blocking conformational cycling by stabilizing and trapping one conformation out of all the steps in a conformational cycle (Pommier and Cherfils, 2005). While we are unsure whether the conformational change occurs as part of a physiological regulatory mechanism or not, the observation that Vps28 is capable of such a structural change is encouraging for the development of small molecule ESCRT-I blockers.
Taken together, the structural and functional results presented here provide a picture of the organization of the ESCRT-I complex. We found that the fundamental unit of the ESCRT-I complex is a 1:1:1 heterotrimer, which appears to be capable of forming weakly interacting dimers in vitro and higher-order oligomers in vivo. The complex is held together by a compact core that comprises only a third of the mass of the complex. From this core, several domains project to bind a variety of partners. The Vps23 UEV domain, which is responsible for binding ubiquitinated cargo and the Vps27 upstream complex, is connected to the core by 160 residues, some predicted to be in flexible linkers and others to form a coiled-coil domain. The Vps28 C-domain, which is responsible for binding ESCRT-II, in contrast, is connected by a linker of less than 20 residues. The Vps28 C-domain is intimately connected to the conformationally labile portion of the Vps28 core domain. It will be important to investigate whether ESCRT-II binding could regulate a conformational change in the ESCRT-I core, and to identify binding partners for the Vps37 N-domain and Vps23 central coiled coil domain. The structural model presented here provides the framework needed for a complete understanding of the functional organization of ESCRT-I.
Experimental Procedures
Expression Vectors
DNAs coding for the appropriate regions of yeast Vps23, Vps28, and Vps37 (Fig. 1A) were amplified by PCR to generate cassettes containing the Shine-Delgarno translational start signal (Hierro et al., 2005) and cloned directly into the polycistronic pST39 vector (Tan, 2001).
Limited Proteolysis, N-terminal Amino Acid Sequencing, and Mass Spectrometry
The EIΔUEV-B complex was digested with varying concentrations of trypsin or endoproteinase Glu-C at 4°C overnight. The digested protein samples were separated on SDS-polyacrylamide gel and transferred to polyvinylidene fluoride membrane, and stained with SimplyBlue™ SafeStain (Invitrogen). Bands were excised and subject to N-terminal amino acid sequencing using a 492cLC Protein Sequencer (Applied Biosystems). Limited proteolysis was also combined with gel filtration to isolate and identify core subcomplexes of ESCRT-I. Proteolyzed samples of EIΔUEV-B were fractionated by passing them over a Superdex 200 column (Pharmacia). Resulting gel filtration fractions were further separated by SDS-PAGE and subsequently sequenced. In some cases, mass spectrometry (MALDI-TOF) analysis was utilized to confirm the identity of the proteins in the associated subcomplexes. MALDI-TOF samples were prepared by mixing 1 μL of protein sample in 49 μL of matrix buffer [0.07% trifluoroacetic acid and 33% acetonitrile]. A matrix solution was prepared by mixing excess sinapinic acid with 30 μL of matrix buffer. One microliter of both sample solution and matrix solution were spotted on a gold plate and were dried at room temperature. The plated samples were then analyzed on a Voyager-DE MALDI-TOF mass spectrometer (Applied Biosystems).
Crystallization and Data Collection
Crystals of the binary-A and B subcomplexes were grown at 4°C by hanging drop vapor diffusion mixing 2 μL of 5 mg/mL protein solution [40 mM Tris (pH 7.4), 120 mM NaCl, 0.02 M DDAO] with 13% PEG 3350, 200 mM MgCl2, and 20% glycerol. Crystals were looped directly from the crystal drop and flash frozen in liquid nitrogen in preparation for low temperature data collection. Se edge MAD data were collected from the binary-A crystals at Stanford Synchrotron Radiation Laboratory beamline 9–2, and data were indexed and reduced using MOSFLM and SCALA (CCP4, 1994). Native data on the binary-B crystals were collected at Advanced Photon Source beamline SER-CAT 22-ID and processed using HKL2000 (HKL Research).
Crystals of the core complex were grown at 25°C by hanging drop vapor diffusion by mixing 2 μL of 6 mg/mL protein solution with equal volume mother liquor [100 mM tri-sodium citrate (pH 5.6), 900 mM lithium sulfate and 500 mM ammonium sulfate]. Streak seeding performed after one day of equilibration was carried out to obtain diffraction quality single crystals, which appeared in 1–2 days. Crystals were cryo-protected in mother liquor supplemented with 25% glycerol and flash frozen in liquid nitrogen. Data were collected at APS beamline 22-ID and indexed and reduced using HKL2000 (HKL Research).
Structure Determination
Phases were calculated for binary-A from a two wavelength (Gonzalez et al., 1999) Se-MAD data set to 3.35 using SOLVE (Terwilliger and Berendzen, 1999). Two selenium sites were located and the resulting electron density map was improved using density modification in RESOLVE (Terwilliger, 2000). The map was traced using the modeling program O and refined using CNS (Brunger et al., 1998). The structure of the binary-B subcomplex was solved by molecular replacement using MOLREP (Vagin and Teplyakov, 1997), using binary-A a search model. Two copies of the complex were found resulting in an R factor of 44.8% and a correlation coefficient of 55.8%. The model was improved by cycles of manual building in O and refined using CNS and REFMAC with TLS parameter refinement (Murshudov et al., 1997).
The structure of the ESCRT-I core complex was determined by molecular replacement using MOLREP with binary-A as a search model. Two solutions were found resulting in an R factor of 53.3% and correlation coefficient of 33.6 %. Subsequent refinement in CNS dropped the R factor and free R factor to 41.2% and 46.8%, respectively. The resulting electron density maps were improved by prime-and-switch phasing (Terwilliger, 2004) using RESOLVE. The prime-and-switch electron density map was sufficient to manually build two copies of the structure of Vps37 absent from the search model as well as substantial reconfiguration of the C-terminus of Vps28. Manual building for this structure was completed utilizing O (Jones et al., 1991) and CNS was used in refinement of the structure.
Microscopy
Living cells expressing the GFP–CPS chimaera were harvested at an A600 of 0.4–0.6, labelled with FM4-64 for vacuolar membrane staining and resuspended in medium for visualization. Visualization of cells was performed on a fluorescence microscope (Axiovert S1002TV; Carl Zeiss MicroImaging, Inc.) equipped with fluorescein isothiocyanate (FITC) and rhodamine filters, captured with a digital camera (Photometrix) and deconvolved using Delta Vision software (Applied Precision Inc.). Results presented were based on observations of >120 cells.
Gel filtration analysis of yeast complexes
For gel filtration analysis, yeast cells were spheroplasted and lysed in PBS containing 0.1 mM AEBSF, 1 og ml−1 pepstatin A, 1 og ml−1 leupeptin, 1 mM benzamide and protease inhibitor cocktail (Complete; Roche Molecular Biochemicals). The lysate was precleared for 5 min at 300g followed by a 100,000g centrifugation for 1 hour at 4°C. The following lysate was loaded onto a Sephacryl S300 column (16/60; Amersham Life Sciences) and separated with PBS. Fractions were analysed with western blotting using anti-HA, anti-Flag monoclonal antibodies and anti-Vps23 polycolonal antibody.
Pull Down Assay
Samples of the ESCRT-I core, full-length and Vps36 Δ1-393 ESCRT-II complexes, the Vps36 289–385 fragment, and bovine serum albumin (BSA) were mixed at a molar ratio 3:1 with His6-Vps28-C in 200 μl of binding buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 5 mM β– ME). After 1 h incubation at room temperature, 50 μl of Co-Talon resin pre-equilibrated with the binding buffer was added and the mixture was incubated for 2 h. The resin was washed three times with 200 volumes of binding buffer plus 5 mM imidazole to remove unbound material. The bound proteins were eluted by boiling in sample buffer and visualized in SDS 12% polyacrylamide gel electrophoresis using Coomassie blue staining.
Supplementary Material
Acknowledgments
We thank Bridgette Beach for making DNA constructs, Alexander Rusnak for mutant construction and phenotypic analysis, Robert Craigie for assistance with protein sequencing, Gali Prag for assistance with data collection, Yihong Ye, Eric Freed, and Tony Chu for discussions and comments on the manuscript, and the staffs of beamlines 9–2, Stanford Synchrotron Radiation Laboratory (SSRL), and 22-ID, Advanced Photon Source (APS), Argonne National Laboratory (ANL) for assistance with x-ray data collection and for access to beam time. M. S. K. is a PRAT fellow of the NIGMS, NIH. This research was supported by NIH intramural support, NIDDK and IATAP, to J. H. H. and by the Howard Hughes Medical Institute to S. D. E. The SSRL, a national user facility operated by Stanford University on behalf of the U.S. DOE, Office of Basic Energy Sciences, is supported by the DOE, Office of Biological and Environmental Research, and by the NIH, National Center for Research Resources, Biomedical Technology Program, and the National Institute of General Medical Sciences. Use of the APS was supported by the U. S. DOE, Basic Energy Sciences, Office of Science, under Contract No.W-31-109-Eng-38.
Footnotes
Accession numbers
Coordinates have been deposited in the protein data bank for the binary and ternary complexes with accession numbers 2F6M and 2F66, respectively.
References
- Alam SL, Sun J, Payne M, Welch BD, Black BK, Davis DR, Meyer HH, Emr SD, Sundquist WI. Ubiquitin interactions of NZF zinc fingers. Embo J. 2004;23:1411–1421. doi: 10.1038/sj.emboj.7600114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amit I, Yakir L, Katz M, Zwang Y, Marmor MD, Citri A, Shtiegman K, Alroy I, Tuvia S, Reiss Y, et al. Tal, a Tsg101-specific E3 ubiquitin ligase, regulates receptor endocytosis and retrovirus budding. Genes Dev. 2004;18:1737–1752. doi: 10.1101/gad.294904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babst M, Katzmann DJ, Estepa-Sabal EJ, Meerloo T, Emr SD. ESCRT-III: An endosome-associated heterooligomeric protein complex required for MVB sorting. Dev Cell. 2002;3:271–282. doi: 10.1016/s1534-5807(02)00220-4. [DOI] [PubMed] [Google Scholar]
- Babst M, Odorizzi G, Estepa EJ, Emr SD. Mammalian tumor susceptibility gene 101 (TSG101) and the yeast homologue, Vps23p, both function in late endosomal trafficking. Traffic. 2000;1:248–258. doi: 10.1034/j.1600-0854.2000.010307.x. [DOI] [PubMed] [Google Scholar]
- Bache KG, Brech A, Mehlum A, Stenmark H. Hrs regulates multivesicular body formation via ESCRT recruitment to endosomes. J Cell Biol. 2003;162:435–442. doi: 10.1083/jcb.200302131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bache KG, Slagsvold T, Cabezas A, Rosendal KR, Raiborg C, Stenmark H. The growth-regulatory protein HCRP1/hVps37A is a subunit of mammalian ESCRT-I and mediates receptor down-regulation. Mol Biol Cell. 2004;15:4337–4346. doi: 10.1091/mbc.E04-03-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bilodeau PS, Winistorfer SC, Kearney WR, Robertson AD, Piper RC. Vps27-Hse1 and ESCRT-I complexes cooperate to increase efficiency of sorting ubiquitinated proteins at the endosome. J Cell Biol. 2003;163:237–243. doi: 10.1083/jcb.200305007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowers K, Stevens TH. Protein transport from the late Golgi to the vacuole in the yeast Saccharomyces cerevisiae. Biochimica Et Biophysica Acta-Molecular Cell Research. 2005;1744:438–454. doi: 10.1016/j.bbamcr.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr Sect D-Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- CCP4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr A. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Collins BM, McCoy AJ, Kent HM, Evans PR, Owen DJ. Molecular architecture and functional model of the endocytic AP2 complex. Cell. 2002;109:523–535. doi: 10.1016/s0092-8674(02)00735-3. [DOI] [PubMed] [Google Scholar]
- Conibear E. An ESCRT into the endosome. Mol Cell. 2002;10:215–216. doi: 10.1016/s1097-2765(02)00601-9. [DOI] [PubMed] [Google Scholar]
- Demirov DG, Ono A, Orenstein JM, Freed EO. Overexpression of the N-terminal domain of TSG101 inhibits HIV-1 budding by blocking late domain function. Proc Natl Acad Sci U S A. 2002;99:955–960. doi: 10.1073/pnas.032511899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eastman SW, Martin-Serrano J, Chung W, Zang T, Bieniasz PD. Identification of human VPS37C, a component of endosomal sorting complex required for transport-I important for viral budding. J Biol Chem. 2005;280:628–636. doi: 10.1074/jbc.M410384200. [DOI] [PubMed] [Google Scholar]
- Feng GH, Lih CJ, Cohen SN. TSG101 protein steady-state level is regulated posttranslationally by an evolutionarily conserved COOH-terminal sequence. Cancer Res. 2000;60:1736–1741. [PubMed] [Google Scholar]
- Freed EO. The HIV-TSG101 interface: recent advances in a budding field. Trends in Microbiology. 2003;11:56–59. doi: 10.1016/s0966-842x(02)00013-6. [DOI] [PubMed] [Google Scholar]
- Garrus JE, von Schwedler UK, Pornillos OW, Morham SG, Zavitz KH, Wang HE, Wettstein DA, Stray KM, Cote M, Rich RL, et al. Tsg101 and the vacuolar protein sorting pathway are essential for HIV-1 budding. Cell. 2001;107:55–65. doi: 10.1016/s0092-8674(01)00506-2. [DOI] [PubMed] [Google Scholar]
- Goila-Gaur R, Demirov DG, Orenstein JM, Ono A, Freed EO. Defects in human immunodeficiency virus budding and endosomal sorting induced by TSG101 overexpression. J Virol. 2003;77:6507–6519. doi: 10.1128/JVI.77.11.6507-6519.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A, Pedelacq JD, Sola M, Gomis-Ruth FX, Coll M, Samama JP, Benini S. Two-wavelength MAD phasing: in search of the optimal choice of wavelengths. Acta Crystallogr Sect D-Biol Crystallogr. 1999;55:1449–1458. doi: 10.1107/s0907444999006745. [DOI] [PubMed] [Google Scholar]
- Gorden P, Carpentier JL, Cohen S, Orci L. Epidermal growth factor: morphological demonstration of binding, internalization, and lysosomal association in human fibroblasts. Proc Natl Acad Sci U S A. 1978;75:5025–5029. doi: 10.1073/pnas.75.10.5025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenberg J, Stenmark H. The biogenesis of multivesicular endosomes. Nat Rev Mol Cell Biol. 2004;5:317–323. doi: 10.1038/nrm1360. [DOI] [PubMed] [Google Scholar]
- Haglund K, Di Fiore PP, Dikic I. Distinct monoubiquitin signals in receptor endocytosis. Trends BiochemSci. 2003;28:598–603. doi: 10.1016/j.tibs.2003.09.005. [DOI] [PubMed] [Google Scholar]
- Haigler HT, McKanna JA, Cohen S. Direct visualization of the binding and internalization of a ferritin conjugate of epidermal growth factor in human carcinoma cells A-431. J Cell Biol. 1979;81:382–395. doi: 10.1083/jcb.81.2.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heldwein EE, Macia E, Jing W, Yin HL, Kirchhausen T, Harrison SC. Crystal structure of the clathrin adaptor protein 1 core. Proc Natl Acad Sci U S A. 2004;101:14108–14113. doi: 10.1073/pnas.0406102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershko A, Ciechanover A, Varshavsky A. The ubiquitin system. Nat Med. 2000;6:1073–1081. doi: 10.1038/80384. [DOI] [PubMed] [Google Scholar]
- Hicke L. Protein regulation by monoubiquitin. Nat Rev Mol Cell Biol. 2001;2:195–201. doi: 10.1038/35056583. [DOI] [PubMed] [Google Scholar]
- Hicke L, Dunn R. Regulation of membrane protein transport by ubiquitin and ubiquitin-binding proteins. Annu Rev Cell Dev Biol. 2003;19:141–172. doi: 10.1146/annurev.cellbio.19.110701.154617. [DOI] [PubMed] [Google Scholar]
- Hierro A, Kim J, Hurley JH. Polycistronic expression and purification of the ESCRT-II endosomal trafficking complex. Methods Enzymol. 2005;403 doi: 10.1016/S0076-6879(05)03027-2. in press. [DOI] [PubMed] [Google Scholar]
- Hierro A, Sun J, Rusnak AS, Kim J, Prag G, Emr SD, Hurley JH. Structure of the ESCRT-II endosomal trafficking complex. Nature. 2004;431:221–225. doi: 10.1038/nature02914. [DOI] [PubMed] [Google Scholar]
- Hochstrasser M. Evolution and function of ubiquitin-like protein-conjugation systems. Nat Cell Biol. 2000;2:E153–E157. doi: 10.1038/35019643. [DOI] [PubMed] [Google Scholar]
- Hurley JH, Emr SD. The ESCRT complexes: structure mechanism of a membrane-trafficking network. Annu Rev Biophys Biomolec Struct. 35 doi: 10.1146/annurev.biophys.35.040405.102126. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Zou JY, Cowan SW, Kjeldgaard M. Improved methods for building protein models in electron- density maps and the location of errors in these models. Acta Crystallogr Sect A. 1991;47:110–119. doi: 10.1107/s0108767390010224. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Babst M, Emr SD. Ubiquitin-dependent sorting into the multivesicular body pathway requires the function of a conserved endosomal protein sorting complex, ESCRT-I. Cell . 2001;106:145–155. doi: 10.1016/s0092-8674(01)00434-2. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Odorizzi G, Emr SD. Receptor downregulation and multivesicular-body sorting. Nat Rev Mol Cell Biol. 2002;3:893–905. doi: 10.1038/nrm973. [DOI] [PubMed] [Google Scholar]
- Katzmann DJ, Stefan CJ, Babst M, Emr SD. Vps27 recruits ESCRT machinery to endosomes during MVB sorting. J Cell Biol. 2003;162:413–423. doi: 10.1083/jcb.200302136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. HIV-I and Ebola virus encode small peptide motifs that recruit Tsg101 to sites of particle assembly to facilitate egress. Nat Med. 2001;7:1313–1319. doi: 10.1038/nm1201-1313. [DOI] [PubMed] [Google Scholar]
- Martin-Serrano J, Zang T, Bieniasz PD. Role of ESCRT-I in retroviral budding. J Virol. 2003;77:4794–4804. doi: 10.1128/JVI.77.8.4794-4804.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morita E, Sundquist WI. Retrovirus budding. Annu Rev Cell Dev Biol. 2004;20:395–425. doi: 10.1146/annurev.cellbio.20.010403.102350. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum- likelihood method. Acta Crystallogr Sect D-Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Odorizzi G, Babst M, Emr SD. Fab1p PtdIns(3)P 5-kinase function essential for protein sorting in the multivesicular body. Cell. 1998;95:847–858. doi: 10.1016/s0092-8674(00)81707-9. [DOI] [PubMed] [Google Scholar]
- Pickart CM. Mechanisms underlying ubiquitination. Annu Rev Biochem. 2001;70:503–533. doi: 10.1146/annurev.biochem.70.1.503. [DOI] [PubMed] [Google Scholar]
- Pommier Y, Cherfils J. Interfacial inhibition of macromolecular interactions: nature's paradigm for drug discovery. Trends Pharmacol Sci. 2005;26:138–145. doi: 10.1016/j.tips.2005.01.008. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Alam SL, Davis DR, Sundquist WI. Structure of the Tsg101 UEV domain in complex with the PTAP motif of the HIV-1 p6 protein. Nat Struct Biol. 2002;9:812–817. doi: 10.1038/nsb856. [DOI] [PubMed] [Google Scholar]
- Pornillos O, Higginson DS, Stray KM, Fisher RD, Garrus JE, Payne M, He GP, Wang HE, Morham SG, Sundquist WI. HIV Gag mimics the Tsg101-recruiting activity of the human Hrs protein. J Cell Biol. 2003;162:425–434. doi: 10.1083/jcb.200302138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raiborg C, Rusten TE, Stenmark H. Protein sorting into multivesicular endosomes. Curr Opin Cell Biol. 2003;15:446–455. doi: 10.1016/s0955-0674(03)00080-2. [DOI] [PubMed] [Google Scholar]
- Scarlata S, Carter C. Role of HIV-1 Gag domains in viral assembly. Biochimica Et Biophysica Acta-Biomembranes. 2003;1614:62–72. doi: 10.1016/s0005-2736(03)00163-9. [DOI] [PubMed] [Google Scholar]
- Sigismund S, Polo S, Di Fiore PP. Signalling from Internalized Growth Factor Receptors. 2004. Signaling through monoubiquitination; pp. 149–185. [DOI] [PubMed] [Google Scholar]
- Slagsvold T, Aasland R, Hirano S, Bache KG, Raiborg C, Trambaiano D, Wakatsuki S, Stenmark H. Eap45 in mammalian ESCRT-II binds ubiquitin via a phosphoinositide-interacting GLUE domain. J Biol Chem. 2005;280:19600–19606. doi: 10.1074/jbc.M501510200. [DOI] [PubMed] [Google Scholar]
- Strack B, Calistri A, Craig S, Popova E, Gottlinger HG. AIP1/ALIX is a binding partner for HIV-1 p6 and EIAV p9 functioning in virus budding. Cell. 2003;114:689–699. doi: 10.1016/s0092-8674(03)00653-6. [DOI] [PubMed] [Google Scholar]
- Stuchell MD, Garrus JE, Muller B, Stray KM, Ghaffarian S, McKinnon R, Krausslich HG, Morham SG, Sundquist WI. The human endosomal sorting complex required for transport (ESCRT-I) and its role in HIV-1 budding. J Biol Chem. 2004;279:36059–36071. doi: 10.1074/jbc.M405226200. [DOI] [PubMed] [Google Scholar]
- Sundquist WI, Schubert HL, Kelly BN, Hill GC, Holton JM, Hill CP. Ubiquitin recognition by the human TSG101 protein. Mol Cell. 2004;13:783–789. doi: 10.1016/s1097-2765(04)00129-7. [DOI] [PubMed] [Google Scholar]
- Tan S. A Modular Polycistronic Expression System for Overexpressing Protein Complexes in Escherichia coli. Protein Expr Purif. 2001;21:224–234. doi: 10.1006/prep.2000.1363. [DOI] [PubMed] [Google Scholar]
- Teo H, Perisic O, Gonzalez B, Williams RL. ESCRT-II, an endosome-associated complex required for protein sorting: Crystal structure and interactions with ESCRT-III and membranes. Dev Cell. 2004a;7:559–569. doi: 10.1016/j.devcel.2004.09.003. [DOI] [PubMed] [Google Scholar]
- Teo H, Veprintsev DB, Williams RL. Structural insights into endosomal sorting complex required for transport (ESCRT-I) recognition of ubiquitinated proteins. J Biol Chem. 2004b;279:28689–28696. doi: 10.1074/jbc.M400023200. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC. Maximum-likelihood density modification. Acta Crystallogr Sect D-Biol Crystallogr. 2000;56:965–972. doi: 10.1107/S0907444900005072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terwilliger TC. Using prime-and-switch phasing to reduce model bias in molecular replacement. Acta Crystallogr Sect D-Biol Crystallogr. 2004;60:2144–2149. doi: 10.1107/S0907444904019535. [DOI] [PubMed] [Google Scholar]
- Terwilliger TC, Berendzen J. Automated MAD and MIR structure solution. Acta Crystallogr Sect D-Biol Crystallogr. 1999;55:849–861. doi: 10.1107/S0907444999000839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vagin AA, Teplyakov A. MOLREP: an automated program for molecular replacement. J Appl Crystallogr. 1997;30:1022–1025. [Google Scholar]
- VerPlank L, Bouamr F, LaGrassa TJ, Agresta B, Kikonyogo A, Leis J, Carter CA. Tsg101, a homologue of ubiquitin-conjugating (E2) enzymes, binds the L domain in HIV type 1 Pr55(Gag) Proc Natl Acad Sci U S A. 2001;98:7724–7729. doi: 10.1073/pnas.131059198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Schwedler UK, Stuchell M, Muller B, Ward DM, Chung HY, Morita E, Wang HE, Davis T, He GP, Cimbora DM, et al. The protein network of HIV budding. Cell. 2003;114:701–713. doi: 10.1016/s0092-8674(03)00714-1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.