

Abstract
The purpose of this study was to investigate (1) whether ischemia-reperfusion increased the content of heat shock protein 72 (Hsp72) transcripts and (2) whether myocardial content of Hsp72 is increased by ischemic preconditioning so that they can be considered as end effectors of preconditioning. Twelve male minipigs (8 protocol, 4 sham) were used, with the following ischemic preconditioning protocol: 3 ischemia and reperfusion 5-minute alternative cycles and last reperfusion cycle of 3 hours. Initial and final transmural biopsies (both in healthy and ischemic areas) were taken in all animals. Heat shock protein 72 messenger ribonucleic acid (mRNA) expression was measured by a semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR) method using complementary DNA normalized against the housekeeping gene cyclophilin. The identification of heat shock protein 72 was performed by immunoblot. In our “classic” preconditioning model, we found no changes in mRNA hsp72 levels or heat shock protein 72 content in the myocardium after 3 hours of reperfusion. Our experimental model is valid and the experimental techniques are appropriate, but the induction of heat shock proteins 72 as end effectors of cardioprotection in ischemic preconditioning does not occur in the first hours after ischemia, but probably at least 24 hours after it, in the so-called “second protection window.”
INTRODUCTION
Ischemic heart disease is the leading cause of morbidity and mortality in the developed world. Thus, in addition to implementing programs to prevent vascular risk factors, it is urgent to search for effective treatments that limit infarction size during the acute phase of coronary occlusion, preserving the viability of ischemic myocardium. Since Jennings and Reimer showed in 1983 that reperfusion was essential to protect ischemic myocardium, thrombolytic therapies gained widespread acceptance. Reperfusion reduced infarction size, but did not suppress infarction, and myocardial cells started to die in a few minutes before thrombus dissolution was achieved by mechanisms not well known even today. Murry et al studied in 1986 the potential existence of a mechanism that slowed adenosine triphosphate (ATP) depletion and was involved in heart protection during a subsequent ischemic episode. These authors showed that four 5-minute cycles of ischemia with intermittent reperfusions protected the heart from a subsequent episode of sustained ischemia, infarction size being reduced by up to 75%. This method for increasing ischemia tolerance was called “cardiac ischemic preconditioning”, and has been shown in all animal species studied to date. However, protection by preconditioning is lost when ischemia lasts longer than 3 hours. Two preconditioning models are described in terms of extension of the protection seen: (1) the “classical preconditioning,” in which the preconditioning state is very transient and only lasts 1–2 hours (Murry et al 1991; Sack et al 1993) and (2) the second protection window (Marber et al 1993), less powerful than the previous one, but longer, that appears from 12 to 72 hours after the preconditioning stimulus (Baxter et al 1997). Both models share pathophysiological similarities. In contrast to the initial view of ischemia as characterized by an energy deficiency, the decisive role of active cellular response to ischemic aggression is universally accepted today. The signal translation systems involved in this response and modulating its consequences are now better understood (Garcia-Dorado 2004): activation of programmed cell death pathways (Eefting et al 2004), changes in the NO/ cGMP pathway (Schulz et al 2004), activation of different protein kinase cascades (Amstrong 2004), and changes in expression of certain genes (Chi and Karliner 2004). It has indeed been shown that hypoxia and reoxygenation alter the pattern of heart proteins, mainly through gene expression changes, changes in messenger ribonucleic acid (mRNA) stability, translation rates, posttranslational protein changes, and degradation; an increase in reactive oxygen species (ROS) also occurs during reperfusion. As a result, cardiac myocytes try to protect themselves by upregulating the levels of antioxidant proteins and stress or heat shock proteins (Hsps) (Polla 1988; Madamanchi et al 2001). The heat shock response (HSR) was described by Ritossa after noting that exposure of salivary glands from Drosophila larvae to high temperatures caused a bulging in the giant chromosomes of these cells (Ritossa 1962). It is now known that these puffs correspond to sites with a strong transcriptional activity for a group of proteins initially called heat shock proteins (Tissières et al 1974), whose cytoprotective role has been widely documented (Voellmy 1996; Burel et al 1997). Multiple reports have subsequently shown that any aggressive agent that alters the protein structure of a cell may induce these proteins (Morimoto et al 1997). Studies on the functionality of the different Hsps have shown that they have an essential role in the processes leading to adequate folding of other cell proteins, acting as molecular chaperones (Hightower and Hendershot 1997), to transport of mitochondrial precursor proteins from the cytosol into the mitochondrion (particularly HSP70) (Hartl and Hayer-Hartl 2002), and in the degradation system of wrongly folded proteins (Friant et al 2003). An altered gene expression of hsps has also been detected in different pathological conditions (Fuller et al 1994; Pockley 2002; Wallin et al 2002; Xu 2002; Kitamura and Nomura 2003). It may therefore be more appropriate to call them stress proteins based on their induction as a universal adaptative response to aggression. The applied nomenclature (Hsps) is primarily derived from the triggering factor leading to occurrence of these proteins.
Hsp72 is cardioprotective, as shown by studies with a myocyte cell line transfected with Hsp72 gene in which cells overexpressing Hsp72 showed significantly higher resistance against hypoxic stress (Heads et al 1995). The cardioprotective effect is also shown by experiments with transgenic animals: the salutary effects of hsp70 transgene expression on metabolic recovery are accompanied by enhanced recovery of contractile function, which expresses Hsp72 in myocardium and shows a great increase in their functional recovery with a decreased infarction size after experimental induction of ischemia and reperfusion (Hutter et al 1996; Radford et al 1996). In addition, an increase in Hsp content caused by hyperthermia before application of an ischemic insult accelerates functional recovery of the stunned heart after the ischemic insult (Cornelussen et al 2001; Latchman 2001; McCormick et al 2003). All of the foregoing suggests that an increased Hsp expression may mediate in the protection conferred by ischemic preconditioning, and that Hsps may be, at least in part, end effectors of such cardioprotection. This function is not restricted to HSP70; other Hsps have been involved in myocardial protection after ischemia-reperfusion, particularly HSP27, which would interact with Akt maintaining the kinase in an active conformation state (Efthymiou et al 2004), HSP60, HSP90, and αβ-crystallin (Chi and Karliner 2004).
The purpose of this study was to investigate (1) whether ischemia-reperfusion increased the content of hsp72 transcripts and (2) whether myocardial content of Hsp72 is increased by ischemic preconditioning so that they may be considered to be end effectors of preconditioning.
MATERIALS AND METHODS
Experiments were conducted at the Unit of Experimental Medicine and Surgery of Hospital General Universitario Gregorio Marañón, Madrid, Spain, (authorized facility number EX 017-U) as per order of August 4, 1989 (BOCM of August 24). Animal handling complied with Spanish (BOE 233/1988) and European committee guidelines for the care and use of laboratory animals (86/609 EEC).
Experimental design
Twelve male Maryland strain miniature pigs with a mean weight of 40 kg, homozygous (dd) for an allele of the major histocompatibility complex, were used in this study (Sach et al 1976). The animals were randomized to one of the following groups: sham group 1 (n = 4); temperature was controlled during surgery using external heat sources. After the instrumentation phase, thoracotomy and dissection of anterior descending (AD) coronary artery were performed; these animals did not undergo ischemic preconditioning. Preconditioned group 2 with controlled temperature (n = 8). After the instrumentation phase, thoracotomy, and dissection of AD coronary artery, the animals were subject to ischemic preconditioning (Fig 1).
Fig 1.

Design of the experimental protocol of cardiac ischemic preconditioning in minipigs
Animals were preanesthetized with ketamine and xylazine, followed by intubation. Complete anesthesia and relaxation were achieved and maintained by injecting repeated boluses of pentobarbital (15 mg/kg) and pancuronium (0.2 mg/kg). An adjustable thermal blanket was placed under the animals to maintain their temperature in the required range (37–39°C) throughout the experiment. Thoracotomy was performed through a median sternotomy, and the pericardium was opened to expose the heart. The AD artery was then dissected in its proximal one-third, passing a tie around it for subsequent manipulations in myocardial perfusion. The following signals were monitored: electrocardiogram (ECG), arterial pressure using a catheter inserted through the right femoral artery, central venous pressure, pulmonary artery pressure, pulmonary capillary pressure, cardiac output measurements using a Swan-Ganz inserted through the right femoral vein, and cardiac, esophageal, and rectal temperature. A PIII computer at 1000 Hz connected to an acquisition system with a 16-channel A-D acquisition card (Keithley Instruments, Cleveland, OH, USA) and a virtual instrumentation software (TestPoint; Capital Equipment, Billerica, MA, USA) both developed at the department, was used for monitoring and recording bioelectric signals.
The preconditioning protocol consisted of 3 ischemia periods of 5 minutes each by complete AD coronary artery occlusion, separated by 3 reperfusion periods also lasting 5 minutes, except for the last reperfusion period, which lasted 3 hours until the final sample was taken. Parameters analyzed to verify the effectiveness of coronary occlusion were as follows: heart rate increase (with or without sporadic extrasystole); ischemic changes in the ECG tracing; changes in blood pressure and pulmonary artery pressure; significant decrease in cardiac output; and obvious dyskinesia in the ischemic area (Figs 2–6). Before the ischemia series, a myocardial biopsy was performed (in the anterior side of left ventricle) with a “tru-cut” biopsy needle to collect a baseline sample. Final samples were obtained after 3 hours since the last reperfusion also by means of biopsies in the ischemic and nonischemic areas of left ventricle. Once the experiments were completed, animals were killed in compliance with the applicable regulations for euthanasia of animals. The “risk area” was verified on the heart extracted from the animal and perfused with methylene blue.
Fig 2.

A representative graphic showing ECG, blood pressure (BP), and pulmonary artery pressure (PAP) of a sham swine at initial sample time
Immediately after extraction, biopsies were washed first with saline and then in phosphate-buffered saline (PBS) with sodium orthovanadate (100 mM), at 4°C, and were kept in liquid nitrogen until processed.
Hsp72 transcript expression
For RNA isolation, samples kept in liquid nitrogen were thawed at 4°C and washed with physiological saline, and RNA was preserved in RNAlater ICE at −40°C overnight. RNAlater ICE was then removed and RNA was extracted twice with Tri-Reagent and chloroform according to the manufacturer's specifications (Chomezynski 1993). RNA samples were resuspended in H2O, incubated for 10 minutes at 57°C, and quantified spectrophotometrically at 260 nm. They all showed an OD260/OD280 ratio ranging from 1.8 to 2.0, ensuring their purity. RNA quality was also checked by electrophoresis in 1% agarose gel labeled with ethidium bromide at 1 μg/mL.
A 2-step semiquantitative reverse transcriptase-polymerase chain reaction (RT-PCR) method was used to measure hsp72 mRNA expression in myocardium samples (Meadus 2003). RNA (250 ng) was reverse-transcribed in 50 μL of reaction solution (Access RT-PCR System; Promega, Madison, WI, USA): 1 μL of dNTPs (ATP, CTP, GTP, TTP); 10 μL of reaction buffer; 0.75 mM Mg+2; 3.3 μL of PCR primers hsp72 (GenBank accession no. NT_007592): sense, 5′-CTCCAGCATCCGACAAGAAGC-3′, and antisense, 5′-ACGGTGTTGTGGGGGTTCAGG-3′; stock concentration (7.5 μM/100 μL); 0.1 U/μL avian myeloblastosis virus (AMV) reverse transcriptase; 0.1 U/ μL Thermus flavus (Tfl) polymerase. RT-PCR was performed in a thermal iCycler (Bio-Rad Laboratories, Hercules, CA, USA) cycler; primary program for RT-PCR initially started with a cycle of 45 minutes at 48°C for reverse transcription, 2 minutes at 94°C for denaturation, followed by 12 cycles of 94°C/30 seconds, 60°C/1 minute, and 68°C/2 minutes. Cyclophilin primers (GenBank accession no. AY008846) were added when 18 cycles were remaining in the same conditions: sense, 5′-ACCGTCTTCTTCGACATCG-3′; antisense, 5′-CAACCACTCAGTCTTGGC-3′. The last extension was kept at 68° for 7 minutes. PCR products were electrophoresed on 2% agarose gels in TAE buffer, stained with ethidium bromide (10 μg/ mL), and photographed on top of a 280-nm ultraviolet (UV) light box. The gel images were digitally captured with a charge-coupled device (CCP) camera and analyzed with Scill-Image software. RT-PCR values are presented as a ratio of the hsp72 gene signal (234 pb) divided by the cyclophilin signal (330 pb).
Quantification of Hsp72 expression
Samples were homogenized in radioimmunoprecipitation assay (RIPA) lysis buffer supplemented with protease and phosphatase inhibitors (phenylmethylsulfonyl fluoride (PMSF) [1 mM], sodium orthovanadate [1 mM], leupeptin [10 μg/mL], aprotinine [10 μg/mL], and pepstatin [10 μg/mL]) using a Potter S homogenizer at 4°C. Lysates were centrifuged in a LB-60 ultracentrifuge (Beckman, Fullerton, CA, USA) at 61 500 × g for 1 hour at 4°C. The sediment was discarded, and the supernatant or soluble fraction was collected.
Protein concentration was measured by the Lowry microassay method (Lowry et al 1951), using the Bio-Rad protein assay kit against an albumin standard curve (0– 10 μg/mL) and a Bio-Rad 3550 Microplate Reader.
Identical aliquot of all samples (50 μg) was denatured by adding sample buffer (Laemmli 1970) (2% sodium dodecyl sulfate (SDS), 10% glycerol, 62.5 mM Tris-HCl, pH 6.8, 0.01% bromophenol blue, 5% 2-mercaptoethanol), boiled for 5 minutes, and centrifuged at 600 × g. Samples were separated by electrophoresis in 10% polyacrylamide gels; when electrophoresis was completed, samples were transferred to polyvinylidene difluoride (PVDF) membranes (Immobilon; Millipore, Bedford, MA, USA).
For identification of myocardial Hsp72 expression, nonspecific bindings of PVDF membranes were blocked by incubation with 3% skimmed powdered milk (w/v) in PBS-Tween 20, 0.05% (v/v). Membranes were then washed 3 times with 0.05% PBS-Tween 20 (v/v), and subsequently incubated with monoclonal antibody (Ab) (StressGen Biotechnologies, Victoria, Canada; SPA 810, HSP72), diluted 1:1000 in 0.05% PBS-Tween 20 (v/v)-0.5% bovine serum albumin (w/v). They were washed 3 times and incubated with a second anti-mouse IgG Ab biotinylated. The signal was amplified by incubation with horseradish peroxidase streptavidin. Membranes were developed using 4-chloro-1-naphthol as substrate. Identification of the tested proteins was completed based on their molecular weight, taking as reference the colored bands of the standards, which were run parallel to the samples. Positive controls were as follows: recombinant human Hsp72 protein (StressGen Biotechnologies; NSP-555) and heat shocked swine heart. Band images were digitized with a SCANJET-II CX scanner (Hewlett-Packard, Palo Alto, CA, USA) with 400 pixels per inch resolution and processed using SCIL-Image (TNO-TNP) software. Total gray levels of test samples were normalized to the average gray levels of control samples.
Statistical analysis
RT-PCR signals were averaged from at least 3 replicates, and Western blot data were generated from 3 replicate runs. Data were expressed as means ± standard error of the mean (SEM). To compare groups, the Mann-Whitney U-test (2-tailed) was used. The significance threshold was set at P < 0.05. All tests were performed using the SPSS, version 12.0, statistical package (SPSS, Chicago, IL, USA).
RESULTS
Ischemic preconditioning model
The model used in our experiment is valid for effective achievement of ischemia in the left ventricular area to be tested, based on ECG changes seen before and after ischemia, with a marked widening of the QRS complex and T wave inversion, and changes in the morphology of both systemic blood pressure (BP) and pulmonary artery pressure (PAP) recording waves, both showing a significant increase after the last 3-hour reperfusion, by 150% in BP recordings and by 200% in PAP recordings, and obvious dyskinesia in the ischemic area (Figs 2–6). This model has been shown effective to reduce the infarct size (Sanz et al 1995).
Myocardial expression of hsp72 mRNAs
The results of RT-PCR with the internal standard housekeeping gene of cyclophilin show no increased in mRNA hsp72 levels after the short ischemia periods and subsequent reperfusion periods, the last one lasting up to 3 hours, in any of the studied groups (Fig 7).
Fig 7.
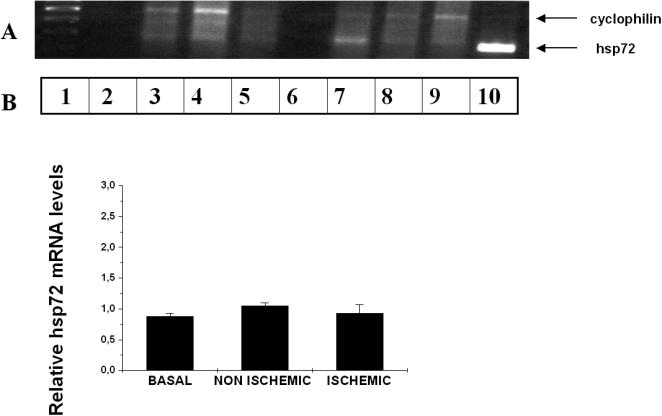
(A) Representative gel image for relative RT-PCR for cyclophilin (330 pb) and HSP72 genes (234 pb) in pig preconditioning myocardium. Lane 1, Molecular weights markers; 2, negative control; 3, baseline sample; 4, nonischemic sample; 5, ischemic sample; 6, negative control; 7, baseline sample; 8, nonischemic sample; 9, ischemic sample; 10, positive control (myocardium sample from swine subjected to total body hyperthermia). (B) Bar graph showing relative Hsp72 mRNA levels in minipig myocardium subjected to ischemia-reperfusion. Densitometric values were corrected to cyclophilin mRNA levels as described in the material and methods section. Results expressed as means (plus minus) standard error of mean. There is no significant increase in Hsp72 mRNA levels after the short ischemia periods and subsequent reperfusion period
Myocardial expression of Hsp72
In our cardiac ischemic preconditioning model, no changes could be shown in Hsp72 content in the preconditioned compared to baseline myocardium (Fig 8).
Fig 8.
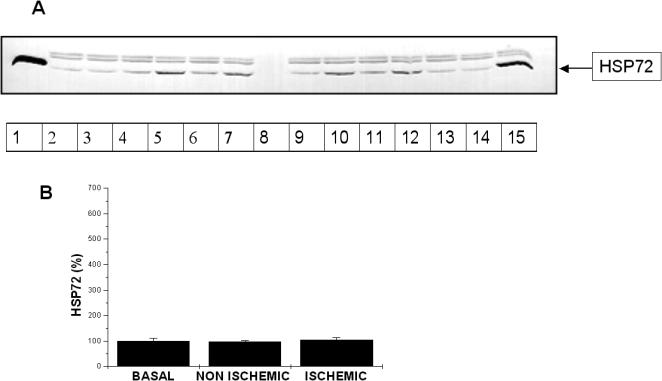
(A) A representative image of a Hsp72 immunoblot in preconditioned myocardium. Lane 1, Positive control, recombinant human Hsp72 protein; 2, baseline sample; 3, nonischemic sample; 4, ischemic sample; 5, baseline sample; 6, nonischemic sample; 7, ischemic sample, 8, protein molecular weight markers; 9, baseline sample; 10, nonischemic sample; 11, ischemic sample; 12, baseline sample; 13, nonischemic sample; 14, ischemic sample; 15, positive control: myocardium sample from swine subjected to total body hyperthermia. The 2 upper bands belong to nonspecific binding of second antibody as we demonstrated performing an immunoblot using only the second antibody. (B) Bar graph showing accumulation of HSP72 in minipig myocardium subjected to ischemic preconditioning. Total gray levels of test samples were normalized to the average gray levels of control samples as described in Materials and Methods. Results are expressed as means ± SEM. There is no significant increase in Hsp72 amount after the short ischemia periods and subsequent reperfusion period
DISCUSSION
The heart is an organ particularly susceptible to hypoxia, because it has only a limited reserve of high energy phosphates. Both hypoxia severity and duration determine cardiac response to a decreased oxygen supply. Hypoxia and oxidative stress induce biochemical and functional changes, despite which the heart attempts to maintain its function to counteract oxygen tension changes (Anaya-Prado et al 2002). Hypoxia and reoxygenation alter the pattern of cardiac proteins through changes in their gene expression, mRNA stability, translation rate, posttranslational modifications, and protein degradation (Piacentini and Karliner 1999). This results in an attempt by ischemic myocardiocytes to protect themselves from ischemia-reperfusion, by up-regulating the levels of stress proteins (Cornelussen et al 2003) and antioxidants (Becker 2004).
Classical studies (Mehta et al 1988) showed induction of a mRNA encoding for a translation product with a molecular weight and isoelectric point similar to HSP70 in the hearts of dogs and rabbits subjected to ischemia. Knowton et al described in 1991 an ischemic preconditioning protocol consisting of 4 cycles of 5 minutes of ischemia/5 minutes of reperfusion in the anterior coronary artery of rabbits, after which an increase in mRNAHsp70 was detected only 1 hour after ischemia, and an increase in HSP70 was found as early as at 2 hours, although levels peaked at 24 hours. Another study using a different preconditioning model in pigs similarly concluded that increases in mRNAHsp72 and Hsp72 occur in the 2 hours following ischemia, with maximum levels reached at 24 hours (Sun et al 1995). It has also been shown that during the second protection window, that is, 24 hours after classical preconditioning, changes occur in protein activity and transcriptional activity, such as increased transcription of several genes, including the hsp72 genes. Hsp72 is synthesized within 3 hours of the ischemic stimulus mainly in cardiac muscle cells. At 24 hours, Hsp72 increase is maintained, and infarction size is significantly reduced (Das et al 1992; Marber et al 1993; Heads et al 1995; Tanaka et al 1998). Because of all of these experiments, Hsps have been proposed as end effectors of ischemic preconditioning of myocardium.
In the experiment conducted in this study, our work focused on Hsps 70 because they are the most widely studied and best documented family of Hsps; despite this, no changes could be detected in mRNA Hsp72 levels or in the myocardial content of Hsp72 after the preconditioning maneuver, suggesting that early protection from “classical” preconditioning should not be related to Hsps70 in the pig animal model (Hughes et al 2003). The suitability of the experimental techniques used had been previously shown, because our group had successfully used them in the same animal model subjected to total body hyperthermia using external heat sources. A 200% increase in HSP72 myocardium content was found after body temperature was maintained at 42°C for 30 minutes, with final sampling 3.5 hours later (data not shown) while mRNA content increased by 700% (Peñaranda 2003). It should be noted in this regard that no changes were also shown in these 2 parameters studied in the control group, subjected to the same anesthetic and surgical stress as the preconditioned group, and in contradiction to our own previous findings in humans (Guisasola et al 2004; Chana 2005).
From the results obtained in our experimental model, we may first conclude that the model is valid and experimental techniques are adequate, but induction of Hsps70 as end effectors of cardioprotection in ischemic preconditioning does not occur in the first few hours following ischemia, but maybe at least 24 hours after ischemia in the so-called “second protection window” (Joyeux-Faure et al 2003). Our facilities did not allow us to maintain the animal model selected, minipigs, for 24 hours under the described surgical conditions with an open thorax in order to take additional samples at that time. However, low-molecular-weight Hsps, HSP27 and αβ-crystallin, have been reported to be the first line of defense against sublethal stress (Vander-Heide 2002; Louapre 2005). Both have been implicated also in the second window and could act via the cytoskeletal mechanism (Yellon and Downey 2003). Although it has been shown that pharmacological induction of delayed preconditioning can occur using an adenosine A1 receptor agonist, the resulting protection has been linked to the smaller HSP of 27 kDa rather than Hsp72 (Dana et al 2000).
So, in summary, it has been demonstrated that three 5-minute episodes of ischemia separated by 5-minute periods of reperfusion triggers profound and almost immediate protection from severe prolonged ischemia (Murry et al 1991). Although some evidence suggests that this brief ischemia likely triggers expression of Hsp70 (Polla 1988; Knowton et al 1991; Sun et al 1995), this study shows that levels of Hsp70 mRNA and protein are unchanged. This study provides evidence that this early protection is unlikely to be due to Hsp70, even though Hsp70 is strongly associated with myocardial protection (Marber et al 1995; Plumier et al 1995; Chiu et al 2003). This study clearly suggests that myocardial protection is a complex phenomenon.
There are still multiple questions to be answered, and more extensive and specific experiments are required to establish the exact role of the different Hsps in cardioprotection after ischemic preconditioning. Such knowledge would lead to potential future therapeutic approaches intended to be ultimately used in clinical practice. The challenge now is to identify pharmacological and/or gene therapy procedures that can efficiently increase Hsp expression in the intact heart in vivo without causing harmful side effects that would be unacceptable in human patients, and without using the stressful procedures originally leading to identification of Hsps and that gave them their name.
Fig 3.

A representative graphic showing ECG, blood pressure (BP), and pulmonary artery pressure (PAP) of a sham swine at final sample time. There are no changes in ECG tracing, blood pressure, and pulmonary artery pressure with respect to the initial time
Fig 4.

A representative graphic showing ECG, blood pressure (BP), and pulmonary artery pressure (PAP) of a protocol swine before ischemia series
Fig 5.

A representative graphic showing ECG, blood pressure (BP), and pulmonary artery pressure (PAP) of a protocol swine after coronary occlusion and last reperfusion period. Obvious ECG changes are seen such as widening of the QRS complex and T wave inversion, morphological changes and blood pressure increase, and morphological changes and pulmonary artery pressure increase after myocardial ischemic preconditioning
Fig 6.
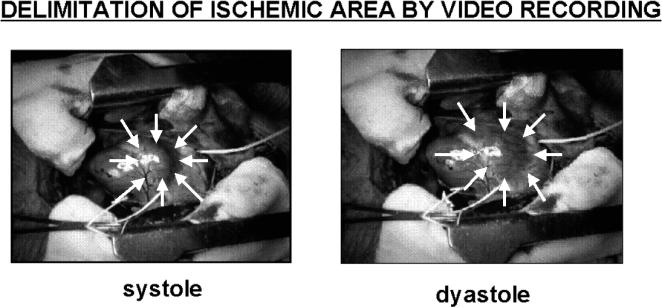
Visualization of the akinetic ischemic area by video recording: arrows have been put over the right ventricle to highlight the area of obvious contraction during systole. This area is clearly dilated during diastole
Acknowledgments
We thank María Jesús Sánchez for her essential technical collaboration. This study was supported in part by Grant FIS 03/1308. The study was approved by the Clinical Research Ethics Committee of the Hospital General Universitario Gregorio Marañon, Madrid, Spain.
REFERENCES
- Anaya-Prado R, Toledo-Pereyra LH, Lentsch AB, Ward PA. Ischemia/reperfusion injury. J Surg Res. 2002;105:248–258. doi: 10.1006/jsre.2002.6385.0022-4804(2002)105[0248:RI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Armstrong SC. Protein kinase activation and myocardial ischemia/reperfusion injury. Cardiovasc Res. 2004;61:427–436. doi: 10.1016/j.cardiores.2003.09.031.0008-6363(2004)061[0427:PKAAMR]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Baxter GRF, Goma FM, Yellon DM. Characterization of the infarct-limiting effect of delayed preconditioning, timecourse and dose-dependency studies in rabbit myocardium. Basic Res Cardiol. 1997;92:159–167. doi: 10.1007/BF00788633.0300-8428(1997)092[0159:COTIEO]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Becker L. New concepts in reactive oxygen species and cardiovascular reperfusion. Physiol Cardiovasc Res. 2004;61:461–470. doi: 10.1016/j.cardiores.2003.10.025. [DOI] [PubMed] [Google Scholar]
- Burel C, Mezger V, Pinto M, Rallu M, Trigon S, Morange M. Mammalian heat shock protein families. Expression and functions. Experientia. 1992;48:629–634. doi: 10.1007/BF02118307.0014-4754(1992)048[0629:MHSPFE]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Chana F 2005 Proteínas de choque térmico en las artroplastias totales de rodilla. Thesis, Facultad de Medicina, Universidad Complutense de Madrid. [Google Scholar]
- Chi NC, Karliner JS. Molecular determinants of responses to myocardial ischemia/reperfusion injury, focus on hypoxia-inducible and heat shock factors. Cardiovasc Res. 2004;61:437–447. doi: 10.1016/j.cardiores.2003.11.033.0008-6363(2004)061[0437:MDORTM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Chomezynski P. A reagent for the single-step simultaneus isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques. 1993;15:532–537.0736-6205(1993)015[0532:ARFTSS]2.0.CO;2 [PubMed] [Google Scholar]
- Cjiu JH, Tsou MT, Tung HH, Tai CH, Tsai SK, Chih CL, Ling JG, Wu CW. Preconditioned somatothermal stimulation on median nerve territory increases myocardial heat shock protein 70 and protects rat hearts against ischemia-reperfusion injury. J Thorac Cardiovasc Surg. 2003;125:678–685. doi: 10.1067/mtc.2003.29.0022-5223(2003)125[0678:PSSOMN]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Cornelussen RN, Vanagt WY, Prinzen FW, Snoeckx LH. Proteins involved in salvage of the myocardium. Adv Exp Med Biol. 2003;543:277–291. doi: 10.1007/978-1-4419-8997-0_20.0065-2598(2003)543[0277:PIISOT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Cornelussen RN, van Nieuwenhoven FA, Snoeckx LH. Presence of heat shock protein 72 in cardiomyocytes after heat stress. Circulation. 2001;104:E123.0009-7322(2001)104[E123:POHSPI]2.0.CO;2 [PubMed] [Google Scholar]
- Dana A, Skarli M, Papakrivopoulou J, Yellon DM. Adenosine A(1) receptor induced delayed preconditioning in rabbits, induction of p38 mitogen-activated protein kinase activation and Hsp27 phosphorylation via a tyrosine kinase- and protein kinase C-dependent mechanism. Circ Res. 2000;86:989–997. doi: 10.1161/01.res.86.9.989.0009-7330(2000)086[0989:AARIDP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Das DK, Prasad MR, Lu D, Jones RM. Preconditioning of heart by repeated stunning. Adaptative modification of antioxidative defense system. Cell Mol Biol. 1992;38:7739–7749.0145-5680(1992)038[7739:POHBRS]2.0.CO;2 [PubMed] [Google Scholar]
- Eefting F, Rensing B, Wigman J, Pannekoek WJ, Liu WM, Cramer MJ, Lips DJ, Doevendans PA. Role of apoptosis in reperfusion injury. Cardiovasc Res. 2004;61:414–426. doi: 10.1016/j.cardiores.2003.12.023.0008-6363(2004)061[0414:ROAIRI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Efthymiou CA, Mocanu MM, de Belleroche J, Wells DJ, Latchmann DS, Yellon DM. Heat shock protein 27 protects the heart against myocardial infarction. Basic Res Cardiol. 2004;99:392–394. doi: 10.1007/s00395-004-0483-6.0300-8428(2004)099[0392:HSPPTH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Friant S, Meier KD, Diezman H. Increased ubiquitin-dependent degradation can replace the essential requirement for heat shock protein induction. EMBO J. 2003;22:3783–3791. doi: 10.1093/emboj/cdg375.1460-2075(2003)022[3783:IUDCRT]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller KJ, Issels RD, Slosman DO, Guillet JG, Soussi T, Polla BS. Cancer and the heat shock response. Eur J Cancer. 1994;30:1884–1891. doi: 10.1016/0959-8049(94)00362-9.0959-8049(1994)030[1884:CATHSR]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Garcia-Dorado D. Myocardial reperfusion injury, a new view. Cardiovasc Res. 2004;61:363–364. doi: 10.1016/j.cardiores.2003.12.020.0008-6363(2004)061[0363:MRIANV]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Guisasola MC, Ramos R, Fernández-Quero L, Suárez A, García-Barreno P. Proteínas de choque térmico en estrés quirúrgico, toracotomía vs herniorrafia. Cir Esp. 2004;75:350–355.0009-739X(2004)075[0350:PDCTEE]2.0.CO;2 [Google Scholar]
- Hartl FU, Hayer-Hartl M. Molecular chaperones in the cytosol, from nascent chain to folded protein. Science. 2002;295:1852–1858. doi: 10.1126/science.1068408.0193-4511(2002)295[1852:MCITCF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Heads RJ, Latchman DS, Yellon DM. Stable high level expression of a transfected human HSP70 gene protects a heart-derived muscle cell line against thermal stress. J Mol Cell Cardiol. 1994;26:695–699. doi: 10.1006/jmcc.1994.1084.0022-2828(1994)026[0695:SHLEOA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Heads RJ, Latchman DS, Yellon DM. Differential stress protein mRNA expression during early ischaemic precondiotining in the rabbit heart and its relationship to adenosine receptor function. J Mol Cell Cardiol. 1995;27:2133–2148. doi: 10.1016/s0022-2828(95)91299-1.0022-2828(1995)027[2133:DSPMED]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hightower LE, Hendershot LM. Molecular chaperones and the heat shock response at Cold Spring Harbor. Cell Stress Chaperones. 1997;2:1–11. doi: 10.1379/1466-1268(1997)002<0001:mcaths>2.3.co;2.1466-1268(1997)002[0001:MCATHS]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes GC, Post MJ, Simons K, Annex BH. Translational physiology, porcine models of human coronary artery disease, implications for preclinical trials of therapeutic angiogenesis. J Appl Physiol. 2003;94:1689–1701. doi: 10.1152/japplphysiol.00465.2002.8750-7587(2003)094[1689:TPPMOH]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Hutter JJ, Mestril R, Tam EK, Sievers RE, Dillmann WH, Wolfe CL. Overexpression of heat shock protein 72 in transgenic mice decreases infarct size in vivo. Circulation. 1996;94:1408–1411. doi: 10.1161/01.cir.94.6.1408.0009-7322(1996)094[1408:OOHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Jennings RB, Reimer KA 1983 Factors involved in salvaging ischemic myocardium, effect of reperfusion of arterial blood. Circulation. 68(Suppl 1). I-25–I-36. [PubMed] [Google Scholar]
- Joyeux-Faure M, Arbaud C, Godin-Ribuot D, Ribuot C. Heat stress preconditioning and delayed myocardial protection, what is new. Cardiovasc Res. 2003;60:469–477. doi: 10.1016/j.cardiores.2003.08.012.0008-6363(2003)060[0469:HSPADM]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Kitamura Y, Nomura Y. Stress proteins and glial functions, possible therapeutic targets for neurodegenerative disorders. Pharmacol Ther. 2003;97:35–53. doi: 10.1016/s0163-7258(02)00301-7.0163-7258(2003)097[0035:SPAGFP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Knowlton AA, Brecher P, Apstein CS. Rapid expression of heat shock protein in the rabitt after brief cardiac ischemia. J Clin Invest. 1991;87:139–147. doi: 10.1172/JCI114963.0021-9738(1991)087[0139:REOHSP]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;23:271–276. doi: 10.1038/227680a0.1476-4687(1970)023[0271:COSPDT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Latchman DS. Heat shock proteins and cardiac protection. Cardiovasc Res. 2001;51:637–646. doi: 10.1016/s0008-6363(01)00354-6.0008-6363(2001)051[0637:HSPACP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Louapre P, Grongnet JF, Tanguay RM, David JC. Effects of hypoxia on stress proteins in the piglet heart at birth. Cell Stress Chaperones. 2005;10:17–23. doi: 10.1379/CSC-74R.1.1466-1268(2005)010[0017:EOHOSP]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lowry OH, Rosebrough NJ, Farr AI, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275.0021-9258(1951)193[0265:PMWTFP]2.0.CO;2 [PubMed] [Google Scholar]
- Madamanchi NR, Patterson C, Runge MS. Reactive oxygen species regulate heat-shock protein 70 via JAK/STAT pathway. Arterioscler Thromb Vasc Biol. 2001;21:321–326. doi: 10.1161/01.atv.21.3.321.1079-5642(2001)021[0321:ROSRHP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Marber MS, Latchman DS, Walker JM, Yellon DM. Cardiac stress protein elevation 24 hours after brief ischaemia or heat stress is associated with resistance to myocardial infarction. Circulation. 1993;88:1264–1272. doi: 10.1161/01.cir.88.3.1264.0009-7322(1993)088[1264:CSPEHA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Marber MS, Mestril R, Chi SH, Sayen MR, Yellon DM, Dillmann WH. Overexpression of the rat inducible 70-kD heat stress protein in a transgenic mouse increases the resistance of the heart to ischemic injury. J Clin Invest. 1995;9:1446–1456. doi: 10.1172/JCI117815.0021-9738(1995)009[1446:OOTRIK]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCormick PH, Chen G, Tlerney S, Kelly CJ, Bouchier-Hayes DJ. Clinically relevant thermal preconditioning attenuates ischemia-reperfusion injury. J Surg Res. 2003;109:24–30. doi: 10.1016/s0022-4804(02)00035-5.0022-4804(2003)109[0024:CRTPAI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Meadus WJ. A semi-quantitative RT-PCR method to measure the in vivo effect of dietary conjugated linoleic acid on porcine muscle PPAR gene expression. Biol Proced Online. 2003;5:20–28. doi: 10.1251/bpo43.1480-9222(2003)005[0020:ASRMTM]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehta HB, Popovich BK, Dillmann WH. Ischemia induces changes in the level of mRNAs coding for stress protein 71 and creatine kinase M. Circ Res. 1988;63:512–517. doi: 10.1161/01.res.63.3.512.0009-7330(1988)063[0512:IICITL]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Morimoto RI, Kline MP, Bimston DN, Cotto JJ. The heat-shock response, regulation and function of heat-shock proteins and molecular chaperones. Essays Biochem. 1997;32:17–29.0071-1365(1997)032[0017:THRRAF]2.0.CO;2 [PubMed] [Google Scholar]
- Murry CE, Jennings RB, Reimer KA. Preconditioning with ischemia, a delay of lethal cell injury in ischemic myocardium. Circulation. 1986;74:1124–1136. doi: 10.1161/01.cir.74.5.1124.0009-7322(1986)074[1124:PWIADO]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Murry CE, Richard VJ, Jennings RB, Reimer KA. Myocardial protection is lost before contractile function recovers from ischemic preconditioning. Am J Physiol. 1991;260:H796–H804. doi: 10.1152/ajpheart.1991.260.3.H796.0002-9513(1991)260[H796:MPILBC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Peñaranda MA 2003 Proteínas de choque térmico en la hipertermia corporal global y el precondicionamiento isquémico cardiaco. Modelos animal y celular en cerdo miniatura. Thesis, Facultad de Veterinaria. Universidad Complutense de Madrid. [Google Scholar]
- Piacentini L, Karliner SJ. Altered gene expression during hypoxia and reoxygenation of the heart. Pharmacol Ther. 1999;83:21–37. doi: 10.1016/s0163-7258(99)00010-8.0163-7258(1999)083[0021:AGEDHA]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Plumier C, Ross BM, Currie RW, Angelidis CE, Kazlaris H, Kollias G, Pagoulatos GN. Transgenic mice expressing the human heat shock protein 70 have improved post-ischemic myocardial recovery. J Clin Invest. 1995;95:1854–1860. doi: 10.1172/JCI117865.0021-9738(1995)095[1854:TMETHH]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pockley AG. Heat shock proteins, inflammation, and cardiovascular disease. Circulation. 2002;105:1012–1017. doi: 10.1161/hc0802.103729.0009-7322(2002)105[1012:HSPIAC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Polla BS. A role for heat shock proteins in inflammation. Immunol Today. 1989;9:134–137. doi: 10.1016/0167-5699(88)91199-1.0167-5699(1989)009[0134:ARFHSP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Radford NB, Fina M, and Benjamin IJ. et al. 1996 Cardioprotective effects of 70-kDa heat shock protein in transgenic mice. Proc Natl Acad Sci U S A. 93:2339–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritossa FM. A new puffing patttern induced by temperature shock and DNP in Drosophila. Experientia. 1962;18:571–573.0014-4754(1962)018[0571:ANPPIB]2.0.CO;2 [Google Scholar]
- Sachs DH, Leight G, Cone J, Scharz S, Stuart L, Rosemberg S. Transplantation in miniature swine. I. Fixation of the major histocompatibility complex. Transplantation. 1976;22:559–567. doi: 10.1097/00007890-197612000-00004.0041-1337(1976)022[0559:TIMSIF]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sack S, Morí M, Arras M, Schwarz ER, Schaper W. Ischemic preconditioning, time course of renewal in the pig. Cardiovasc Res. 1993;27:551–555. doi: 10.1093/cvr/27.4.551.0008-6363(1993)027[0551:IPTCOR]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sanz E, García-Dorado D, and Oliveras J. et al. 1995 Dissociation between anti-infarct effect and anti-edema effect of ischemic preconditioning. Am J Physiol. 268:H233–H241. [DOI] [PubMed] [Google Scholar]
- Schult R, Kelm M, Heusch G. Nitric Oxide in ischemia/reperfusion injury. Cardiovasc Res. 2004;61:402–413. doi: 10.1016/j.cardiores.2003.09.019.0008-6363(2004)061[0402:NOIRI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Sun JZ, Tang XL, Knowlton AA, Park SW, Qiu Y, Bolli R. Late preconditioning against myocardial stunning. An endogenous protective mechanism that confers resistance to postischemic dysfunction 24 h after brief ischemia in conscious pigs. J Clin Invest. 1995;95:388–403. doi: 10.1172/JCI117667.0021-9738(1995)095[0388:LPAMSA]2.0.CO;2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka MF, Fujiwara H, and Yamasaki K. et al. 1998 Expression of heat shock protein after ischemic preconditioning in rabbit hearts. Jpn Circ J. 62:512–516. [DOI] [PubMed] [Google Scholar]
- Tissières A, Mitchell HK, Tracy VM. Protein synthesis in salivary glands of Drosophila melanogaster, relation to chromosome puffs. J Mol Biol. 1974;84:389–398. doi: 10.1016/0022-2836(74)90447-1.0022-2836(1974)084[0389:PSISGO]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Vander Heide RS. Increased expression of HSP27 protects canine myocytes from simulated ischemia-reperfusion injury. Am J Physiol. 2002;282:H935–H941. doi: 10.1152/ajpheart.00660.2001.0002-9513(2002)282[H935:IEOHPC]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Voellmy R 1996 Sensing stress and responding to stress. In: Stress-Inducible Cellular Responses, ed Feige U, Morimoto RI, Yahara I, Polla BS. Birkhäuser Verlag, Basel, Switzerland, 121–137. [DOI] [PubMed] [Google Scholar]
- Wallin RPA, Lundqvist A, Moré SH, Bonin A, Kiessling R, Ljunggren HG. Heat-shock proteins as activators of the innate immune system. Trends Immunol. 2002;23:130–135. doi: 10.1016/s1471-4906(01)02168-8.1471-4906(2002)023[0130:HPAAOT]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Xu Q. Role of heat shock proteins in atherosclerosis. Arterioscler Thromb Vasc Biol. 2002;22:1547–1559. doi: 10.1161/01.atv.0000029720.59649.50.1079-5642(2002)022[1547:ROHSPI]2.0.CO;2 [DOI] [PubMed] [Google Scholar]
- Yellon DM, Downey JM. Preconditioning the myocardium, from cellular physiology to clinical cardiology. Physiol Rev. 2003;83:1113–1151. doi: 10.1152/physrev.00009.2003.0031-9333(2003)083[1113:PTMFCP]2.0.CO;2 [DOI] [PubMed] [Google Scholar]