

Abstract
Expansion of trinucleotide repeats is associated with a growing number of human diseases. The mechanism and timing of expansion of the repeat tract are poorly understood. In humans, trinucleotide repeats show extreme meiotic instability, and expansion of the repeat tract has been suggested to occur in the germ-line mitotic divisions or postmeiotically during early divisions of the embryo. Studies in model organisms have indicated that polymerase slippage plays a major role in the repeat tract instability and meiotic instability is severalfold higher than the mitotic instability. We show here that meiotic instability of the CAG/CTG repeat tract in yeast is associated with double-strand break (DSB) formation within the repeated sequences, and that the DSB formation is dependent on the meiotic recombination machinery. The DSB repair results in both expansions and contractions of the CAG repeat tract.
Expansion of trinucleotide repeats has been shown to be associated with a growing number of human diseases (1). Several of these genetic diseases are associated with alterations of the CAG/CTG (hereafter CAG) repeat tract length. The expansion mutation is highly dependent on the repeat tract length: the longer the repeat tract, the greater the possibility for expansion (2–4). Such mutational changes are dynamic, since the mutated region can undergo further changes in subsequent generations and during the lifespan of an individual.
Two mechanisms have been used to explain the expansion of trinucleotide repeats: unequal genetic recombination and error in DNA replication caused by polymerase slippage (5, 6). In the recombination model, unequal crossing-over or gene conversion between triplet repeats either on sister chromatids or on homologs results in an altered tract length. In the slippage model, the replicating strand becomes dissociated, and then it misaligns during reassociation. This misalignment causes the formation of an unpaired sequence either on the template strand or on the nascent strand. A failure to repair the unpaired sequence will result in a DNA molecule containing a larger or smaller number of repeats after the next round of DNA replication.
Several studies using model organisms indicate that replication slippage plays a major role in the repeat tract instability (7–14). The latter model is also supported by the in vitro observations that CAG repeats can form hairpin structures, with the CTG hairpin being more stable than the CAG hairpin (15–18). Recent studies with diploid yeast strains containing heterozygous trinucleotide repeat-insertion mutations suggest that trinucleotide repeats in single-stranded DNA are likely to form hairpin structures in vivo (19). During DNA replication, the hairpin formation on the template strand or on the newly synthesized strand would produce a deletion or an expansion of the repeat tract, respectively. This hypothesis is consistent with the observation that CAG-repeat instability depends on the direction in which the replication fork proceeds through the repeat tract (8, 9, 13, 20).
The repeat tract instability is also higher in the rad27-deletion mutant, which fails to process Okazaki fragments (12, 14, 21, 22). A failure to process the Okazaki fragments may facilitate hairpin formation on the newly synthesized strand. Unprocessed hairpin structures would cause an alteration in the repeat tract length after the next round of DNA replication. In addition, a double-strand break (DSB) in the DNA can be generated by endonucleolytic cleavage of the secondary structure (10, 23, 24). Alternatively, stalled or slowed DNA replication because of the presence of unprocessed Okazaki fragments can give rise to DSBs (25, 26). These DSBs can be repaired by homologous recombination. Consistent with this proposal is the observation that the replication fork is stalled by a long trinucleotide repeat tract (27). Recently, Freudenreich et al. (21) have shown that long CAG repeat tracts are often sites of chromosome breakage in yeast when DNA replication is slowed.
Although replication-slippage appears to be a major contributing factor for repeat tract instability, the role of recombination cannot be ruled out. The instability of minisatellites in humans (28, 29) and the rDNA genes (30) and CUP1 repeats (31) in Saccharomyces cerevisiae appear to be mediated by the recombination mechanism. In bacteria, CAG repeat instability in a two-plasmid system has been shown to require recombination functions (32). Recently, Richard et al. (33) have demonstrated that artificially induced DSBs in the repeated DNA increases the rate of trinucleotide repeat tract instability.
Most experiments in model organisms were designed to investigate the mitotic instability of the repeat tract. Little is known about meiotic instability of the repeat tract. In humans, trinucleotide repeats show extreme meiotic instability, and expansion of the repeat tract has been suggested to occur in the germ-line mitotic divisions or postmeiotically during early divisions of the embryo (34, 35). Recently, Cohen et al. (36) have shown that instability of CAG repeats in yeast is severalfold higher during meiosis compared with mitosis.
In the present study, we designed experiments to investigate whether meiotic instability of trinucleotide repeats is due to DSB formation. Our results indicate that meiotic instability of the CAG repeat tract in yeast occurs by DSB formation within the repeated sequences, and the DSB formation depends on the meiotic recombination machinery. The repair of DSBs leads to both expansion and contraction of the repeat tract. In addition, tract alterations also occur as a postmeiotic event during early divisions of the spore cell.
Materials and Methods
Yeast Strains.
All yeast strains were derived from AS4 (α trp1-1 arg4-17 tyr7-1 ura3 ade6) and AS13 (a leu2-Bst ura3-52 ade6) background (37). AS4 × AS13 diploids show a high level of meiotic recombination at the HIS4 locus (24, 37). The his4-79 and his4-64 mutant alleles were introduced into AS13 and AS4 chromosomes, respectively, by a two-step transplacement procedure. The rad50S mutation was introduced into the chromosome by using the plasmid pNKY349 as described previously (24). The spo11 deletion mutation was introduced by the one-step transplacement procedure using the plasmid pGB518, where the entire SPO11 coding region is replaced by the hisG–URA3–hisG cassette. Haploid and diploid strains used in this study were as follows: CJY39 (AS13, his4-79); CJY47 (AS4, his4-64); DNY277 (AS13, his4-260); DNY115 (AS13, rad50S × AS4, rad50S); CJY42 (CJY39, rad50S × AS4, rad50S); CJY48 (CJY39 × CJY47); CJY55 (CJY48 but homozygous for rad50S); CJY68 (CJY42 but homozygous for spo11); and DNY278 (DNY277, rad50S × AS4, rad50S).
Media and Genetic Techniques.
Standard genetic methods and media were used (38). Diploid strains were sporulated in 1% potassium acetate as described in ref. 24. All other media were as described by Rose et al. (38).
Plasmids.
All insertions were made at the SalI site within the coding sequence of the HIS4 gene. The plasmid pCJ1, which contained a portion of the S. cerevisiae HIS4 gene interrupted with 80 CAG repeats at the SalI site, was constructed as follows: a PCR product containing the CAG repeats was obtained from genomic DNA of a mouse that is transgenic for a mutant exon 1 of the human Huntington's disease (HD) gene carrying 116 CAG repeats, using primers TRP1 (5′-CCGCTCGAGATGAAGGCCTTCGAGTCCCTCAAGTCCTTC-3′) and TRP2 (5′-CCGCTCGAGGGCGGCTGAGGAAGCTGAGGA-3′) (39). Both primers had a XhoI linker at the 5′ end. The PCR product was digested with XhoI and ligated into SalI-digested pDN9 (37). The plasmid pDN9 had an XhoI to BglII fragment containing most of the HIS4 coding region ligated into BamHI- and SalI-digested YIp5. The CAG repeat tract also had a CGG triplet as the 15th repeat. AS13 and AS4 were transformed with SnaBI-linearized pCJ1. The resulting strains, CJY39 and CJY47, contained 79 and 64 CAG repeats at the HIS4 locus, respectively, and the final sequence of the insertion at the his4-79 allele site was 5′-TCGAGATGAAGGCCTTCGAGTCCCTCAAGTCCTTC(CAG)14CGG(CAG)64CCGCCGCCGCCGCCGCCGCCGCCGCCGCCTCCTCAGCTTCCTCAGCCGCCCTCGA-3′. The change in the repeat number from 80 to 79 and 64 indicates instability during the integration process. The control strain DNY278 had a heterozygous 260-bp insertion of a nonrepeated sequence at the SalI site of the HIS4 gene. The nonrepeated sequence was obtained from the bacterial insertion element IS50 by using primers 5′-CCGCTCGAGCAGAGGGTAGTGAAGCCATGCAGGA-3′ (240–265 of the IS50L sequence) and 5′-TGGCCTCGAGCAAGAGAACGGAGTG-3′ (479–495 of IS50L).
PCR and Gel Electrophoresis of Spore Colony DNA.
The his4-79/his4-64 diploid strain was sporulated and tetrads were dissected on YPD plates (yeast extract/peptone/dextrose; see ref. 38). After 3 days of growth at 30°C, a portion of the spore colony was used for PCR amplification using primers CJO3 (5′-CCAAAGGAGCGTGTTGTTGTGGAAGAG-3′ +288 to +314 in the HIS4 coding sequence) and CJO4 (5′-GGCCATTGCCAGAAGTTTCACCCTTGA-3′ +583 to +609 of HIS4), which flank the SalI site in the HIS4 gene. Reactions were carried out in 50 μl containing 50 pmol of each primer, 10% (vol/vol) DMSO, AM buffer [67 mM Tris⋅HCl, pH 8.8/16.6 mM (NH4)2SO4/2 mM MgCl2 and 0.17 mg/ml BSA], 200 μM dNTPs, and 2.5 units of Taq DNA polymerase (Perkin–Elmer). A small amount of the spore colony was suspended in 10 μl of water and denatured at 94°C for 5 min and then mixed with the remaining reagents. PCR parameters were set at 94°C for 1 min, 59°C for 1 min, and 72°C for 2 min for 35 cycles. PCR products were separated on 2.2% MetaPhor agarose gels (FMC BioProducts).
Physical Analysis of Meiotic DNA.
Meiotic DNA was isolated as described in ref. 24 and digested with PvuII. The resulting fragments were separated on a 0.8% agarose gel. The DNA was transferred to a nylon membrane, which was then hybridized with a 32P-labeled probe. The amount of DNA was quantitated by using a Molecular Dynamics PhosphorImager.
PCR Analysis of Mitotic Repeat Tract Instability.
The repeat tract length of isolated single colonies was determined by PCR analysis as described for the spore colonies. A portion of the colony was also inoculated into YPD for DNA isolation. DNA was isolated after overnight growth at 30°C and analyzed by Southern hybridization.
Results
Long CAG Repeat Tracts Induce DSBs During Meiosis.
As mentioned earlier, trinucleotide repeats, like inverted repeats, have been shown to form hairpin structures in vitro, and in vivo results are consistent with the in vitro studies (15–19). Hairpin structures are substrates for several structure-specific nucleases, the action of which may lead to DSB formation in DNA. In S. cerevisiae, long palindromic sequences have been shown to form DSBs during meiosis (24). Long inverted repeats may undergo cruciform extrusion at a frequency higher than the short palindromes and are then cleaved by structure-specific nucleases to generate DSBs. Because expansion of trinucleotide repeats is also length dependent (2–4), we examined whether long trinucleotide repeat tracts are able to induce DSBs in yeast. We constructed a his4 mutant allele (his4-79) by inserting 79 copies of CAG repeats within the HIS4 coding sequence. The diploid strain, heterozygous for the his4-79 allele, was sporulated, and meiotic DNA was analyzed physically and compared with that of the HIS4/HIS4 diploid strain. Both of these strains were homozygous for the rad50S mutation, which prevents processing of DSBs.
DSBs are initiators of nearly all meiotic recombination in yeast (40), and most meiotic recombination at the HIS4 locus occurs because of DSBs at the HIS4 promoter (site I, Fig. 1A) (41). A PvuII digestion of DNY115 (HIS4/HIS4 rad50S/rad50S) DNA generates a 2.4-kb band that contains most of the HIS4 gene and a portion of the BIK1 gene (Fig. 1A). A similar digestion of CJY42 (HIS4/his4-79 rad50S/rad50S) DNA would produce 2.4-kb and ≈2.7-kb fragments (resulting from the wild-type and the his4-79 chromosome, respectively). The results are shown in Fig. 1B. Both DNY115 and CJY42 produced bands (≈1.9 kb) characteristic of the DSBs at the HIS4 promoter. CJY42 DNA produced an extra band, and the size (≈1.5 kb) of the band suggests that breaks had occurred at the CAG repeats (Fig. 1B). In addition, the band caused by DSBs at the triplet repeat was broad, suggesting that breaks had occurred at multiple sites within the repeats. DSBs at the CAG-insertion site are expected to produce two fragments, ≈1.5 kb and ≈1.2 kb. Only the ≈1.5 kb was observed; the smaller fragment could not be observed because of short homology with the probe and the smeary band pattern.
Figure 1.

Physical analysis of DSB formation in wild-type and in his4 insertion-mutant strains. (A) Partial restriction map of the HIS4-BIK1 region. The boxes indicate the coding regions and the arrows indicate the direction of transcription. All insertions were made at the SalI site within the HIS4 coding region. Abbreviations: Bg, BglII; H, HindIII; P, PvuII; S, SalI; Sc, SacI; X, XhoI. (B) DSB formation in a HIS4/HIS4 strain and in strains containing HIS4/his4-79 heterozygous or his4-79/his4-64 homozygous CAG insertions. All diploid strains were homozygous for the rad50S mutation. DNA was isolated at different times (hours) after induction of meiosis and digested with PvuII. The XhoI–BglII fragment (probe right) was used as a probe. The number above each lane indicates the time (in hours) of sample collection. Site I represents the DSB site at the HIS4 promoter. Site II indicates the DSB site at the CAG insertion. PvuII digestion of meiotic DNA generates a 2428-bp fragment for DNY115, 2428- and 2751-bp fragments (from HIS4 and his4-79 chromosome, respectively) for CJY42, and 2751- and 2706-bp fragments (from his4-79 and his4-64 chromosome, respectively) for CJY55. A similar digestion of DNY278 DNA produces 2428- and 2688-bp fragments.
Normally, trinucleotide repeat tracts are present at allelic positions in a given pair of homologs. To mimic the natural situation, a diploid strain was constructed in which one homolog had 79 repeats and the other had 64 repeats. There were more breaks in his4-79/his4-64 cells compared with HIS4/his4-79 cells because breaks were occurring on both homologs (Fig. 1B and Table 1). About 5.6–7.4% of total meiotic DNA had DSBs at the CAG-insertion site (Table 1). The amount of DSBs at the promoter was reduced because of competition between the two recombination-initiation sites (42–44).
Table 1.
Amount of DSBs formed in different his4 mutants
| Diploid genotype | Amount of DSBs,* % of total meiotic DNA
|
|
|---|---|---|
| HIS4 promoter | CAG-insertion site | |
| HIS4/HIS4 | 2.5–4.6 | 0 |
| HIS4/his4-79 | 1.4–1.6 | 1.8–2.6 |
| his4-79/his4-64 | 1.2–1.7 | 5.6–7.4 |
DNA was isolated at different times (hours) from cells undergoing meiosis. The amount of DSBs was determined from the 24-hr sample. DSBs at the HIS4 promoter in the heterozygous diploid represent breaks only on the wild-type chromosome. DSBs at the HIS4 promoter on the mutant chromosome could not be seen because the band ran along with the wild-type parental fragment. In a HIS4/HIS4 diploid strain, the amount of DSBs varies from 2% to 5% of total meiotic DNA (41). The range of DSBs were taken from three independent experiments.
DSBs Are Formed Within the CAG Repeats.
To determine whether the DSBs are formed within the trinucleotide repeats or outside the CAG repeats, we digested the 24-hr sample of CJY55 (his4-79/his-64 rad50S/rad50S) with PvuII and compared the sizes of the fragments with those derived from the 0-hr sample of DNY115 digested with PvuII and SalI. If the breaks had occurred on one side of the CAG repeats, one of the two fragments would be equal to or smaller than the control fragments derived from DNY115 DNA. The results shown in Fig. 2 indicate that both fragments generated by the DSB were larger than the control fragments, suggesting that DSBs are forming within the trinucleotide repeat tract.
Figure 2.
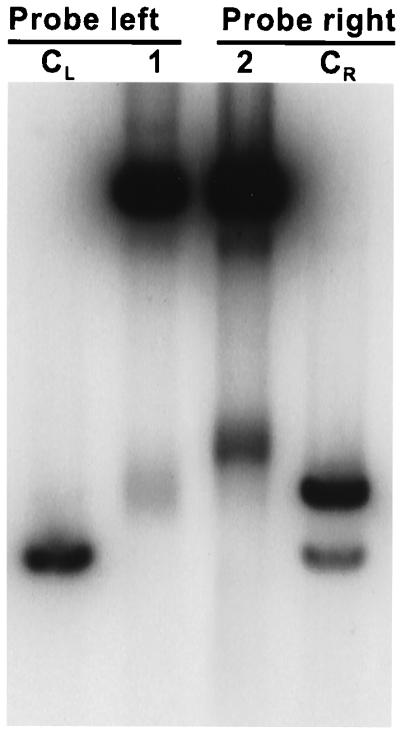
Physical mapping of DSB sites. Lane CL and lane CR had 0-hr DNY115 DNA digested with PvuII and SalI. Lanes 1 and 2 had PvuII-digested CJY55 DNA collected at 24 hr. Lanes CL and 1 were probed with SacI-SalI fragment (probe left, Fig. 1A), and lanes CR and 2 were probed with XhoI–BglII fragment (probe right, Fig. 1A).
DSBs at the Insertion Site Are CAG-Repeat-Specific and SPO11-Dependent.
The HIS4 locus in our strain background has a high rate of meiotic recombination. It is possible that the insertion of any sequence at a site that is primed for recombination initiation would create a DSB site. To eliminate this possibility, we inserted a 260-bp nonrepeated DNA fragment at the same SalI site within the HIS4 coding sequence to create the his4-260 allele. The diploid strain DNY278 (HIS4/his4-260 rad50S/rad50S) was sporulated and the meiotic DNA was analyzed physically, and the results are shown in Fig. 1B. A PvuII digestion of the DNY278 DNA failed to exhibit DSBs at the site of insertion, suggesting that DSBs observed in CJY55 are CAG-repeat-specific.
To determine whether the CAG-repeat-induced DSBs depend on the meiotic recombination machinery, we made a spo11 derivative (CJY68) of the HIS4/his4-79 diploid strain. Spo11, a putative topoisomerase II-like transesterase that remains attached to the 5′ end of the DSB site, is believed to catalyze the DSB formation (40). When CJY68 meiotic DNA was analyzed physically, no bands characteristic of DSBs either at the HIS4 promoter or at the CAG-insertion site could be detected (Fig. 1B).
DSB Repair Results in CAG-Repeat Tract Alterations.
The observation that CAG repeats induced DSBs prompted us to investigate whether the repair of breaks leads to CAG-repeat tract alterations in the his4-79/his4-64 meiotic products. The his4-79/his4-64 diploid strain (CJY48) was sporulated and the tetrads were dissected. The CAG-repeat tract length in the spore colonies was monitored by PCR analysis and the results are shown in Table 2. The PCR results were confirmed by Southern hybridization (Fig. 3). Trinucleotide repeats exhibited a high level of meiotic instability. Both expansions and contractions were observed. However, contractions were more frequent than expansions. Among 103 unselected tetrads, 25% had a 4+:4− segregation pattern (following the nomenclature of eight-spore-producing organisms) and 75% had aberrant segregation patterns. These aberrant events can be divided into five classes (Table 2).
Table 2.
Different types of trinucleotide repeat tract alterations
| Class | Segregation pattern* | Number of repeats† |
|---|---|---|
| Class I (18) | 79:79:79:64 (9) | |
| 79:64:64:64 (9) | ||
| Class II (25) | 79:79:64:n1 (13) | 79:79:64:45; 79:79:64:39 |
| 79:64:64:n1 (12) | 79:64:64:37; 86:79:64:64 | |
| Class III (24) | 79:64:n1:n2 (2) | 79:64:45:40; 79:64:55:24 |
| 79:79:n1:n2 (2) | 140:79:79:73 | |
| 64:64:n1:n2 (1) | ||
| 79:64:n1:n1 (3) | ||
| 79:79:n1:n1 (3) | 79:79:53:53; 79:79:47:47 | |
| 64:64:n1:n1 (4) | 64:64:27:27; 64:64:22:22 | |
| 79:79:79:n1 (5) | 79:79:79:44 | |
| 64:64:64:n1 (4) | ||
| Class IV (8) | 79:n1:n1:n2 (1) | |
| 64:n1:n1:n2 (4) | 102:102:80:64; 64:35:35:29 | |
| 79:n1:n2:n3 (1) | ||
| 64:n1:n2:n3 (2) | 109:103:80:64 | |
| Class V (2) | 79:79:79:79 (1) | |
| 64:64:64:64 (1) |
Tract alterations were determined by PCR analysis of a portion of the spore colony and DNA sequencing of the PCR product. The letter n represents an altered repeat number. The largest expansion and contraction were from 64 repeats to 140 repeats and 79 repeats to 22 repeats, respectively. Postmeiotic events were observed in all classes. Similar events were also observed by Cohen et al. (36). Contraction events of intermediate sizes and most of the postmeiotic events were not sequenced. The numbers in parentheses indicate the number of events observed in each class.
† Not all PCR products were sequenced. Only the sequenced events are shown here.
Figure 3.

Southern analysis of CAG repeat tract instability in the DNA from spore colonies. Tetrads were dissected on YPD plates, and the spore colony was used for PCR analysis of a 640-bp fragment that covers all of the CAG repeats and a portion of the flanking HIS4 sequence. Another portion was inoculated into YPD broth for DNA isolation. DNA from the spore colonies was digested with XhoI and HindIII before running on a 0.8% agarose gel. The DNA was then transferred onto a nylon membrane and probed with XhoI–HindIII fragment as a probe. Representative tetrads of different classes are shown here. P1 and P2 indicate parental his4-79 and his4-64 fragments, respectively. Tetrad 1 indicates a 4+:4− segregation, tetrads 2–5 are examples of aberrant segregation patterns, and tetrad 6 is an example of a postmeiotic event. Tetrad 2 had a 2:6 event, tetrads 3 and 4 are examples of meiotic expansions, and tetrad 5 had a contraction. Expansions and contractions were observed with approximately equal frequency among the postmeiotic events. Both expansions and contractions were confirmed by DNA sequencing. PCR analysis of the repeat tract length identified only large expansions or contractions (changes involving more than 2–4 repeats). The tract length changes smaller than 2 repeats would not be detected by our assay system.
Class I tetrads include typical 6+:2− and 2+:6− (considering his4-79 as the + allele and his4-64 as the − allele) gene conversion events and were observed without any disparity. Two tetrads in this class had four spores of original sizes, but one spore colony had a mixture of parental and a different size (79:79:79, 62:64 and 79:64:64:64, 68). These two tetrads represent a postmeiotic event in which the changes had occurred after meiosis during early divisions of the spore cell. Postmeiotic tetrads were observed in both normal and aberrant events. Such events were also observed by Cohen et al. (36). Class II tetrads, the major class among aberrants, had three spore colonies of the parental sizes, and one had a nonparental size that resulted from a single break in one of the four chromatids. Class III tetrads represent double events in which DSBs have occurred on two chromatids, the repair of which resulted in either contraction or expansion. Class IV includes tetrads that had either multiple meiotic events or a combination of both mitotic and meiotic events. The last class simply represent 8+:0− or 0+:8− events.
In the class III tetrads, parental:n1:n1 events could arise because of mitotic events. To monitor mitotic instabilities, we performed PCR analysis of 84 colonies of his4-79/his4-64 diploid cells. PCR results were confirmed by Southern hybridization (Fig. 4). A total of 3 colonies (3.5%) showed deletions in the CAG tract length. These results also suggest that most meiotic tract alterations observed in our system were caused by DSB repair, and meiotic instability is severalfold higher than the mitotic instability.
Figure 4.

Analysis of mitotic instability of the CAG-repeat tract. DNA samples derived from isolated single colonies were analyzed by Southern hybridization as described in the legend of Fig. 3.
Discussion
Previous in vitro and in vivo studies have demonstrated that single-stranded DNA containing disease-associated trinucleotide repeats can form hairpin structures (15–19). Hairpin and cruciform or double-hairpin structures are substrates for several structure-specific nucleases in vivo. Nag and Kurst (24) previously demonstrated that long inverted repeats, which also have the potential to form hairpin structures in vivo, induce DSBs during meiosis in yeast. We have examined whether trinucleotide repeats, like inverted repeats, induce DSBs during meiosis. Our results indicate the following: (i) CAG repeats induce DSBs during meiosis. (ii) Meiotic DSBs are SPO11 dependent. (iii) The rate of meiotic instability is much higher than that of the mitotic instability. (iv) DSB repair results in both expansion and contraction of the repeat tract length.
Our results and those obtained by Moore et al. (19) suggest that CAG-repeat-induced DSB formation depends on the length of the trinucleotide repeat tract. A tract length consisting of 10 repeats fails to induce DSBs (19), whereas 64 and 79 repeats induce DSBs. The repeat tract sizes used in our system were well over the threshold size (1). Consequently, tract length alterations were expected if yeast and humans are to follow the same mechanism of repeat instability. The trinucleotide expansion in humans occurs in two stages (2, 3, 33). First, a moderate expansion increases the repeat tract length to a premutation state in individuals who are not affected by the disease. In the next step, a large jump from the unstable premutation state to full mutation occurs in the affected individuals. It is likely that the moderate increase in the allele size in normal individuals occurs by a replication slippage mechanism, and once a threshold size is achieved a large jump in allele size occurs by DSB formation.
Trinucleotide-repeat-induced DSBs depend on the meiotic recombination machinery, as these breaks were SPO11 dependent. Spo11-mediated meiotic DSBs usually occur at open chromatin structures (45, 46). Although in vitro studies have suggested that CAG repeats form stable chromatin structures (47), it is possible that open chromatin structures are formed when they are present at the HIS4 locus. An alternative explanation is that during meiosis, a certain fraction of DNA containing CAG repeats is extruded into cruciform conformation because of changes in the chromatin structure (45, 46, 48). This cruciform structure is then cleaved by Spo11-dependent structure-specific nucleases.
The repair of the DSBs leads to repeat tract length alterations resulting in both expansions and contractions, although contractions were more frequent than expansions. The spectrum of events observed in our experiments is very similar to that observed by Cohen et al. (36). The 2:6 and 6:2 events might have occurred because of DSBs at the HIS4 promoter. The rest of the aberrants are likely to be caused by DSBs within the CAG repeats. Class III events are likely to be caused by two DSBs. However, several of the class III tetrads (e.g., 79:64:n1:n2) could also be explained by gene conversion associated with crossover events. The 79:79:n1:n1 and 64:64:n1:n1 tetrads appear to be caused by a mitotic event. Because mitotic instability in our strain background was low (Fig. 4), we assume that the majority of these events were caused by multiple DSBs. This conclusion is also supported by 79:64:n1:n1 events. Class IV tetrads could result from more than two breaks or are a result of both meiotic and mitotic events.
Forty-nine tetrads had contraction events (deleting from 57 to 11 repeats; Table 2), including 4 postmeiotic events, and 11 had expansions, including 7 postmeiotic events. The largest meiotic expansion was from 64 copies to 140 copies (class III in Table 2). Our results indicate that 27% of total spores had one conversion event, assuming that class III and class V tetrads were due to double events. About 82% of total breaks were at the CAG-insertion site and 18% were at the HIS4 promoter (Table 1). On the basis of 35% sporulation efficiency (49), we calculate that 7.7% and 1.7% of total meiotic DNA should have had breaks at the CAG-insertion site and at the promoter region, respectively. These values are close to the observed values (Table 1).
The DSB repair could occur with either the sister chromatid or the homolog used as a template. However, the repair appears to occur by a nonreciprocal gene conversion event, since there was not a single tetrad that had the reciprocal products. In our system, because of the lack of flanking markers, homolog gene conversion associated with crossover events could not be monitored. Note that the repeat tract length alterations in human diseases do not appear to occur on crossover chromosomes (50). About 10% of tract size alterations observed in our system were expansions and the rest were contractions. Our results can be explained as follows: most, if not all, contraction events are caused by an intramolecular DSB-repair event (Fig. 5), whereas expansion occurs when the homolog or the sister chromatid is used as a template for the DSB repair. We favor this model because DSB repair does not appear to involve crossover chromosomes and, as a result, the tract size alterations occur on the broken chromosomes. A similar mechanism for the trinucleotide repeat instability was proposed by other investigators (6, 33, 51).
Figure 5.

DSB-induced contractions and expansions of the trinucleotide repeat tract. (A) Contraction of the repeat tract can occur as an intramolecular event. After DSB formation, exonucleolytic cleavage of the 5′-containing strand leaves two complementary single strands containing 3′ ends. Annealing of these complementary strands followed by DNA synthesis and ligation will lead to a contraction event. (B) An intermolecular event (as described in the synthesis-dependent strand-annealing model of ref. 52) may cause either an expansion or a contraction of the repeat tract, depending on the site of strand invasion on the intact chromatid. Only the expansion is shown here.
In summary, results presented here indicate that meiotic instability of the CAG repeat tract occurs by the DSB repair mechanism. In humans, trinucleotide repeat expansion has been suggested to occur during germ-line mitotic divisions or postmeiotically during early divisions of the embryo. We showed here that most large expansions occur by DSB repair during meiosis in yeast, which shares several features of gametogenesis with higher eukaryotes. These results also explain why transmission between generations is necessary to reach the mutation stage in humans.
Acknowledgments
We thank the Wadsworth Center Molecular Genetics core facility for synthesizing oligonucleotides and rapid sequencing of the PCR products. We also thank Kevin Manley for amplifying the CAG repeats from mouse DNA; Craig Giroux for the spo11-deletion plasmid; and Dennis Livingston, Marlene Belfort, Pam Swiatek, and Anne Messer for their comments on the manuscript. This work was supported by National Institutes of Health Grant GM56266.
Abbreviation
- DSB
double-strand break
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
Article published online before print: Proc. Natl. Acad. Sci. USA, 10.1073/pnas.040460297.
Article and publication date are at www.pnas.org/cgi/doi/10.1073/pnas.040460297
References
- 1.Ashley T, Warren S T. Annu Rev Genet. 1995;29:703–728. doi: 10.1146/annurev.ge.29.120195.003415. [DOI] [PubMed] [Google Scholar]
- 2.Bates G P, Lehrach H. BioEssays. 1994;16:277–284. doi: 10.1002/bies.950160411. [DOI] [PubMed] [Google Scholar]
- 3.Richards R I, Sutherland G R. Cell. 1992;70:709–712. doi: 10.1016/0092-8674(92)90302-s. [DOI] [PubMed] [Google Scholar]
- 4.Leeflang E P, Zhang L, Tavare S, Hubert R, Srinidhi J, MacDonald M E, Myers R H, de Young M, Wexler N S, Gusella J F, et al. Hum Mol Genet. 1995;4:1519–1526. doi: 10.1093/hmg/4.9.1519. [DOI] [PubMed] [Google Scholar]
- 5.McMurray C T. Chromosoma. 1995;104:2–13. doi: 10.1007/BF00352220. [DOI] [PubMed] [Google Scholar]
- 6.Sia E A, Jinks-Robertson S, Petes T D. Mutat Res. 1997;383:61–70. doi: 10.1016/s0921-8777(96)00046-8. [DOI] [PubMed] [Google Scholar]
- 7.Schweitzer J K, Livingston D M. Hum Mol Genet. 1997;6:349–355. doi: 10.1093/hmg/6.3.349. [DOI] [PubMed] [Google Scholar]
- 8.Maurer D J, O'Callaghan B L, Livingston D M. Mol Cell Biol. 1996;16:6617–6622. doi: 10.1128/mcb.16.12.6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Freudenreich C H, Stavenhagen J B, Zakian V A. Mol Cell Biol. 1997;17:2090–2098. doi: 10.1128/mcb.17.4.2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gordenin D A, Kunkel T A, Resnick M A. Nat Genet. 1997;16:116–118. doi: 10.1038/ng0697-116. [DOI] [PubMed] [Google Scholar]
- 11.Miret J J, Pessoa-Brandao L, Lahue R S. Mol Cell Biol. 1997;17:3382–3387. doi: 10.1128/mcb.17.6.3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kokoska R J, Sefanovic L, Tran H T, Resnick M A, Gordenin D A, Petes T D. Mol Cell Biol. 1998;18:2779–2788. doi: 10.1128/mcb.18.5.2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wells R D, Bacolla A, Bowater R P. Results Probl Cell Differ. 1998;21:133–165. doi: 10.1007/978-3-540-69680-3_4. [DOI] [PubMed] [Google Scholar]
- 14.White P J, Borts R H, Hirst M C. Mol Cell Biol. 1999;19:5675–5684. doi: 10.1128/mcb.19.8.5675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chen X, Mariappan S V S, Catasti P, Ratliff R, Moyzis R, Laayoun A, Smith S S, Bradbury E M, Gupta G. Proc Natl Acad Sci USA. 1995;92:5199–5203. doi: 10.1073/pnas.92.11.5199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gacy A M, Goellner G, Juranic N, Macura S, McMurray C T. Cell. 1995;81:533–540. doi: 10.1016/0092-8674(95)90074-8. [DOI] [PubMed] [Google Scholar]
- 17.Pearson C E, Sinden R R. Biochemistry. 1997;35:5041–5053. doi: 10.1021/bi9601013. [DOI] [PubMed] [Google Scholar]
- 18.Mitas M. Nucleic Acids Res. 1997;25:2245–2253. doi: 10.1093/nar/25.12.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Moore H, Greenwell P W, Liu C-P, Arnheim N, Petes T D. Proc Natl Acad Sci USA. 1999;96:1504–1509. doi: 10.1073/pnas.96.4.1504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miret J J, Pessoa-Brandao L, Lahue R S. Proc Natl Acad Sci USA. 1998;95:12438–12443. doi: 10.1073/pnas.95.21.12438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Freudenreich C H, Kantrow S M, Zakian V A. Science. 1998;279:853–856. doi: 10.1126/science.279.5352.853. [DOI] [PubMed] [Google Scholar]
- 22.Schweitzer J K, Livingston D M. Hum Mol Genet. 1998;7:69–74. doi: 10.1093/hmg/7.1.69. [DOI] [PubMed] [Google Scholar]
- 23.Leach D R F. BioEssays. 1994;16:893–900. doi: 10.1002/bies.950161207. [DOI] [PubMed] [Google Scholar]
- 24.Nag D K, Kurst A. Genetics. 1997;146:835–847. doi: 10.1093/genetics/146.3.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tishkoff D X, Filosi N, Gaida G M, Kolodner R D. Cell. 1997;88:253–263. doi: 10.1016/s0092-8674(00)81846-2. [DOI] [PubMed] [Google Scholar]
- 26.Michel B, Ehrlich S D, Uzest M. EMBO J. 1997;16:430–438. doi: 10.1093/emboj/16.2.430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Samadashwily G, Raca G, Mirkin S M. Nat Genet. 1997;17:298–304. doi: 10.1038/ng1197-298. [DOI] [PubMed] [Google Scholar]
- 28.Buard J, Jeffreys A. Nat Genet. 1997;15:327–328. doi: 10.1038/ng0497-327. [DOI] [PubMed] [Google Scholar]
- 29.Debrauwere H, Buard J, Tessier J, Aubert D, Vergnaud G, Nicolas A. Nat Genet. 1999;23:367–371. doi: 10.1038/15557. [DOI] [PubMed] [Google Scholar]
- 30.Petes T D. Cell. 1980;19:765–774. doi: 10.1016/s0092-8674(80)80052-3. [DOI] [PubMed] [Google Scholar]
- 31.Welch J W, Maloney D H, Fogel S. Mol Gen Genet. 1990;222:304–310. doi: 10.1007/BF00633833. [DOI] [PubMed] [Google Scholar]
- 32.Jakupciak J P, Wells R D. J Biol Chem. 1999;33:23468–23479. doi: 10.1074/jbc.274.33.23468. [DOI] [PubMed] [Google Scholar]
- 33.Richard G-F, Dujon B, Haber J E. Mol Gen Genet. 1999;261:871–882. doi: 10.1007/s004380050031. [DOI] [PubMed] [Google Scholar]
- 34.Malter H E, Iber J C, Willemsen R, de Graaff E, Tarleton J C, Leisti J, Warren S T, Oostra B A. Nat Genet. 1997;15:165–169. doi: 10.1038/ng0297-165. [DOI] [PubMed] [Google Scholar]
- 35.Leeflang E P, Tavare S, Marjoram P, Neal C O, Srinidhi J, MacDonald M E, de Young M, Wexler N S, Gusella J F, Arnheim N. Hum Mol Genet. 1999;8:173–183. doi: 10.1093/hmg/8.2.173. [DOI] [PubMed] [Google Scholar]
- 36.Cohen H, Sears D D, Zenvirth D, Hieter P, Simchen G. Mol Cell Biol. 1999;19:4153–4158. doi: 10.1128/mcb.19.6.4153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nag D K, White M A, Petes T D. Nature. 1989;340:318–320. doi: 10.1038/340318a0. [DOI] [PubMed] [Google Scholar]
- 38.Rose M, Winston F, Hieter P, editors. Methods in Yeast Genetics: A Laboratory Course Manual. Plainview, NY: Cold Spring Harbor Lab. Press; 1990. [Google Scholar]
- 39.Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies S W, Bates G P. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- 40.Paques F, Haber J E. Microbiol Mol Biol Rev. 1999;63:349–404. doi: 10.1128/mmbr.63.2.349-404.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fan Q-Q, Xu F, Petes T D. Mol Cell Biol. 1995;15:1679–1688. doi: 10.1128/mcb.15.3.1679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wu T-C, Lichten M. Genetics. 1995;140:55–66. doi: 10.1093/genetics/140.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xu L, Kleckner N. EMBO J. 1995;14:5115–5128. doi: 10.1002/j.1460-2075.1995.tb00194.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fan Q-Q, Xu F, Petes T D. Genetics. 1997;145:661–670. doi: 10.1093/genetics/145.3.661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wu T-C, Lichten M. Science. 1994;263:515–518. doi: 10.1126/science.8290959. [DOI] [PubMed] [Google Scholar]
- 46.Fan Q-Q, Petes T D. Mol Cell Biol. 1996;16:2037–2043. doi: 10.1128/mcb.16.5.2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wang Y-H, Amirhaeri S, Kang S, Wells R D, Griffith J D. Science. 1994;265:669–671. doi: 10.1126/science.8036515. [DOI] [PubMed] [Google Scholar]
- 48.Ohta K, Shibata T, Nicolas A. EMBO J. 1994;13:5754–5763. doi: 10.1002/j.1460-2075.1994.tb06913.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nag D K, Scherthan H, Rockmill B, Bhargava J, Roeder G S. Genetics. 1995;141:75–86. doi: 10.1093/genetics/141.1.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jeffreys A J, Allen M J, Armour J A, Collick A, Dubrova Y, Fretwell N, Guram T, Jobling M, May C A, Neil D L, et al. Electrophoresis. 1995;16:1577–1585. doi: 10.1002/elps.11501601261. [DOI] [PubMed] [Google Scholar]
- 51.Paques F, Leung W-Y, Haber J E. Mol Cell Biol. 1998;18:2045–2054. doi: 10.1128/mcb.18.4.2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nassif N, Penney J, Pal S, Engels W R, Gloor G B. Mol Cell Biol. 1994;14:1613–1625. doi: 10.1128/mcb.14.3.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]