

Abstract
Drosophila visual signaling is one of the fastest G-protein-coupled transduction cascades, because effector and modulatory proteins are organized into a macromolecular complex (“transducisome”). Assembly of the complex is orchestrated by inactivation no afterpotential D (INAD), which colocalizes the transient receptor potential (TRP) Ca2+ channel, phospholipase Cβ, and eye protein kinase C (eye-PKC), for more efficient signal transduction. Eye-PKC is critical for deactivation of vision. Moreover, deactivation is regulated by the interaction between INAD and TRP, because abrogation of this interaction in InaDp215 results in slow deactivation similar to that of inaCp209 lacking eye-PKC. To elucidate the mechanisms whereby eye-PKC modulates deactivation, here we demonstrate that eye-PKC, via tethering to INAD, phosphorylates TRP in vitro. We reveal that Ser982 of TRP is phosphorylated by eye-PKC in vitro and, importantly, in the fly eye, as shown by mass spectrometry. Furthermore, transgenic expression of modified TRP bearing an Ala substitution leads to slow deactivation of the visual response similar to that of InaDp215. These results suggest that the INAD macromolecular complex plays an essential role in termination of the light response by promoting efficient phosphorylation at Ser982 of TRP for fast deactivation of the visual signaling.
Keywords: Drosophila, INAD, PKC, TRP, vision, phosphorylation
Introduction
Drosophila visual transduction is a G-protein-coupled signaling pathway that provides a model system for understanding the molecular basis of signal transduction in the vertebrate nervous systems. Drosophila visual signaling is initiated with the activation of rhodopsin by light. Activated rhodopsin, via a Gq heterotrimeric protein, stimulates phospholipase Cβ (PLCβ) named no-receptor potential A (NORPA) (Bloomquist et al., 1988). NORPA hydrolyzes PIP2 (phosphatidylinositol 4,5-bisphosphate) to inositol 1,4,5-trisphosphate (IP3) and 1,2-diacylglycerol (DAG), which leads to opening of the transient receptor potential (TRP) Ca2+ and TRP-like channels, and depolarization of photoreceptors (Niemeyer et al., 1996; Reuss et al., 1997). The key second messenger that activates the TRP Ca2+ channel is thought to be either DAG or its lipid metabolites (Chyb et al., 1999; Raghu et al., 2000a), whereas IP3 does not appear to play a role (Montell, 1999; Raghu et al., 2000b). DAG may have a dual function, because it also activates the eye-specific protein kinase C (eye-PKC) essential for deactivation of the light response (Ranganathan et al., 1991; Smith et al., 1991).
Drosophila visual signaling is one of the fastest G-protein-coupled transduction cascades (Zuker, 1996). The fast kinetics of vision is partly attributable to the formation of a macromolecular complex containing TRP Ca2+ channel (Huber et al., 1996b; Shieh and Zhu, 1996; Chevesich et al., 1997), NORPA (Shieh et al., 1997; Tsunoda et al., 1997; van Huizen et al., 1998), and eye-PKC (Tsunoda et al., 1997; Adamski et al., 1998; Kumar and Shieh, 2001). This complex is organized by inactivation no afterpotential D (INAD) (Shieh and Niemeyer, 1995), a scaffolding protein with five PDZ (postsynaptic density-95/disc large/zonula occludens-1) domains. INAD regulates the subcellular localization and stability of these three proteins. Flies lacking INAD exhibit a profound reduction of the light response (Tsunoda et al., 1997).
To gain a better understanding of how the INAD complex modulates the kinetics of vision, we and others have shown that the INAD–TRP interaction is required for normal deactivation of the light response, because a loss of the interaction leads to slow deactivation in InaDp215 flies (Shieh and Niemeyer, 1995; Henderson et al., 2000). In addition, the INAD–eye-PKC interaction is essential for the in vivo activity of eye-PKC, because expression of modified eye-PKC that does not interact with INAD, fails to rescue inaCp209 flies lacking eye-PKC (Adamski et al., 1998). Indeed, two proteins in the complex, INAD and TRP, were found to be phosphorylated in vitro by eye-PKC (Huber et al., 1996a, 1998; Liu et al., 2000).
Here, we report the identification and functional characterization of an eye-PKC phosphorylation site in TRP. We show that TRP is phosphorylated at Ser982 by eye-PKC and this phosphorylation depends on INAD in vitro. By differential mass spectrometry (MS), we confirm that Ser982 of TRP is phosphorylated in vivo by eye-PKC. Moreover, we demonstrate that transgenic flies lacking this phosphorylation site display a slow deactivation phenotype similar to that of InaDp215. Our results indicate that INAD is critical for deactivation of visual signaling by positioning eye-PKC in close proximity to TRP, to facilitate its phosphorylation at Ser982.
Materials and Methods
Preparation of fly head extracts.
Approximately 100 μl of young wild-type, inaD1, or InaDp215 fly heads were homogenized with 1 ml of extraction buffer or EB (50 mm Tris-HCl, pH 8.0, 150 mm NaCl, 1% Triton X-100, and a mixture of protease inhibitors). Head homogenates were incubated at 4°C with constant agitation for 1 h. The mixture was then centrifuged for 10 min (12,000 × g), and the supernatant was used for the in vitro complex-dependent kinase assay. Protein concentrations were determined by BCA (Pierce, Rockford, IL).
In vitro complex-dependent kinase assay. This assay consists of a glutathione S-transferase (GST) pull-down followed by an in vitro kinase assay. GST fusion proteins were immobilized to glutathione-agarose beads and incubated with 500 μl of fly head extract (total protein concentration, 3.5–5 μg/ml) for 1 h at 4°C. After incubation, GST fusion proteins and associated proteins were recovered by centrifugation and washed three times with EB. For the kinase assay, the GST fusion protein mixture was washed once with kinase reaction buffer or RB (50 mm Tris-HCl, pH 8.0, 10 mm MgCl2, 5 mm 2-mercaptoethanol, 0.1 mm DTT, 0.4 mm EGTA, 0.7 mm CaCl2) and incubated at 30°C with 50 μl RB containing phorbol myristate acetate (PMA) (1 μm), 3 μCi of carrier-free [γ-32P]ATP in the presence of 100 μm cold ATP. SDS-PAGE loading buffer (2×) was added to terminate the kinase reactions. Samples were then subjected to SDS-PAGE (6 or 10%) followed by Western blotting or Coomassie blue staining. Dried and stained gels were subject to autoradiography or PhosphorImager analysis to quantify phosphorylation of fusion proteins. PMA, 12-(2-cyanoethyl)-6,7,12,13-tetrahydro-13-methyl-5-oxo-5H-indolo[2,3-a]pyrrolo[3,4-c]carbazole (Go6976), and recombinant human PKCα were obtained from Calbiochem (San Diego, CA). [γ-32P]ATP (6000 Ci/mmol) was from PerkinElmer (Boston, MA). Glutathione-agarose beads were from Pierce. The synthetic peptide was purchased from Sigma-Genosys (Woodlands, TX).
Liquid chromatography-MS analysis.
Liquid chromatography (LC)-MS was performed by the Proteomics Laboratory in the Vanderbilt Mass Spectrometry Research Center. Approximately 14 pmol of TRP was excised from SDS-PAGE gels for in-gel digestion with either trypsin or chymotrypsin (Ham, 2005). The resulting peptides were separated by reverse-phase HPLC that is coupled directly with automatic tandem MS (LC-MS) using a ThermoFinnigan LTQ ion trap mass spectrometer equipped with a Thermo MicroAS autosampler and Thermo Surveyor HPLC pump, Nanospray source, and Xcalibur 1.4 instrument control. MS/MS scans were acquired using an isolation width of 2 m/z, an activation time of 30 ms, and activation Q of 0.250 and 30% normalized collision energy using 1 microscan and ion time of 100 for each scan. The mass spectrometer was tuned before analysis using the synthetic peptide TpepK (AVAGKAGAR). Typical tune parameters were as follows: spray voltage of 1.8 kV, a capillary temperature of 150°C, a capillary voltage of 50 V, and tube lens of 100 V. Initial analysis was performed using data-dependent scanning in which one full MS spectra, using a full mass range of 400–2000 amu, were followed by three MS/MS spectra. Incorporated into the method was a data-dependent scan for the neutral loss of phosphoric acid or phosphate (−98, −80), such that if these masses were found, an MS/MS/MS of the neutral loss ion was performed. Peptides were identified using a cluster compatible version SEQUEST algorithm (Yates et al., 1995), using a Drosophila subset of proteins from the nonredundant database from the National Center for Biotechnology Information (NCBI). Sequest searches are done on a high speed, multiprocessor Linux cluster in the Advanced Computing Center for Research. In addition to using the SEQUEST algorithm to search for phosphorylation on serines or threonines, the data were also analyzed using the Pmod algorithm (Hansen et al., 2005). All possible modified peptides were verified by manual inspection of the spectra.
P-element-mediated germline transformation.
Wild-type and modified trp cDNA were subcloned into a modified pCaSpeR 4 vector (Thummel and Pirrotta, 1992) that contains Drosophila hsp70 promoter without the hsp70 3′ trailer region. The P-element construct and a transposase plasmid (“wings-clipped”) were injected into y[1] w[67c23] embryos (CBRC Transgenic Drosophila Core, Massachusetts General Hospital/Harvard Medical School, Charlestown, MA). Flies with the transgene integrated into the second or third chromosome were selected and made homozygotes in the trpp301 background for additional analysis.
Electroretinogram recordings.
Electroretinogram (ERG) recordings were performed using red-eye young flies (1–3 d of age) that were reared in a 12 h light/dark cycle. The flies were anesthetized by carbon dioxide and immobilized using nondrying modeling clay. Glass electrodes were filled with physiological saline (0.7% NaCl). White light stimulation (light intensity, 4.45 mW) was delivered by a fiber optic light source (Oriel, Stratford, CT) and attenuated using absorptive nd filters (Newport, Irvine, CA). Signals were amplified by means of a WPI Dam 50 differential amplifier (World Precision Instruments, Sarasota, FL), displayed on an oscilloscope. Data were digitalized and analyzed using AxonScope 9.0 software (Molecular Devices, Sunnyvale, CA).
Statistical analysis.
All bar graph data were analyzed with GraphPad (San Diego, CA) Prism 4.0 software one-way ANOVA. Data represent the means ± SEM, unless otherwise noted, from several independent experiments.
Results
The C-terminal tail of TRP contains PKC phosphorylation sites
To investigate the regulation of TRP by eye-PKC, we first identified potential eye-PKC phosphorylation sites in TRP. TRP consists of six transmembrane domains with both N and C termini localized intracellularly. By NetPhos 2.0 (http://www.cbs.dtu.dk/services/NetPhos/) and Prosite (http://www.expasy.ch/prosite/) software using the PKC consensus sequence motif (S/T)-X-(R/K), we found 16 putative phosphorylation sites in TRP with 14 present within the C-terminal sequence (Fig. 1A). Because the C-terminal tail of TRP has been implicated in gating and regulation of the channel, phosphorylation of this region may serve to switch on/off the channel activity. To investigate whether any of the putative PKC sites are bona fide PKC phosphorylation sites, we generated GST fusion proteins containing different intracellular regions of TRP and subjected them to in vitro kinase assays. As positive and negative controls, we used a fusion protein containing full-length INAD and GST alone, respectively. We first determined whether a recombinant PKCα could phosphorylate these fusion proteins because both PKCα and eye-PKC belong to the conventional PKC family. Indeed, we found that TRP906–1275 containing the last 370 residues of TRP including the six putative PKC sites became phosphorylated by PKCα (Fig. 1B), whereas TRP1–367, which contains two PKC sites did not (data not shown). Sequences spanning TRP657–905 failed to produce stable fusion proteins in Escherichia coli and therefore were not tested. The stoichiometry of TRP906–1275 phosphorylation by PKCα was ∼0.55 mol phosphate/moles fusion protein. These findings indicated that TRP906–1275 contains one PKC phosphorylation site.
Figure 1.

The C-terminal tail of TRP is phosphorylated by recombinant PKCα. A, Distribution of the 16 putative PKC phosphorylation sites in TRP. B, Phosphorylation of TRP906–1275 and INAD by PKCα. Lane 1, No substrate; lane 2, GST; lane 3, INAD; lane 4, TRP906–1275. The asterisks indicate INAD degradation products. The protein standards (in kilodaltons) are denoted on the left.
Phosphorylation of TRP906–1275 by eye-PKC is dependent on INAD in vitro
Next we investigated whether TRP906–1275 can be phosphorylated by eye-PKC. In Drosophila photoreceptors, eye-PKC and TRP form a macromolecular complex by tethering to INAD. To obtain eye-PKC, immobilized GST fusion proteins containing TRP906–1275 were incubated with wild-type fly head extracts to compete with endogenous TRP for retrieval of the INAD complex, including eye-PKC. The resulting complex was recovered by centrifugation and used for in vitro kinase assays. We found that TRP906–1275 pulled down INAD and eye-PKC, and became phosphorylated after the addition of a PKC activator, PMA, by this complex-dependent kinase assay (Fig. 2A, lane 2). To demonstrate that the observed phosphorylation of TRP906–1275 is dependent on INAD, we used fly extracts prepared from inaD1 and InaDp215. inaD1 is a loss-of-function allele of inaD (Tsunoda et al., 1997), whereas InaDp215 expresses a modified protein resulting in a loss of the TRP–INAD association (Shieh and Zhu, 1996). As shown in Figure 2A (lanes 3–6), both extracts failed to support TRP906–1275 phosphorylation by the complex-dependent kinase assay. In both cases, phosphorylation was diminished because TRP906–1275 was unable to isolate INAD and consequently, eye-PKC, from these two extracts (Fig. 2A, middle and bottom).
Figure 2.

TRP is phosphorylated in vitro by eye-PKC in a complex-dependent manner. A, Phosphorylation of TRP906–1275 using wild-type (wt), inaD1, and InaDp215 fly head extracts. Recovery of INAD (middle) and eye-PKC (bottom) was detected by Western blotting (WB). Lanes 1, 3, and 5, GST; lanes 2, 4, and 6, TRP906–1275. B, Reduced phosphorylation of a modified TRP906–1275 containing an Asp substitution at Val1266 by the complex-dependent kinase assay (top). The corresponding Coomassie-stained SDS-PAGE gel is shown in the middle. Recovery of eye-PKC was detected by Western blotting (bottom). Lane 1, GST; lane 2, TRP906–1275; lane 3, TRP906–1275, V1266D. C, Time course of the TRP phosphorylation using wild-type fly head extracts. Recovery of eye-PKC by wild-type TRP was detected by Western blotting (top). GST (▪); TRP906–1275 (▾); TRP906–1275, V1266D (▴). All quantifications were performed as described in Materials and Methods (n = 3). D, TRP906–1275, V1266D remains a substrate for recombinant PKCα (top). The corresponding Coomassie-stained SDS-PAGE gel is shown at the bottom. Lane 1, GST; lane 2, TRP906–1275; lane 3, TRP906–1275, V1266D.
To further support the role of INAD in directing eye-PKC to TRP, we investigated phosphorylation of a modified TRP906–1275 containing an Asp substitution at Val1266, which has been previously shown to disrupt the interaction between TRP and INAD (Shieh and Zhu, 1996; Li and Montell, 2000). We found that phosphorylation of TRP906–1275, V1266D was greatly reduced (Fig. 2B,C), because this modified TRP failed to recruit INAD and, consequently, eye-PKC (Fig. 2B, bottom). Importantly, this modified TRP remained an excellent substrate for recombinant human PKCα (Fig. 2D). Together, these results indicate that the phosphorylation of TRP906–1275 by endogenous eye-PKC in vitro is dependent on the interaction between TRP and INAD.
To demonstrate that eye-PKC is involved in phosphorylation of TRP906–1275, we show that this phosphorylation is abolished in the presence of a specific conventional PKC inhibitor Go6976 (5 μm) (Fig. 3A). To further confirm that eye-PKC is responsible for the observed phosphorylation of TRP906–1275, we performed the complex-dependent kinase assay using extracts from inaCp209 (Pak, 1979), which lacks endogenous eye-PKC (Smith et al., 1991). As expected, phosphorylation of TRP906–1275 and full-length INAD was greatly reduced, by 82 and 86%, respectively (Fig. 3B) (mean ± SEM; TRP906–1275, 17.77 ± 2.5%; INAD, 13.81 ± 4.13%; n = 3). The absence of phosphorylation is attributable to a lack of eye-PKC recovery when inaCp209 extracts were used (Fig. 3B, middle). These findings indicate that eye-PKC is indeed responsible for phosphorylation of TRP906–1275.
Figure 3.
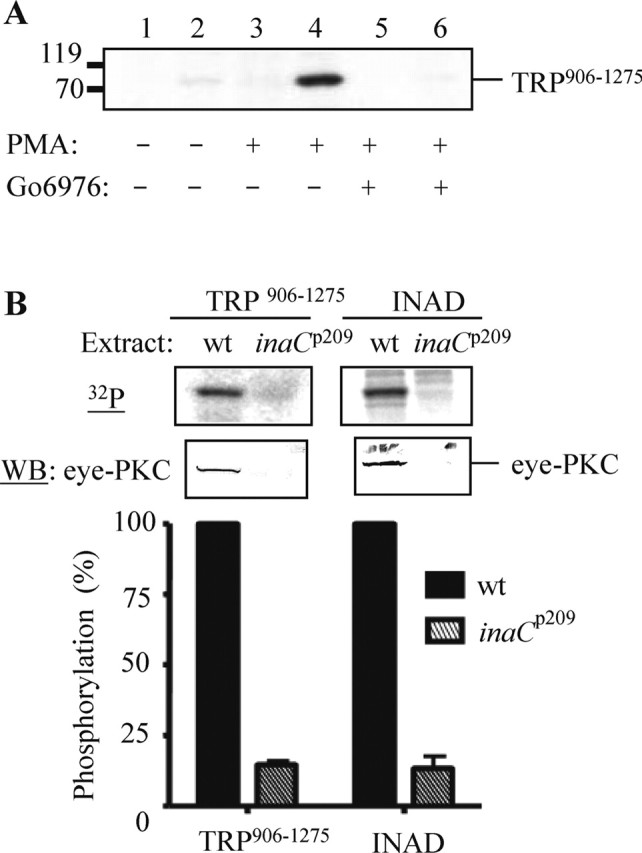
TRP906–1275 is phosphorylated in vitro by eye-PKC. A, Phosphorylation of TRP906–1275 was eliminated by a specific conventional PKC inhibitor, Go6976. Lanes 1, 3, and 5, GST; lanes 2, 4, and 6, TRP906–1275. B, inaCp209 extracts failed to promote the complex-dependent phosphorylation of TRP and INAD. The incorporated radioactivity was normalized to protein levels obtained by densitometry (Odyssey analysis software), and the relative phosphorylation was presented as mean ± SEM (n = 3). WB, Western blot.
TRP is phosphorylated at Ser982 in vitro
To investigate which of the six putative PKC sites in TRP906–1275 is phosphorylated by eye-PKC, we examined phosphorylation of two shorter TRP fusion proteins that contain one (TRP1157–1275) or four (TRP1030–1275) predicted PKC sites. We found that TRP1030–1275 and TRP1157–1275 displayed a drastic reduction of phosphorylation by 78 and 91%, respectively (Fig. 4A) (mean ± SEM; TRP1030–1275, 22.09 ± 7.78%; TRP1157–1275, 8.33 ± 5.43%; n = 3), by eye-PKC (Fig. 4B). Interestingly, these two fusion proteins were also not phosphorylated by recombinant PKCα (Fig. 4C). These data indicate that Ser958 or Ser982, the two sites present in TRP906–1275 but not in TRP1030–1275, are the potential phosphorylation sites for both eye-PKC and recombinant PKCα.
Figure 4.

Mapping the PKC phosphorylation site in TRP906–1275. A, The complex-dependent phosphorylation of three TRP fusion proteins: TRP906–1275, TRP1030–1275, and TRP1157–1275. One relevant autoradiography and its corresponding Coomassie-stained proteins are shown. B, Eye-PKC is recovered by all three TRP fusion proteins. Lane 1, GST; lane 2, TRP906–1275; lane 3, TRP1030–1275; lane 4, TRP1157–1275. WB, Western blot. C, Phosphorylation of TRP fusion proteins by PKCα. Lane 1, No substrate; lane 2, GST; lane 3, TRP906–1275; lane 4, TRP1030–1275; lane 5, TRP1157–1275. The asterisks denote the location of each TRP fusion protein. D, TRP is phosphorylated in vitro at Ser982 by PKCα. Lane 1, TRP906–1275; lane 2, TRP906–1275, S958A; lane 3, TRP906–1275, S982A; lane 4, TRP906–1275, S958A, S982A. All quantifications were done as described previously (n = 3). All data are presented as mean ± SEM.
To investigate whether Ser958 or Ser982 is indeed the primary PKC phosphorylation site, we generated a modified TRP906–1275 containing Ala substitutions at these two sites and performed in vitro kinase assays. We found that Ala substitution of Ser982 resulted in a marked decrease of phosphorylation by PKCα, whereas a similar substitution at Ser958 did not (Fig. 4D) (mean ± SEM; TRP906–1275, 100%; TRP906–1275, S958A, 80.47 ± 22.24%; TRP906–1275, S982A, 29.46 ± 2.84%; n = 3). Consistently, fusion proteins containing both mutations (S958A and S982A) also exhibited a drastically reduced phosphorylation (Fig. 4D) (mean ± SEM; TRP906–1275, S958A, S982A, 18.88 ± 6.93%; n = 3).
To further confirm that Ser982 of TRP is a PKC phosphorylation site, we obtained a synthetic peptide, ALRAS982VKNVD, spanning Ser982 of TRP, and used it for in vitro kinase assays. This oligopeptide was a substrate of recombinant human PKCα with a Km of 263.1 μm and a Vmax of 17.35 pmol/min. Together, these data indicate that Ser982 represents a major in vitro PKC phosphorylation site in TRP906–1275.
TRP is phosphorylated in vivo at Ser982 by eye-PKC
Once we established that Ser982 of TRP was phosphorylated in vitro by eye-PKC, we investigated whether Ser982 is phosphorylated in vivo by eye-PKC using LC-MS analysis. First, we isolated the INAD complexes from light-adapted wild-type flies via immunoprecipitation, using anti-INAD antibodies. The proteins in the INAD complexes were separated by SDS-PAGE and visualized by staining with Colloidal Blue. The 145 kDa protein band corresponding to TRP was excised (Fig. 5A), digested “in-gel” with trypsin or chymotrypsin, and the resulting peptide mixture was subjected to LC-MS analysis. Peptide fragments were analyzed and identified by a cluster compatible version SEQUEST algorithm (Yates et al., 1995), using a subset of Drosophila proteins from the NCBI database. We obtained ∼70% amino acid coverage of TRP (Fig. 5B) including sequences spanning Ser982. We also used collision induced dissociation (CID), which fragments peptides such that the fragmentation pattern can be used to discern the amino acid sequence and the exact site(s) of phosphorylation. By CID analysis, we identified and confirmed the amino acid sequence of the peptide RAS982VKNVDEKS-GADGKPGTM and revealed the presence of a phosphate group at Ser982. Moreover, we also found the spectra of the unmodified peptide as well as both the doubly and triply charged phosphopeptides (Fig. 5C,D). Importantly, only the unphosphorylated peptide RAS982VKNVDEKSGADGKPGTM was detected in TRP isolated from inaCp209 flies. Based on these data, we conclude that TRP is phosphorylated in vivo at Ser982 by eye-PKC.
Figure 5.

TRP is phosphorylated in vivo at Ser982 as revealed by LC-MS analysis. A, Colloidal Blue staining of a gel from which TRP was recovered after immunoprecipitation (IP). B, The sequence coverage of TRP was ∼70%, including 11 putative PKC sites. The solid lines denote tryptic and chymotryptic peptides identified by MS. Putative PKC phosphorylation sites are coded blue, Ser982 is coded red, and Lys-Pro repeats are coded green. C, Low-energy CID spectrum of the doubly charged phosphopeptide Arg980-Met999. The spectrum was zoomed in fivefold because of a very prominent neutral loss of ion. D, A low-energy CID spectrum of the triply charged phosphopeptide spanning Arg980-Met999. The insets show the sequence of the phosphopeptide; the black lines denote the identified cleavages. Fragment ions are labeled according to the accepted nomenclature. b-ions are coded blue, y-ions are coded red, and precursor ions are coded green. Spectra from the MS/MS/MS analysis of the neutral loss of phosphoric acid ions confirmed the sequence and the site of phosphorylation (data not shown).
trpS982A displays slow deactivation of the visual response
To gain insight into the functional significance of TRP phosphorylation at Ser982, we generated and characterized transgenic flies expressing a modified trp, trpS982A, in which the phosphorylation site is eliminated. As a control, we also generated transgenic flies expressing a wild-type trp (trpwt). The expression of wild-type or modified trp was under the control of the hsp70 promoter, and the function of TRP was analyzed in a null genetic background (trpp301) (Montell and Rubin, 1989; Shieh and Zhu, 1996). We first determined whether the modified TRP is stably expressed by Western blotting. Indeed, we observed that the TRP protein in trpS982A flies reaches a steady-state concentration similar to that of wild-type flies [Oregon-R (OR)] or transgenic flies expressing a wild-type trp, trpwt (Fig. 6A). It appears that basal transcription driven by the hsp70 promoter is sufficient for transcription of trp leading to wild-type level of TRP in trpwt and trpS982A flies.
Figure 6.

Biochemical and electrophysiological characterization of transgenic flies lacking the phosphorylation site at Ser982. A, Western blotting (WB). The expression of TRP in the fly head or body was analyzed. B, ERG analysis. Shown are representative ERG recordings of wild-type (OR), trpp301, trpwt, trpS982A, inaCp209, and InaDp215 flies after stimulation of a 2 s pulse of the brightest light stimulus (log I/I0 = 0). C, A histogram that compares half-repolarization times (n = 5; mean ± SEM) at different light stimuli. The stimuli were 2 s white lights without any attenuation (log I/I0 = 0, where I represents stimulus intensity used and I0 represents maximum stimulus intensity available) or attenuated by 1 (log I/I0 = −1), 2 (log I/I0 = −2), or 3 (log I/I0 = −3) log units. The half-repolarization time is the time required to reach 50% of repolarization, as diagrammatically depicted for trpS982A flies in B. Two independent transgenic lines for both trpwt and trpS982A were used for quantification. ***p = 0.001.
Next, we characterized the visual electrophysiology by ERG for gaining insight into the in vivo activity of the modified TRP. ERG is an extracellular recording of the compound eye. Briefly, red-eye flies were dark-adapted for 2 min, and then given a 2 s white light stimulation. Using this stimulation paradigm, wild-type flies displayed the characteristic ERG waveform consisting of fast depolarization, maintained depolarization, and fast repolarization components (Fig. 6B). In contrast, trpp301 flies displayed the initial fast depolarization but lacked the maintained component, and therefore the membrane potential returned gradually to baseline. This abnormal phenotype of trpp301 was completely rescued by transgenic expression of wild-type trp (Fig. 6B). Remarkably, transgenic expression of trpS982A rescued the trpp301 phenotype but with delayed deactivation kinetics (Fig. 6B). Close inspection of the deactivation kinetics in ERG revealed two subcomponents: a fast and a slow component. The fast subcomponent occurs immediately after light termination and achieves over 50% repolarization. The fast subcomponent is followed by the slow subcomponent, which eventually returns the potential to baseline. It appears that trpS982A flies exhibit defects in the fast subcomponent.
To further characterize trpS982A flies, we examined their visual response to various intensities of light over 4 log units. We show that the prolonged deactivation kinetics is more prominent during the brightest light stimulation (log I/I0 = 0): the half-repolarization time of trpS982A is approximately twofold longer than that of wild-type flies (Fig. 6C) (mean ± SEM; wild type, 0.801 ± 0.119 s; trpwt, 0.842 ± 0.064 s; trpS982A, 1.668 ± 0.253 s; n = 5). In contrast, the amplitude of the ERG responses in trpS982A was comparable with that of trpwt flies (mean ± SEM; trpwt, 18.008 ± 0.95 mV; trpS982A, 20.71 ± 2.73 mV; n = 5), indicating that activation of visual signaling is not affected in trpS982A. These results demonstrate that expression of trpS982A leads to slow deactivation of visual response, which is likely attributable to a loss of eye-PKC phosphorylation of the modified TRP.
We compared the deactivation kinetics of trpS982A with that of inaCp209 flies that lack eye-PKC. Interestingly, inaCp209 exhibited prolonged deactivation kinetics similar to trpS982A, in response to bright light stimuli. However, inaCp209 also shows defects in deactivation at lower light intensities: the half-repolarization time was at least twofold longer than that of wild type, regardless of the light intensity used (mean ± SEM; log I/I0 = 0, inaCp209, 2.216 ± 0.1 s; log I/I0 = −1, wild type, 0.708 ± 0.087 s, trpwt, 0.721 ± 0.209 s, trpS982A, 0.938 ± 0.182 s, inaCp209, 1.852 ± 0.053 s; log I/I0 = −2, wild type, 0.491 ± 0.044 s, trpwt, 0.501 ± 0.139 s, trpS982A, 0.626 ± 0.102 s, inaCp209, 1.493 ± 0.091 s; log I/I0 = −3, wild type, 0.490 ± 0.087 s, trpwt, 0.410 ± 0.091 s, trpS982A, 0.337 ± 0.048 s, inaCp209, 1.219 ± 0.1 s; n = 5). These results indicate that the deactivation defect in inaCp209 is more complex than that of trpS982A and suggest that phosphorylation of additional PKC sites in TRP or other substrates may be responsible for the fast deactivation of the visual response.
We also investigated the deactivation kinetics of trpS982A in comparison with that of InaDp215 (Pak, 1979). InaDp215 contains a modified INAD, INADM442K, which fails to associate with TRP (Shieh and Zhu, 1996). The lack of the TRP–INAD interaction leads to a slow recovery of the visual response (Shieh and Zhu, 1996). We found that InaDp215 displayed an ERG phenotype similar to that of trpS982A with deactivation defects that manifested at bright light stimulation. Moreover, as for trpS982A, the deactivation kinetics of InaDp215 at low light intensities were indistinguishable from wild type (Fig. 6C) (mean ± SEM; half-repolarization time for InaDp215, log I/I0 = 0, 1.432 ± 0.064 s; log I/I0 = −1, 0.815 ± 0.045 s; log I/I0 = −2, 0.374 ± 0.052 s; log I/I0 = −3, 0.398 ± 0.041 s; n = 5). These results indicate that the slow recovery of InaDp215 may be attributable to a loss of eye-PKC phosphorylation in TRP.
Together, our biochemical and electrophysiological analyses demonstrate that phosphorylation of TRP at Ser982 by eye-PKC is important for the rapid deactivation of visual signaling in Drosophila.
Discussion
Reversible phosphorylation modulates the dynamics of signal transduction by transiently altering activities of signaling proteins. Members of the conventional PKC family (Newton, 1995), which are activated by Ca2+ and DAG, are capable of phosphorylating a wide variety of protein substrates for temporal and spatial regulation of signaling processes (Violin and Newton, 2003). In Drosophila, eye-PKC is involved in the negative regulation of visual signaling, because inaCp209 flies lacking eye-PKC display abnormal desensitization, slow deactivation, and defects in light adaptation (Smith et al., 1991; Hardie et al., 1993). Eye-PKC is anchored to a macromolecular complex by tethering to INAD (Tsunoda et al., 1997; Adamski et al., 1998). Interaction with INAD enhances the stability of eye-PKC as well as targets eye-PKC to the rhabdomeres of photoreceptors (Tsunoda et al., 2001), in which visual signaling occurs. Importantly, the in vivo function of eye-PKC is regulated by interaction with INAD. Previously, it was shown that eye-PKC phosphorylates TRP in vitro (Huber et al., 1998; Liu et al., 2000). In the present study, we investigated the molecular basis of TRP phosphorylation by eye-PKC.
To mimic eye-PKC phosphorylation of TRP in vitro, we designed a complex-dependent kinase assay. We demonstrated that the in vitro complex-specific phosphorylation of TRP is regulated by the presence of the INAD-interacting domain in TRP, as well as the existence of INAD in the fly extracts. We showed that extracts lacking either eye-PKC or INAD fail to support TRP phosphorylation. Similarly, extracts prepared from InaDp215 that expresses a modified INAD devoid of the TRP binding (Shieh and Zhu, 1996), are not able to promote TRP phosphorylation. Together, these findings indicate that INAD targets eye-PKC to its substrates, similar to RACK (receptor for activated C kinase) (Liu et al., 2000). By the complex-dependent kinase assay, we identified Ser982 of TRP as an eye-PKC phosphorylation site. Moreover, we analyzed TRP isolated from flies by LC-MS and found that Ser982 of TRP is indeed phosphorylated in vivo by eye-PKC, because phosphorylated peptides encompassing Ser982 of TRP were present in wild-type, but absent in inaCp209 flies.
Next, we investigated the in vivo functional contribution of phosphorylation by characterizing transgenic flies expressing a modified TRP bearing an Ala substitution at Ser982 (trpS982A). Remarkably, these transgenic flies displayed prolonged deactivation kinetics in response to bright light stimuli, indicating that phosphorylation of TRP at Ser982 by eye-PKC is involved in inactivation of TRP, leading to fast deactivation. A model of the TRP regulation by eye-PKC is proposed (Fig. 7). TRP is an integral part of the INAD complex and is opened by light. After light termination, the visual response is rapidly deactivated. Although molecular mechanisms underlying deactivation remain elusive, Ca2+ is known to play a vital role in response termination (Hardie, 1991; Ranganathan et al., 1991, 1994; Peretz et al., 1994). The increased intracellular Ca2+ (primarily mediated by TRP) and DAG activate eye-PKC, which, in turn, phosphorylates TRP at Ser982. Phosphorylation of TRP leads to a rapid inactivation of the channel on cessation of the light stimulation (Fig. 7B), without affecting the interaction between TRP and INAD (data not shown). How does phosphorylation influence the TRP channel activity? Ser982 is located within the Lys-Pro-rich region of TRP (Fig. 5B), which may function in TRP gating (Sinkins et al., 1996). We speculate that phosphorylation at Ser982 may induce a conformational change in the pore domain, which in turn leads to a rapid closure and inactivation of TRP. Phosphorylation has been linked directly to conformational changes that play key roles in the regulation of ion channels (Dulhanty and Riordan, 1994). It is also possible that phosphorylation of TRP at Ser982 affects the interaction with some yet-unidentified proteins that may be important for the modulation of the TRP channel activity.
Figure 7.

A model of the TRP regulation by eye-PKC-mediated phosphorylation at Ser982. A, INAD facilitates phosphorylation of TRP at Ser982 by eye-PKC, by positioning eye-PKC in close proximity to TRP. B, Resting TRP channels (I) become open in response to light stimulation (II), leading to a massive influx of Ca2+ that results in depolarization of the photoreceptor cells. Meanwhile, increases in the concentrations of intracellular Ca2+ and DAG activate eye-PKC, which, in turn, phosphorylates TRP (III). When light stimulation is off, phosphorylated TRP rapidly becomes inactivated (IV). It is likely that eye-PKC-mediated phosphorylation at Ser982 of TRP facilitates the conformational changes associated with the closure of the channel. To return the TRP channels to the resting state, dephosphorylation with the participation of protein phosphatases may occur. Shown at the bottom is a representative ERG of a wild-type fly.
In the absence of eye-PKC-mediated phosphorylation of TRP, deactivation of visual signaling is slower as observed in inaCp209 or trpS982A. We found that inaCp209 displays a more complex deactivation defect, whereas trpS982A exhibits prolonged deactivation only in response to bright light. These findings suggest that, in addition to TRP, eye-PKC phosphorylates other substrates for efficient termination of the light response. Indeed, eye-PKC has been shown to phosphorylate INAD (Huber et al., 1996a; Liu et al., 2000), but the functional relevance of this phosphorylation is not known. Furthermore, Gu et al. (2005) reported that eye-PKC is required for the Ca2+-dependent inhibition of NORPA. NORPA is part of the INAD complex; however, it is not known to be phosphorylated by eye-PKC. Gu et al. (2005) also showed that the Ca2+-dependent inactivation of the light-induced current was unaltered in inaCp209. This finding suggests the existence of a parallel Ca2+-dependent mechanism in inaCp209 by which TRP is inactivated or of an upregulation of a Ca2+-dependent mechanism that activates other kinases (Matsumoto et al., 1994) to compensate for the loss of eye-PKC in inaCp209.
Importantly, trpS982A displays slow deactivation kinetics similar to that of InaDp215. InaDp215 was isolated by Pak (1979) based on the ina (inactivation no afterpotential) phenotype elicited by ERG. By whole-cell recordings, Shieh and Niemeyer (1995) showed that InaDp215 exhibits slow deactivation kinetics. However, Tsunoda et al. (1997) reported a delay in latency of the quantum bump and proposed that activation was affected in the InaDp215 mutant. To resolve this discrepancy, Henderson et al. (2000) reexamined the mutant and concluded that the primary defect in InaDp215 is prolonged deactivation and not slow activation. InaDp215 expresses INADM442K, which fails to associate with TRP (Shieh and Niemeyer, 1995). How does a loss of INAD–TRP interaction lead to abnormal deactivation of visual signaling? It is likely that the lack of the INAD–TRP interaction prevents the recruitment of TRP to the INAD complex and, consequently, eye-PKC-mediated regulation. Indeed, both trpS982A and InaDp215 exhibit similar deactivation defects, indicating that the molecular basis underlying the slow deactivation defect in InaDp215 is attributable to a lack of negative regulation of the TRP channel by eye-PKC. Together, these findings suggest that formation of the INAD complex is essential for fast deactivation of the visual response by promoting phosphorylation of TRP by eye-PKC. Moreover, Ser982 may be the sole eye-PKC phosphorylation site in TRP, because trpS982A and InaDp215 display similar deactivation defects. A loss of INAD–TRP interaction was previously investigated in transgenic flies expressing modified TRP in which the INAD-interacting domain was deleted (trpΔ1272). Li and Montell (2000) reported a reduced light response with normal deactivation kinetics in trpΔ1272. These authors proposed that the suppression of the delayed termination, which is attributable to a reduced eye-PKC level in trpΔ1272 is probably masked by a concomitant decrease in TRP and INAD levels (Li and Montell, 2000).
To date, many proteins related to Drosophila TRP have been discovered in both invertebrates and vertebrates. These TRP ion channels are subdivided into seven subfamilies (TRPC, TRPV, TRPM, TRPN, TRPA, TRPP, and TRPML) (Ramsey et al., 2006). Drosophila TRP belongs to the TRPC subfamily. Members of the TRPC subfamily are also activated by receptor-induced activation of phospholipase C (Montell et al., 2002) and therefore may be regulated by PKC. Indeed, phosphorylation of the TRPC channels by PKC appears important for modulating the channel activity. For example, the PKC-mediated phosphorylation of TRPC1 was shown to contribute to its SOC (store operated channel) activation, triggering Ca2+ entry into endothelial cells (Ahmmed et al., 2004). In contrast, PKC-mediated phosphorylation was demonstrated to inhibit the activity of TRPC3 in HEK 293 cells (Trebak et al., 2005) and of TRPC6 in PC12D neuronal cells (Kim and Saffen, 2005). In both cases, TRPC3 and TRPC6 are activated by DAG, whereas DAG also turns on PKC. The authors proposed that timing is important because the channels are activated by DAG more rapidly than they are inhibited by DAG-activated PKC (Kim and Saffen, 2005). Heterologously expressed TRPC7 was also shown to be regulated by PKC: inhibition of PKC prolonged inactivation of the channel (Shi et al., 2004). Moreover, PKC phosphorylation of heterologously expressed TRPC5 resulted in desensitization of this channel, a process that was dependent on both extracellular and intracellular Ca2+ concentrations (Zhu et al., 2005).
In conclusion, here we uncover the molecular mechanism underlying the complex-dependent phosphorylation of TRP by eye-PKC and its role in fast deactivation of vision. Specifically, we show that eye-PKC phosphorylates TRP at Ser982 in vitro and in vivo. Importantly, phosphorylation of TRP facilitates rapid inactivation of the channel because transgenic flies bearing an Ala substitution at Ser982 display prolonged deactivation kinetics of the light response. Significantly, this slow deactivation defect is similar to that observed in InaDp215 in which TRP fails to associate with INAD. Our findings provide insights into the mechanistic basis of slow deactivation in InaDp215, suggesting that INAD plays a critical role in targeting eye-PKC to TRP for rapid deactivation of the visual signaling. Together, these data indicate that the INAD macromolecular complex is important for deactivation of the visual response by directing eye-PKC to TRP. Furthermore, PKC-mediated phosphorylation of TRP at Ser982 leads to fast deactivation of vision by promoting inactivation of the TRP channel.
Footnotes
This work was supported by National Institutes of Health Grants EY09743 (B.-H.S.) and EY08126 (Vanderbilt University). We thank Dr. William Pak for inaCp209 flies, Dr. Charles Zuker for inaD1 flies, Dr. Eric Ward for help with genetic crosses, and Dr. Ning Wang for help with ERG recordings. We thank Drs. John Exton, Brian Wadzinski, Louis DeFelice, Joy Purdy, and Lisan Parker for critical reading of this manuscript. We thank Li Peng for excellent technical support. We also thank Salisha Hill and Kristin Cheek for their technical support in the Proteomics Laboratory.
References
- Adamski FM, Zhu MY, Bahiraei F, Shieh BH (1998). Interaction of eye protein kinase C and INAD in Drosophila. Localization of binding domains and electrophysiological characterization of a loss of association in transgenic flies. J Biol Chem 273:17713–17719. [DOI] [PubMed] [Google Scholar]
- Ahmmed GU, Mehta D, Vogel S, Holinstat M, Paria BC, Tiruppathi C, Malik AB (2004). Protein kinase Cα phosphorylates the TRPC1 channel and regulates store-operated Ca2+ entry in endothelial cells. J Biol Chem 279:20941–20949. [DOI] [PubMed] [Google Scholar]
- Bloomquist BT, Shortridge RD, Schneuwly S, Perdew M, Montell C, Steller H, Rubin G, Pak WL (1988). Isolation of a putative phospholipase C gene of Drosophila, norpA, and its role in phototransduction. Cell 54:723–733. [DOI] [PubMed] [Google Scholar]
- Chevesich J, Kreuz AJ, Montell C (1997). Requirement for the PDZ domain protein, INAD, for localization of the TRP store-operated channel to a signaling complex. Neuron 18:95–105. [DOI] [PubMed] [Google Scholar]
- Chyb S, Raghu P, Hardie RC (1999). Polyunsaturated fatty acids activate the Drosophila light-sensitive channels TRP and TRPL. Nature 397:255–259. [DOI] [PubMed] [Google Scholar]
- Dulhanty AM, Riordan JR (1994). Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry 33:4072–4079. [DOI] [PubMed] [Google Scholar]
- Gu Y, Oberwinkler J, Postma M, Hardie RC (2005). Mechanisms of light adaptation in Drosophila photoreceptors. Curr Biol 15:1228–1234. [DOI] [PubMed] [Google Scholar]
- Ham A-J (2005). Proteolytic digestion protocols. In: The encyclopedia of mass spectrometry, Vol 2, Biological applications, Pt A, Peptides and proteins (Caprioli RM, Gross ML, eds) pp. 10–17. Oxford: Elsevier.
- Hansen BT, Davey SW, Ham AJ, Liebler DC (2005). P-Mod: an algorithm and software to map modifications to peptide sequences using tandem MS data. J Proteome Res 4:358–368. [DOI] [PubMed] [Google Scholar]
- Hardie RC (1991). Whole-cell recordings of the light induced current in dissociated photoreceptors: evidence for feedback by calcium permeating the light-sensitive channels. Proc R Soc Lond B Biol Sci 245: pp. 203–210. [Google Scholar]
- Hardie RC, Peretz A, Suss-Toby E, Rom-Glas A, Bishop SA, Selinger Z, Minke B (1993). Protein kinase C is required for light adaptation in Drosophila photoreceptors. Nature 363:634–637. [DOI] [PubMed] [Google Scholar]
- Henderson SR, Reuss H, Hardie RC (2000). Single photon responses in Drosophila photoreceptors and their regulation by Ca2+ J Physiol (Lond) 524:179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber A, Sander P, Paulsen R (1996a). Phosphorylation of the InaD gene product, a photoreceptor membrane protein required for recovery of visual excitation. J Biol Chem 271:11710–11717. [DOI] [PubMed] [Google Scholar]
- Huber A, Sander P, Gobert A, Bahner M, Hermann R, Paulsen R (1996b). The transient receptor potential protein (Trp), a putative store-operated Ca2+ channel essential for phosphoinositide-mediated photoreception, forms a signaling complex with NorpA, InaC and InaD. EMBO J 15:7036–7045. [PMC free article] [PubMed] [Google Scholar]
- Huber A, Sander P, Bahner M, Paulsen R (1998). The TRP Ca2+ channel assembled in a signaling complex by the PDZ domain protein INAD is phosphorylated through the interaction with protein kinase C (ePKC). FEBS Lett 425:317–322. [DOI] [PubMed] [Google Scholar]
- Kim JY, Saffen D (2005). Activation of M1 muscarinic acetylcholine receptors stimulates the formation of a multiprotein complex centered on TRPC6 channels. J Biol Chem 280:32035–32047. [DOI] [PubMed] [Google Scholar]
- Kumar R, Shieh BH (2001). The second PDZ domain of INAD is a type I domain involved in binding to eye protein kinase C. Mutational analysis and naturally occurring variants. J Biol Chem 276:24971–24977. [DOI] [PubMed] [Google Scholar]
- Li HS, Montell C (2000). TRP and the PDZ protein, INAD, form the core complex required for retention of the signalplex in Drosophila photoreceptor cells. J Cell Biol 150:1411–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Parker LL, Wadzinski BE, Shieh BH (2000). Reversible phosphorylation of the signal transduction complex in Drosophila photoreceptors. J Biol Chem 275:12194–12199. [DOI] [PubMed] [Google Scholar]
- Matsumoto H, Kurien BT, Takagi Y, Kahn ES, Kinumi T, Komori N, Yamada T, Hayashi F, Isono K, Pak WL, Jackson KW, Tobin SL (1994). Phosrestin I undergoes the earliest light-induced phosphorylation by a calcium/calmodulin-dependent protein kinase in Drosophila photoreceptors. Neuron 12:997–1010. [DOI] [PubMed] [Google Scholar]
- Montell C (1999). Visual transduction in Drosophila. Annu Rev Cell Dev Biol 15:231–268. [DOI] [PubMed] [Google Scholar]
- Montell C, Rubin GM (1989). Molecular characterization of the Drosophila trp locus: a putative integral membrane protein required for phototransduction. Neuron 2:1313–1323. [DOI] [PubMed] [Google Scholar]
- Montell C, Birnbaumer L, Flockerzi V (2002). The TRP channels, a remarkably functional family. Cell 108:595–598. [DOI] [PubMed] [Google Scholar]
- Newton AC (1995). Protein kinase C. Seeing two domains. Curr Biol 5:973–976. [DOI] [PubMed] [Google Scholar]
- Niemeyer BA, Suzuki E, Scott K, Jalink K, Zuker CS (1996). The Drosophila light-activated conductance is composed of the two channels TRP and TRPL. Cell 85:651–659. [DOI] [PubMed] [Google Scholar]
- Pak WL (1979). Study of photoreceptor function using Drosophila mutants. In: Neurogenetics: genetic approaches to the nervous system (Breakfield XO, ed.) pp. 67–99. New York: Elsevier.
- Peretz A, Suss-Toby E, Rom-Glas A, Arnon A, Payne R, Minke B (1994). The light response of Drosophila photoreceptors is accompanied by an increase in cellular calcium: effects of specific mutations. Neuron 12:1257–1267. [DOI] [PubMed] [Google Scholar]
- Raghu P, Usher K, Jonas S, Chyb S, Polyanovsky A, Hardie RC (2000a). Constitutive activity of the light-sensitive channels TRP and TRPL in the Drosophila diacylglycerol kinase mutant, rdgA. Neuron 26:169–179. [DOI] [PubMed] [Google Scholar]
- Raghu P, Colley NJ, Webel R, James T, Hasan G, Danin M, Selinger Z, Hardie RC (2000b). Normal phototransduction in Drosophila photoreceptors lacking an InsP(3) receptor gene. Mol Cell Neurosci 15:429–445. [DOI] [PubMed] [Google Scholar]
- Ramsey IS, Delling M, Clapham DE (2006). An Introduction to TRP channels. Annu Rev Physiol 68:619–647. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Harris GL, Stevens CF, Zuker CS (1991). A Drosophila mutant defective in extracellular calcium-dependent photoreceptor deactivation and rapid desensitization. Nature 354:230–232. [DOI] [PubMed] [Google Scholar]
- Ranganathan R, Bacskai BJ, Tsien RY, Zuker CS (1994). Cytosolic calcium transients: spatial localization and role in Drosophila photoreceptor cell function. Neuron 13:837–848. [DOI] [PubMed] [Google Scholar]
- Reuss H, Mojet MH, Chyb S, Hardie RC (1997). In vivo analysis of the Drosophila light-sensitive channels, TRP and TRPL. Neuron 19:1249–1259. [DOI] [PubMed] [Google Scholar]
- Shi J, Mori E, Mori Y, Mori M, Li J, Ito Y, Inoue R (2004). Multiple regulation by calcium of murine homologues of transient receptor potential proteins TRPC6 and TRPC7 expressed in HEK293 cells. J Physiol (Lond) 561:415–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shieh BH, Niemeyer B (1995). A novel protein encoded by the InaD gene regulates recovery of visual transduction in Drosophila. Neuron 14:201–210. [DOI] [PubMed] [Google Scholar]
- Shieh BH, Zhu MY (1996). Regulation of the TRP Ca2+ channel by INAD in Drosophila photoreceptors. Neuron 16:991–998. [DOI] [PubMed] [Google Scholar]
- Shieh BH, Zhu MY, Lee JK, Kelly IM, Bahiraei F (1997). Association of INAD with NORPA is essential for controlled activation and deactivation of Drosophila phototransduction in vivo. Proc Natl Acad Sci USA 94:12682–12687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkins WG, Vaca L, Hu Y, Kunze DL, Schilling WP (1996). The COOH-terminal domain of Drosophila TRP channels confers thapsigargin sensitivity. J Biol Chem 271:2955–2960. [DOI] [PubMed] [Google Scholar]
- Smith DP, Ranganathan R, Hardy RW, Marx J, Tsuchida T, Zuker CS (1991). Photoreceptor deactivation and retinal degeneration mediated by a photoreceptor-specific protein kinase C. Science 254:1478–1484. [DOI] [PubMed] [Google Scholar]
- Thummel CS, Pirrotta V (1992). New pCaSpeR P element vectors. Drosoph Inf Serv 71:150. [Google Scholar]
- Trebak M, Hempel N, Wedel BJ, Smyth JT, Bird GS, Putney JW Jr (2005). Negative regulation of TRPC3 channels by protein kinase C-mediated phosphorylation of serine 712. Mol Pharmacol 67:558–563. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Sierralta J, Sun Y, Bodner R, Suzuki E, Becker A, Socolich M, Zuker CS (1997). A multivalent PDZ-domain protein assembles signalling complexes in a G-protein-coupled cascade. Nature 388:243–249. [DOI] [PubMed] [Google Scholar]
- Tsunoda S, Sun Y, Suzuki E, Zuker C (2001). Independent anchoring and assembly mechanisms of INAD signaling complexes in Drosophila photoreceptors. J Neurosci 21:150–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Huizen R, Miller K, Chen DM, Li Y, Lai ZC, Raab RW, Stark WS, Shortridge RD, Li M (1998). Two distantly positioned PDZ domains mediate multivalent INAD-phospholipase C interactions essential for G protein-coupled signaling. EMBO J 17:2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Violin JD, Newton AC (2003). Pathway illuminated: visualizing protein kinase C signaling. IUBMB Life 55:653–660. [DOI] [PubMed] [Google Scholar]
- Yates JR III, Eng JK, McCormack AL, Schieltz D (1995). Method to correlate tandem mass spectra of modified peptides to amino acid sequences in the protein database. Anal Chem 67:1426–1436. [DOI] [PubMed] [Google Scholar]
- Zhu MH, Chae M, Kim HJ, Lee YM, Kim MJ, Jin NG, Yang DK, So I, Kim KW (2005). Desensitization of canonical transient receptor potential channel 5 by protein kinase C. Am J Physiol 289:C591–C600. [DOI] [PubMed] [Google Scholar]
- Zuker CS (1996). The biology of vision of Drosophila. Proc Natl Acad Sci USA 93:571–576. [DOI] [PMC free article] [PubMed] [Google Scholar]