

Abstract
Intracellular Mg2+ at physiological concentrations activates mSlo1 BK channels by binding to a metal-binding site in the cytosolic domain. Previous studies suggest that residues E374, Q397, and E399 are important in Mg2+ binding. In the present study, we show that mutations of E374 or E399 to other amino acids, except for Asp, abolish Mg2+ sensitivity. These results further support that the side chains of E374 and E399 are essential for Mg2+ coordination. To the contrary, none of the Q397 mutations abolishes Mg2+ sensitivity, suggesting that its side chain may not coordinate to Mg2+. However, because Q397 is spatially close to E374 and E399, its mutations affect the Mg2+ sensitivity of channel gating by either reducing or increasing the Mg2+ binding affinity. The pattern of mutational effects and the effect of chemical modification of Q397C indicate that Q397 is involved in the Mg2+-dependent activation of BK channels and that mutations of Q397 alter Mg2+ sensitivity by affecting the conformation of the Mg2+ binding site as well as by electrostatic interactions with the bound Mg2+ ion.
INTRODUCTION
BK-type K+ channels are activated by voltage and intracellular Ca2+. This unique property of the BK channel activation makes it important in physiological processes in various cell types (1–15). Because BK channels have a large single-channel conductance, their opening hyperpolarizes membranes with high efficiency, resulting in the closure of voltage-dependent Ca2+ channels and hence decreasing [Ca2+]i. Thus, BK channels provide a negative feedback in controlling the membrane potential and [Ca2+]i. Based on this mechanism, BK channels modulate various physiological processes including neural excitation (5,11,16–19), muscle contraction (8,15,20–23), hearing (24–27), and immunity (28).
Apart from Ca2+, the function of BK channels is also modulated by intracellular Mg2+ (29–40). Mg2+ modulates a variety of Ca2+ and K+ channels to affect the excitability or excitation-contraction coupling in neurons, cardiac myocytes, and smooth muscle cells (41–47). Following central nervous system injury, [Mg2+]i is significantly reduced, contributing to a number of factors including neurotransmitter release and oxidative stress that initiate an autodestructive cascade of biochemical and pathophysiological changes, known as secondary injury, that ultimately result in irreversible tissue damage (48). Pharmacological studies have shown that Mg2+ may be an effective therapeutic agent following neurotrauma to improve survival and motor outcome and to alleviate cognitive deficits (48). Magnesium supplements are also important in the prevention and management of cardiovascular diseases that predispose to hypertension or congestive heart failure (49,50). Because of the importance of BK channels in neurotransmitter release and vascular tone, Mg2+ modulation of BK channels may play a substantial role in these pathophyisological processes.
BK channels are encoded by Slo1 genes (51–56) and possess common structural features of voltage-gated K+ channels. Each Slo1 protein contains a pore domain formed by S5-S6 transmembrane segments and a voltage-sensing domain formed by S0-S4 transmembrane segments (Fig. 1). In addition, a long cytosolic carboxyl terminus forms the Ca2+ and Mg2+ sensing module (57), which may adopt a similar structure as the cytosolic domain of the K+ channel in E. coli and an archeon Ca2+-activated K+ channel, MthK (58,59). Previous studies have shown that site-directed mutations of mSlo1 E374A, E399N, and Q397C either abolish or reduce Mg2+ sensitivity of channel activation (38,40). In models based on the x-ray crystallographic structures of the K+ channel in E. coli (38) and of MthK, these three residues locate closely (Fig. 1) and can form a metal-binding site (38). However, the roles of these residues in Mg2+ sensitivity, such as whether they are the ligands for the Mg2+ binding site, and how the mutations E374A, E399N, or Q397C affected Mg2+ sensitivity are not clear. On the other hand, the putative Mg2+ binding site formed by these residues is located in the cytosolic domain, whereas the activation gate is located in the membrane-spanning domain (Fig. 1). The structural separation between the binding site and the activation gate indicates that Mg2+ binding activates the channel by an allosteric mechanism; i.e., Mg2+ binding may cause a conformational change at the binding site that propagates to the activation gate for channel opening. Such conformational changes have not been explored.
FIGURE 1.

Schematic diagram of the α subunit of BK channels (left) and the homology model of the Mg2+-binding site of mSlo1 channels based on the crystal structure of the MthK channel (58).
In this study we mutated each of the three residues, E374, E399, and Q397, into amino acids of different chemical properties and examined the activation of mutant channels. We found that mutation of E374 or E399 to any amino acid except Asp abolishes Mg2+ sensitivity, a result that is consistent with these two residues being the ligands for the Mg2+ binding site. However, none of the mutations of Q397 abolished Mg2+ sensitivity. Rather, Mg2+ sensitivity decreased when Q397 is mutated to neutral or positively charged residues, whereas it increased when Q397 is mutated to negatively charged residues. These results suggest that the side chain of Q397 may not be the ligand for the binding site but is close to the binding site so that the Mg2+ sensitivity of channel activation can be tuned by mutating Q397 into different amino acids. We also studied the chemical modification of Q397C mutant channels. The results suggest that the mutations alter Mg2+ sensitivity by affecting the conformation of the Mg2+ binding site as well as by electrostatic interactions.
MATERIALS AND METHODS
Clones, mutagenesis, and channel expression
All channel constructs were made from the mbr5 clone of mSlo1 (60) using PCR with Pfu polymerase (Stratagene, La Jolla, CA). The PCR-amplified regions of all mutants were verified by sequencing. RNA was transcribed in vitro with T3 polymerase (Ambion, Austin, TX). We injected 0.05–50 ng of RNA into each Xenopus laevis oocyte 2–6 days before recording.
Electrophysiology
Macroscopic currents were recorded from inside-out patches formed with borosilicate pipettes of ∼1–2 MΩ resistance. Data were acquired using an Axopatch 200-B patch-clamp amplifier (Axon Instruments, Sunnyvale, CA) and pulse acquisition software (HEKA Electronik, Southboro, MA). Records were digitized at 20-μs intervals and low-pass filtered at 10 kHz with the 4-pole Bessel filter built into the amplifier. The pipette solution contained the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl, and 2 MgCl2, pH 7.20. The basal internal solution contains the following (in mM): 140 potassium methanesulfonic acid, 20 HEPES, 2 KCl, and 1 EGTA, pH 7.20. MgCl2 was added to the internal solution to give the appropriate free [Mg2+]i, and 50 μM 18-crown-6-tetracarboxylic acid (18-C-6-T, Sigma-Aldrich, St. Louis, MO) was added to internal solutions to prevent Ba2+ block. Experiments were conducted at room temperature (∼22−24°C).
Structural model
The homology structural model of the mSlo1 channel (βB to βC) based on the crystal structure of the MthK channel (58) was produced using the PyMol molecular graphics system (http://www.pymol.org) (61). E374, Q397, and E399 of the mSlo1 channel correspond to E138, G156, and N158 of the MthK channel. The SWISS-PDB Viewer (62) was used to generate the corresponding mutant structure by substituting Gln for G156 and Glu for N158 and selecting the lowest-energy rotamer.
Analysis and model fitting
Relative conductance was determined by measuring tail current amplitudes at −50 mV for the WT and mutant mSlo1 channels. The conductance-voltage (G-V) relations of the WT and mutant channels were fitted with a Boltzmann equation:
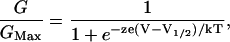 |
(1) |
where z is the number of equivalent charges, V½ is the voltage for a channel in half activation, e is the elementary charge, k is Boltzmann's constant, and T is the absolute temperature. For model fittings of E399D, Q397K, Q397E, and Q397C, the G-V relations of the mutant channels at various [Mg2+]i and 0 [Ca2+]i were fitted with Eq. 2 in Results. Curve fittings were done with Igor Pro software (WaveMetrics, Lake Oswego, OR) using the Levenberg-Marquardt algorithm to perform nonlinear least-squares fits. The means of the data were obtained by averaging from 4 to 16 patches, and error bars represent standard error of means.
Chemical modification
MTSET ([2-(trimethylammonium)ethyl] methanethiosulfonate bromide) was purchased from Toronto Research Chemicals (North York, ON, Canada), dissolved in water at 100 mM, and stored at −80°C. An aliquot of MTSET stock solution was thawed and diluted 500-fold into the appropriate internal solution immediately before use. Currents were recorded after 2.5 min of MTSET treatment and 0.5 min of washing.
RESULTS
We first examined the effects of E374 mutations on the activation of mSlo1 channels. Fig. 2 shows the comparison of the WT mSlo1 activation with that of mutant channels E374R, E374K, E374D, E374A, E374Q, and E374W. It is obvious that the Mg2+ sensitivity of mutant E374A is significantly reduced (Fig. 2 A). Addition of 10 mM Mg2+ resulted in a shift of the G-V relation −67.39 ± 1.86 mV for the WT mSlo1 channel but only −12.51 ± 2.60 mV for E374A (Fig. 2 A). Our previous studies demonstrated that the channel is activated by two independent Mg2+ binding sites, one with high affinity (Kd = 5.46 mM when channel is closed and 2.25 mM when channel is open) and the other with low affinity (Kd = 136.1 mM when channel is closed and 40.1 mM when channel is open). Mutations of E374, E399, or Q397 affect only the high-affinity binding site but have no effects on the low-affinity site (63). We use a model for channel activation (63) to calculate the amount of the G-V shift related to Mg2+ binding to the low-affinity site at 10 mM (Eq. 2 and Table 1). This value is then subtracted from the G-V shift measured experimentally (gray bar in Fig. 2 C), which gives rise to the remaining Mg2+ sensitivity mediated by the high-affinity Mg2+ binding site (black bar in Fig. 2 C). The result shows that mutation E374A completely abolishes Mg2+ sensitivity through the high-affinity Mg2+ binding site (Fig. 2 C). Fig. 2, B and C, shows that another mutation, E374D, does not entirely abolish Mg2+ binding to the site but significantly reduces Mg2+ sensitivity. A 10 mM increase in [Mg2+]i caused a G-V shift of −15.95 ± 3.05 mV for E374D even after subtraction of the effect of the low-affinity Mg2+ site (Fig. 2 C). Among all mutations on E374 that we have studied, only E374D retains Mg2+ sensitivity. Other E374 mutations abolish Mg2+ sensitivity, similar to E374A (Fig. 2 C). Multiple comparison with Tukey's test indicates that the result of E374D in Fig. 2 C is significantly different from the result of the other mutants (> 95% confidence level). These results demonstrate that a carboxylate, or negative charge, in the residue at position 374 is essential for Mg2+ sensitivity. This result is consistent with E374 being part of the high-affinity Mg2+ binding site. Both Asp and Glu residues at position 374 are able to provide coordination for Mg2+ binding, whereas other residues, regardless of their size or hydrophobicity, destroy Mg2+ binding.
FIGURE 2.

Effects of E374 mutations on Mg2+ sensitivity. (A and B) Mg2+-dependent activation of E374A (A) and E374D (B) channels at 0 [Ca2+]i. (Left panels) Current traces recorded at 0 (upper) or 10 mM (lower) [Mg2+]i. Testing potentials were from −30 to 250 mV with 20-mV increments. The holding and repolarizing potentials were −80 and −50 mV, respectively. (Right panels) Mean G-V relations. Gray lines are G-V relations of WT mSlo1 channels for comparison. G-V relations of mutant channels are fitted with the Boltzmann relation (solid lines; see Materials and Methods). (C) G-V shift from 0 to 10 mM [Mg2+]i at 0 [Ca2+]i of WT mSlo1 and mutant channels. V½ is the voltage where the G-V relation is half-maximum. Gray bars are the data measured in experiments, and black bars are the experimental data subtracted by the amount of G-V shift (−12.2 mV) caused by Mg2+ binding to the low-affinity Mg2+ site and calculated from model simulation (see Materials and Methods).
TABLE 1.
Parameters from MWC model fitting to activation data of the WT and mutant mSlo1 channels
| WT | E399D | Q397K | Q397E | Q397C | |
|---|---|---|---|---|---|
| L0 | 9334 | 6857 ± 814 | 1492 ± 187 | 63,525 ± 1010 | 16,525 ± 1410 |
| z | 1.25 | 1.25 | 1.25 | 1.25 | 1.25 |
| KcM1 (mM) | 5.46 | 2.50 ± 0.78 | 6.90 ± 1.43 | 3.01 ± 0.20 | 4.47 ± 0.28 |
| KoM1 (mM) | 2.25 | 2.10 ± 0.65 | 4.99 ± 0.98 | 1.20 ± 0.07 | 2.50 ± 0.14 |
| KcM2 (mM) | 136.1 | 136.1 | 136.1 | 136.1 | 136.1 |
| KoM2 (mM) | 40.1 | 40.1 | 40.1 | 40.1 | 40.1 |
Parameters for the WT channels were taken from Hu et al. (63); z, KcM2, and KoM2 of the mutant channels are fixed to be the same as those of the WT channels. Bold values indicate the parameters that are different between the WT and mutant channels. ± indicates parameter mean ± SE.
We also examined the effects of E399 mutations on the activation of mSlo1 channels. Fig. 3 shows the comparison of the WT mSlo1 activation with that of mutant channels E399R, E399D, E399N, E399Q, and E399W. Similar to E374, mutation of E399 to any amino acid other than Asp destroys Mg2+ sensitivity, indicating that the carboxylate on this residue is also essential for Mg2+ sensitivity. Multiple comparison with Tukey's test indicates that the result of E399D in Fig. 3 C is significantly different from the result of the other mutants (>95% confidence level).
FIGURE 3.

Effects of E399 mutations on Mg2+ sensitivity. (A and B) Mg2+-dependent activation of E399N (A) and E399D (B) channels at 0 [Ca2+]i. (Left panels) Current traces recorded at 0 (upper) or 10 mM (lower) [Mg2+]i. Voltage protocols are similar as in Fig. 2, A and B. (Right panels) Mean G-V relations and fits with the Boltzmann relation (solid lines). Gray lines are G-V relations of WT mSlo1 channels for comparison. (C) G-V shift from 0 to 10 mM [Mg2+]i at 0 [Ca2+]i of WT mSlo1 and mutant channels. Gray and black bars have similar meanings as in Fig. 2 C.
Although Mg2+ sensitivity is retained in both E374D and E399D mutant channels, it is significantly reduced (Fig. 2, B and C, Fig. 3 B and C). In order to examine the Mg2+ sensitivity reduction in more detail, we measured G-V relations of E399D in various [Mg2+]i between 0 and 100 mM at 0 [Ca2+]i and fitted the data with Eq. 2 (Fig. 4 A):
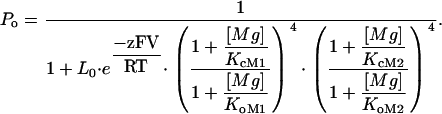 |
(2) |
FIGURE 4.

Effects of mutation E399D on Mg2+ binding. (A) Mean G-V relations of E399D mutant channels at 0 [Ca2+]i and indicated [Mg2+]i. Solid lines are model fittings according to Eq. 2. The parameters obtained from the fitting are listed in Table 1. (B) G-V shifts from 10 to 100 mM [Mg2+]i at 0 [Ca2+]i of WT mSlo1 and mutant channels. For WT mSlo1 channels, the gray bar is data measured in experiments, and the black bar is the experimental data subtracted by the amount of G-V shift (−16.2 mV) caused by Mg2+ binding to the high-affinity Mg2+ site.
Equation 2 is derived from a model of mSlo1 activation by Mg2+ binding to the two different binding sites (63), where Po is the open probability of the channel; L0 is the equilibrium constant between the closed and open states at the voltage of 0 mV when no Ca2+ or Mg2+ is bound (L0 = [C0]/[O0]); z is the number of equivalent gating charges; KcM1, KcM2, KoM1, and KoM2 are the dissociation constants of Mg2+ binding to the high- and low-affinity Mg2+ sites on each subunit when the channel is at closed and open states, respectively; and V, T, F, and R have their usual meanings. Equation 2 does not include Ca2+-dependent activation because [Ca2+]i in these experiments is 0 (Fig. 4). We have demonstrated that the two Mg2+ binding sites act independently in channel activation, and mutations E399N and E374A:E399N do not affect channel activation through the low-affinity binding site (63). Such independence is further demonstrated in Fig. 4 B, which shows that for several other E399 mutations that also destroy Mg2+ sensitivity related to the high-affinity site, the shift of G-V relations in response to an increase of [Mg2+]i from 10 to 100 mM is similar to that of WT mSlo1. Therefore, it is reasonable to assume that the low-affinity binding site is not affected by E399D either, and the parameters related to this binding site in Eq. 2 are the same as in the WT mSlo1 (Table 1). Comparing the parameters of model fitting to the WT and E399D data (Table 1), we find that Mg2+ affinity of the high-affinity binding site is altered by the mutation in both open and closed conformation (KoM1 and KcM1). The model used in the fitting is derived from the MWC model (39,64), in which Mg2+ binds to the open channel with higher affinity and shifts the closed-open equilibrium toward the open conformation by factor c (c = KoM1/KcM1) (64). Table 1 shows that in the open conformation, the affinity of Mg2+ binding may be unaffected by the mutation E399D (KoM1 similar), whereas in the closed conformation, the affinity may even be increased (KcM1 smaller). Thus, the mutant channel has a larger c factor than the WT mSlo1, signifying that activation of the E399D channel is less sensitive to the effects of Mg2+ binding (Figs. 3 C and 4 A).
We then examined the effects of Q397 mutations on the activation of mSlo1 channels. Fig. 5 A shows the comparison of Mg2+-dependent activation of the WT mSlo1 channels with that of mutant channels Q397R, Q397K, Q397D, Q397E, Q397C, and Q397W. Unlike mutations of E374 and E399, most of which abolish Mg2+ sensitivity, none of the mutations of Q397 destroys Mg2+ sensitivity entirely. Most surprisingly, even when the residue is mutated to positively charged side chains (Q397R and Q397K), the channel still retains Mg2+ sensitivity (Fig. 5 A). These results are not consistent with the idea that the side chain of Q397 is the ligand for the Mg2+ binding site. Nevertheless, the results in Fig. 5 A suggest that because the side chain of Q397 is spatially close to E374 and E399, the mutations of Q397 may affect Mg2+ binding if E374 and E399 are part of the Mg2+ binding site, and thus the Mg2+ sensitivity of channel activation. Mutations Q397R, Q397K, Q397C, and Q397W reduce Mg2+ sensitivity of the channel significantly (Fig. 5 A). On the other hand, when Q397 is mutated to negatively charged side chains (Q397E and Q397D), Mg2+ sensitivity is increased (Fig. 5 A). We also measured the G-V relations of Q397C, Q397K, and Q397E mutant channels at various [Mg2+]i levels between 0 and 100 mM at 0 [Ca2+]i, and fitted the data with Eq. 2 (Fig. 5, B–D). The parameters obtained from model fittings are listed in Table 1. Compared to the WT mSlo1 channels, these mutations altered Mg2+ affinity in both the closed and the open conformations of the channel (KcM1 and KoM1), resulting in a smaller c factor (c = KoM1/KcM1) and an increased Mg2+ sensitivity in the case of Q397E and in a larger c factor and a reduced Mg2+ sensitivity in the case of Q397K and Q397C (Fig. 5 and Table 1).
FIGURE 5.

Effects of Q397 mutations on Mg2+ sensitivity of channel activation and on Mg2+ binding. (A) G-V shifts from 0 to 10 mM [Mg2+]i at 0 [Ca2+]i of WT mSlo1 and mutant channels. Gray and black bars have similar meanings as in Fig. 2 C. (B–D) Mean G-V relations of mutant channels Q397K (B), Q397E (C), and Q397C (D) at 0 [Ca2+]i and indicated [Mg2+]i. Solid lines are model fittings according to Eq. 2. The parameters obtained from the fitting are listed in Table 1.
The increase of Mg2+ sensitivity by mutations Q397D and Q397E suggests that a negative charge at position 397 may affect Mg2+ binding by an electrostatic attraction to the bound Mg2+ ion. If this is the case, a positive charge at this position is expected to reduce Mg2+ sensitivity by electrical repulsion. Our experiment results show that mutations Q397R and Q397K indeed reduced Mg2+ sensitivity (Fig. 5, A and B). However, both Q397C and Q397W also reduced Mg2+ sensitivity (Fig. 5 A), suggesting that a conformational change caused by Q397 mutations may also be important to Mg2+ binding. Therefore, it is not clear whether the effect of Q397R and Q397K was caused by an electrostatic interaction or a conformational change. To address this question, we treated Q397C with the Cys-modifying reagent MTSET(+). The covalent addition of a positively charged MTSET(+) to Q397C had no apparent effect on the kinetics of channel gating (Fig. 6 A), but it reduced Mg2+ sensitivity as compared to Q397C before MTSET(+) treatment (Fig. 6 B). These experiments were performed on the double mutation Q397C:C430A to eliminate the effect of MTSET(+) modification of the native C430 (65). C430A alone does not alter Mg2+ sensitivity of the channel, and MTSET(+) treatment of C430A has no effect on Mg2+ sensitivity either (Fig. 6 B), indicating that the effects of MTSET(+) on Q397C:C430A are specifically caused by the modification of Q397C. Fig. 6 B shows that Q397C reduces Mg2+ sensitivity in the background of C430A (10 mM Mg2+ resulted in a shift of the G-V relation −47.76 ± 1.59 mV for Q397C:C430A channels as compared to −70.28 ± 1.68 mV for C430A), which is similar to its effect on the WT mSlo1 (Fig. 5 A). The modification of Q397C by MTSET(+) further reduces Mg2+ sensitivity, supporting the mechanism that electric charges at position 397 may affect Mg2+ sensitivity by an electrostatic interaction with the bound Mg2+ ion.
FIGURE 6.

MTSET(+) modification of Q397C reduces Mg2+ sensitivity of channel activation. (A) Current traces of Q397C:C430A mutant channels recorded at 0 [Mg2+]i and 0 [Ca2+]i before (left) and after (right) MTSET(+) treatment. (B) G-V shifts of C430A and Q397C:C430A mutant channels from 0 to 10 mM [Mg2+]i at 0 [Ca2+]i before and after MTSET(+) treatment. The G-V shifts are subtracted by the amount of G-V shift (−12.2 mV) caused by Mg2+ binding to the low-affinity Mg2+ site.
DISCUSSION
We examined the mechanism of Mg2+-dependent activation of mSlo1 BK channels by studying the effects of mutating E374, E399, and Q397. We found that a carboxylate, or a negative charge, on the side chain of residues 374 and 399 is essential for Mg2+ sensitivity (Figs. 2 and 3). On the other hand, no mutations of Q397 abolished Mg2+ sensitivity, suggesting that the side chain of Q397 may not coordinate to Mg2+ (Fig. 5). However, Q397 is spatially close to E374 and E399 (Fig. 1), so the effects of Q397 mutations may derive from their interference with Mg2+ binding if E374 and E399 indeed form the binding site (Fig. 1). Effects of Q397 mutations and chemical modification of Q397C suggest that the mutations affect Mg2+ binding by altering the conformation of the binding site as well as by electrostatic interactions with the bound Mg2+ ion (Figs. 5 and 6; Table 1).
Based on the homology between Slo1 channels and the K+ channels in E. coli or MthK, E374, E399, and Q397 are located on the surface of a Rossman fold (38,58,59). The relation of these residues is comparable to the organization of the Mg2+ binding site in the response regulator of bacterial chemotaxis, CheY, which is also located on the surface of a Rossman fold, and the bound Mg2+ ion is coordinated by the side-chain carboxylates of two Asp residues (D13 and D57), the backbone carbonyl of Asn-59, and three water molecules (66–68). In mSlo1, mutations of E374 and E399 even to amino acids with an oxygen-containing side chain abolish Mg2+ sensitivity as long as the substituting residue is not an Asp (Figs. 2 and 3). Similarly, mutations D13N and D57N of the CheY's Mg2+ binding site reduce Mg2+ affinity by more than an order of magnitude (69). In addition, the Mg2+ binding sites in mSlo1 or CheY do not discriminate between metals on the basis of their size. For example, the dissociation constants of Mg2+ and Ca2+ binding to mSlo1 at the open state are 2.2–6.0 mM and 0.66 mM, respectively (37,39,63), whereas the dissociation constants of Mg2+ and Ca2+ binding to CheY are 1.0 mM and 0.4 mM, respectively (70). Such a lack of size specificity is in striking contrast to the dissociation constants of Mg2+ and Ca2+ binding to the high-affinity Ca2+ binding site of mSlo1 at the open state, which are 4.73–5.6 mM and 0.75–1.3 μM, respectively (37,39,63,64,71). These properties of the Mg2+ binding site in CheY derive from its coordination scheme in which one hemisphere of the bound ion is coordinated by three protein oxygens, and the other hemisphere is coordinated by three solvent molecules at the protein surface (70). On one hand, the flexible solvent shell can easily vary its coordination number and shape to accommodate bound ions of different sizes; on the other hand, the side-chain carboxylates are required to provide strong coordination (72). Because the Mg2+ binding site in mSlo1 shares the same properties as that in CheY, it may also share a similar coordination scheme, i.e., the two side-chain carboxylates from E374 and E399 and the main-chain carbonyl at Q397 provide oxygens for Mg2+ coordination (38), whereas the side chain of Q397 may not be part of the ligands for the Mg2+ binding site in mSlo1 (Fig. 5). According to this coordination scheme, although mutations E374D and E399D may cause conformational changes of the binding site, the channel retains Mg2+ sensitivity because the flexible solvent shell may vary to accommodate such conformational changes to allow Mg2+ binding.
Mg2+ activates the BK channel by an allosteric mechanism (Fig. 1) (37–40,63); i.e., Mg2+ binding induces a conformational change of the channel protein that eventually opens the activation gate distant from the Mg2+ binding site. In CheY, Mg2+ binding induces a conformational change near the binding site (67,68,73). Conformational changes near the binding site are also observed in many other proteins as a result of metal binding (74–77). In mSlo1, Q397 is close to E374 and E399 (Fig. 1). Because all mutations of Q397 in our study affect Mg2+ sensitivity, this residue is likely involved in the Mg2+-dependent activation mechanism such that the Mg2+ binding induces conformational changes at and around Q397, and reciprocally, Mg2+ binding is also altered by the mutations of Q397.
However, it is hard for a simple conformational change to explain the results that Q397D and Q397E increase Mg2+ sensitivity whereas other mutations reduce Mg2+ sensitivity (Fig. 5 A). When the chemical properties of the substituting side chains of Q397 mutations are compared with their effects on Mg2+ sensitivity, it is clear that the side-chain size (Van der Waals volume: W > R > E > K ≥ Q > D > C) or hydrophobicity (C > W > Q > K > E > D > R) (78) may not decide the effects of mutations on Mg2+ sensitivity (E ≈ D > Q > C > W ≈ R ≈ K) (Fig. 5 A). On the other hand, it is apparent that both Q397E and Q397D contain a negative charge, and both increase Mg2+ sensitivity. When Q397C is modified by MTSET(+), the covalently attached positive charge reduces Mg2+ sensitivity, similar to the effects of mutations Q397R and Q397K (Figs. 5 and 6). Thus, electrostatic interaction plays an important role in the effect of Q397 mutations. The charge introduced to the residue at 397 by mutations affects Mg2+ binding by either electrical repulsion or attraction to the bound Mg2+ ion. Consistent with this mechanism, fittings of the MWC model to G-V relations at various [Mg2+]i suggest that Q397K reduces Mg2+ affinity in both the closed and the open conformations of the channel (KcM1 and KoM1 become larger), whereas Q397E increases Mg2+ affinity (KcM1 and KoM1 become smaller) (Table 1). Such an electrostatic contribution to metal binding by charged surface residues has been reported previously and has been suggested as a common mechanism in metal-binding proteins (79–82). The electrostatic interaction between Q397 mutations and the Mg2+ binding site suggests that Q397 is located close to the Mg2+ binding site, further supporting that E374 and E399 are the ligands for the Mg2+ binding site.
In conclusion, this study further demonstrates that E374 and E399 side chains are part of the Mg2+ binding site. Mg2+ sensitivity of channel activation can be tuned by altering Mg2+ binding through changing the conformation of the binding site per se (E374D and E399D) or the environment of the binding site (Q397 mutations). However, the effect of Q397 mutations on Mg2+-dependent activation requires a functional Mg2+ binding site because a double mutation, E374A:Q397E, had no Mg2+ sensitivity (data not shown). This result also demonstrates that although Q397 is close to the Mg2+ binding site, within the range of electrostatic interactions, it cannot replace E374 in coordination of Mg2+ binding.
Acknowledgments
An abstract of this work was presented at the 49th Annual Meeting of the Biophysical Society.
The mSlo1 clone was kindly provided to us by Larry Salkoff. We thank Kelli McFarland for technical assistance.
This work was supported by National Institutes of Health grant R01-HL70393, the American Heart Association, and the Whitaker Foundation (J.C.). J.C. is Associate Professor of Biomedical Engineering on the Spencer T. Olin Endowment.
Huanghe Yang and Lei Hu contributed equally to this work.
References
- 1.Lewis, R. S., and A. J. Hudspeth. 1983. Voltage- and ion-dependent conductances in solitary vertebrate hair cells. Nature. 304:538–541. [DOI] [PubMed] [Google Scholar]
- 2.Magleby, K. L., and B. S. Pallotta. 1983. Calcium dependence of open and shut interval distributions from calcium-activated potassium channels in cultured rat muscle. J. Physiol. 344:585–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Magleby, K. L., and B. S. Pallotta. 1983. Burst kinetics of single calcium-activated potassium channels in cultured rat muscle. J. Physiol. 344:605–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Petersen, O. H., and Y. Maruyama. 1984. Calcium-activated potassium channels and their role in secretion. Nature. 307:693–696. [DOI] [PubMed] [Google Scholar]
- 5.Storm, J. F. 1987. Action potential repolarization and a fast after-hyperpolarization in rat hippocampal pyramidal cells. J. Physiol. (Lond.). 385:733–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lancaster, B., R. A. Nicoll, and D. J. Perkel. 1991. Calcium activates two types of potassium channels in rat hippocampal neurons in culture. J. Neurosci. 11:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.McManus, O. B. 1991. Calcium-activated potassium channels: regulation by calcium. J. Bioenerg. Biomembr. 23:537–560. [DOI] [PubMed] [Google Scholar]
- 8.Brayden, J. E., and M. T. Nelson. 1992. Regulation of arterial tone by activation of calcium-dependent potassium channels. Science. 256:532–535. [DOI] [PubMed] [Google Scholar]
- 9.Bielefeldt, K., and M. B. Jackson. 1993. A calcium-activated potassium channel causes frequency-dependent action-potential failures in a mammalian nerve terminal. J. Neurophysiol. 70:284–298. [DOI] [PubMed] [Google Scholar]
- 10.Crest, M., and M. Gola. 1993. Large conductance Ca2+-activated K+ channels are involved in both spike shaping and firing regulation in Helix neurones. J. Physiol. 465:265–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Robitaille, R., M. L. Garcia, G. J. Kaczorowski, and M. P. Charlton. 1993. Functional colocalization of calcium and calcium-gated potassium channels in control of transmitter release. Neuron. 11:645–655. [DOI] [PubMed] [Google Scholar]
- 12.Yazejian, B., D. A. DiGregorio, J. L. Vergara, R. E. Poage, S. D. Meriney, and A. D. Grinnell. 1997. Direct measurements of presynaptic calcium and calcium-activated potassium currents regulating neurotransmitter release at cultured Xenopus nerve-muscle synapses. J. Neurosci. 17:2990–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Marrion, N. V., and S. J. Tavalin. 1998. Selective activation of Ca2+-activated K+ channels by co-localized Ca2+ channels in hippocampal neurons. Nature. 395:900–905. [DOI] [PubMed] [Google Scholar]
- 14.Safronov, B. V., and W. Vogel. 1998. Large conductance Ca2+-activated K+ channels in the soma of rat motoneurones. J. Membr. Biol. 162:9–15. [DOI] [PubMed] [Google Scholar]
- 15.Nelson, M. T., and J. M. Quayle. 1995. Physiological roles and properties of potassium channels in arterial smooth muscle. Am. J. Physiol. 268:C799–C822. [DOI] [PubMed] [Google Scholar]
- 16.Adams, P. R., A. Constanti, D. A. Brown, and R. B. Clark. 1982. Intracellular Ca2+ activates a fast voltage-sensitive K+ current in vertebrate sympathetic neurones. Nature. 296:746–749. [DOI] [PubMed] [Google Scholar]
- 17.Lancaster, B., and R. A. Nicoll. 1987. Properties of two calcium-activated hyperpolarizations in rat hippocampal neurones. J. Physiol. (Lond.). 389:187–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Roberts, W. M., R. A. Jacobs, and A. J. Hudspeth. 1990. Colocalization of ion channels involved in frequency selectivity and synaptic transmission at presynaptic active zones of hair cells. J. Neurosci. 10:3664–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robitaille, R., and M. P. Charlton. 1992. Presynaptic calcium signals and transmitter release are modulated by calcium-activated potassium channels. J. Neurosci. 12:297–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wellman, G. C., and M. T. Nelson. 2003. Signaling between SR and plasmalemma in smooth muscle: sparks and the activation of Ca2+-sensitive ion channels. Cell Calcium. 34:211–229. [DOI] [PubMed] [Google Scholar]
- 21.Tanaka, Y., M. Aida, H. Tanaka, K. Shigenobu, and L. Toro. 1998. Involvement of maxi-KCa channel activation in atrial natriuretic peptide-induced vasorelaxation. Naunyn Schmiedebergs Arch. Pharmacol. 357:705–708. [DOI] [PubMed] [Google Scholar]
- 22.Perez, G. J., A. D. Bonev, J. B. Patlak, and M. T. Nelson. 1999. Functional coupling of ryanodine receptors to KCa channels in smooth muscle cells from rat cerebral arteries. J. Gen. Physiol. 113:229–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pluger, S., J. Faulhaber, M. Furstenau, M. Lohn, R. Waldschutz, M. Gollasch, H. Haller, F. C. Luft, H. Ehmke, and O. Pongs. 2000. Mice with disrupted BK channel beta1 subunit gene feature abnormal Ca2+ spark/STOC coupling and elevated blood pressure. Circ. Res. 87:E53–E60. [DOI] [PubMed] [Google Scholar]
- 24.Wu, Y. C., J. J. Art, M. B. Goodman, and R. Fettiplace. 1995. A kinetic description of the calcium-activated potassium channel and its application to electrical tuning of hair cells. Prog. Biophys. Mol. Biol. 63:131–158. [DOI] [PubMed] [Google Scholar]
- 25.Rosenblatt, K. P., Z. P. Sun, S. Heller, and A. J. Hudspeth. 1997. Distribution of Ca2+-activated K+ channel isoforms along the tonotopic gradient of the chicken's cochlea. Neuron. 19:1061–1075. [DOI] [PubMed] [Google Scholar]
- 26.Hudspeth, A. J., and R. S. Lewis. 1988. Kinetic analysis of voltage- and ion-dependent conductances in saccular hair cells of the bull-frog, Rana catesbeiana. J. Physiol. (Lond.). 400:237–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fettiplace, R., and P. A. Fuchs. 1999. Mechanisms of hair cell tuning. Annu. Rev. Physiol. 61:809–834. [DOI] [PubMed] [Google Scholar]
- 28.Ahluwalia, J., A. Tinker, L. H. Clapp, M. R. Duchen, A. Y. Abramov, S. Pope, M. Nobles, and A. W. Segal. 2004. The large-conductance Ca2+-activated K+ channel is essential for innate immunity. Nature. 427:853–858. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Squire, L. G., and O. H. Petersen. 1987. Modulation of Ca2+- and voltage-activated K+ channels by internal Mg2+ in salivary acinar cells. Biochim. Biophys. Acta. 899:171–175. [DOI] [PubMed] [Google Scholar]
- 30.Zamoyski, V. L., V. N. Serebryakov, and R. Schubert. 1989. Activation and blocking effects of divalent cations on the calcium-dependent potassium channel of high conductance. Biomed. Biochim. Acta. 48:S388–S392. [PubMed] [Google Scholar]
- 31.Ferguson, W. B. 1991. Competitive Mg2+ block of a large-conductance, Ca2+-activated K+ channel in rat skeletal muscle. Ca2+, Sr2+, and Ni2+ also block. J. Gen. Physiol. 98:163–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McLarnon, J. G., and D. Sawyer. 1993. Effects of divalent cations on the activation of a calcium-dependent potassium channel in hippocampal neurons. Pflugers Arch. 424:1–8. [DOI] [PubMed] [Google Scholar]
- 33.Zhang, X., E. Puil, and D. A. Mathers. 1995. Effects of intracellular Mg2+ on the properties of large-conductance, Ca2+-dependent K+ channels in rat cerebrovascular smooth muscle cells. J. Cereb. Blood Flow Metab. 15:1066–1074. [DOI] [PubMed] [Google Scholar]
- 34.Morales, E., W. C. Cole, C. V. Remillard, and N. Leblane. 1996. Block of large conductance Ca2+-activated K+ channels in rabbit vascular myocytes by internal Mg2+ and Na+. J. Physiol. 495:701–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wachter, C., and K. Turnheim. 1996. Inhibition of high-conductance, calcium-activated potassium channels of rabbit colon epithelium by magnesium. J. Membr. Biol. 150:275–282. [DOI] [PubMed] [Google Scholar]
- 36.Bringmann, A., F. Faude, and A. Reichenbach. 1997. Mammalian retinal glial (Muller) cells express large-conductance Ca2+-activated K+ channels that are modulated by Mg2+ and pH and activated by protein kinase A. Glia. 19:311–323. [DOI] [PubMed] [Google Scholar]
- 37.Zhang, X., C. R. Solaro, and C. J. Lingle. 2001. Allosteric regulation of BK channel gating by Ca2+ and Mg2+ through a nonselective, low affinity divalent cation site. J. Gen. Physiol. 118:607–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shi, J., G. Krishnamoorthy, Y. Yang, L. Hu, N. Chaturvedi, D. Harilal, J. Qin, and J. Cui. 2002. Mechanism of magnesium activation of calcium-activated potassium channels. Nature. 418:876–880. [DOI] [PubMed] [Google Scholar]
- 39.Shi, J., and J. Cui. 2001. Intracellular Mg2+ enhances the function of BK-type Ca2+ activated K+ channels. J. Gen. Physiol. 118:589–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Xia, X. M., X. Zeng, and C. J. Lingle. 2002. Multiple regulatory sites in large-conductance calcium-activated potassium channels. Nature. 418:880–884. [DOI] [PubMed] [Google Scholar]
- 41.Altura, B. M., B. T. Altura, A. Carella, A. Gebrewold, T. Murakawa, and A. Nishio. 1987. Mg2+-Ca2+ interaction in contractility of vascular smooth muscle: Mg2+ versus organic calcium channel blockers on myogenic tone and agonist- induced responsiveness of blood vessels. Can. J. Physiol. Pharmacol. 65:729–745. [DOI] [PubMed] [Google Scholar]
- 42.Altura, B. M., and R. K. Gupta. 1992. Cocaine induces intracellular free Mg deficits, ischemia and stroke as observed by in-vivo 31P-NMR of the brain. Biochim. Biophys. Acta. 1111:271–274. [DOI] [PubMed] [Google Scholar]
- 43.Matsuda, H., A. Saigusa, and H. Irisawa. 1987. Ohmic conductance through the inwardly rectifying K channel and blocking by internal Mg2+. Nature. 325:156–159. [DOI] [PubMed] [Google Scholar]
- 44.Vandenberg, C. A. 1987. Inward rectification of a potassium channel in cardiac ventricular cells depends on internal magnesium ions. Proc. Natl. Acad. Sci. USA. 84:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.White, R. E., and H. C. Hartzell. 1988. Effects of intracellular free magnesium on calcium current in isolated cardiac myocytes. Science. 239:778–780. [DOI] [PubMed] [Google Scholar]
- 46.Chuang, H., Y. N. Jan, and L. Y. Jan. 1997. Regulation of IRK3 inward rectifier K+ channel by m1 acetylcholine receptor and intracellular magnesium. Cell. 89:1121–1132. [DOI] [PubMed] [Google Scholar]
- 47.Romani, A. M., V. D. Matthews, and A. Scarpa. 2000. Parallel stimulation of glucose and Mg2+ accumulation by insulin in rat hearts and cardiac ventricular myocytes. Circ. Res. 86:326–333. [DOI] [PubMed] [Google Scholar]
- 48.Vink, R., and I. Cernak. 2000. Regulation of intracellular free magnesium in central nervous system injury. Front. Biosci. 5:D656–D665. [DOI] [PubMed] [Google Scholar]
- 49.Laurant, P., and R. M. Touyz. 2000. Physiological and pathophysiological role of magnesium in the cardiovascular system: implications in hypertension. J. Hypertens. 18:1177–1191. [DOI] [PubMed] [Google Scholar]
- 50.Seelig, M. S. 2000. Interrelationship of magnesium and congestive heart failure. Wien. Med. Wochenschr. 150:335–341. [PubMed] [Google Scholar]
- 51.Atkinson, N. S., G. A. Robertson, and B. Ganetzky. 1991. A component of calcium-activated potassium channels encoded by the Drosophila slo locus. Science. 253:551–555. [DOI] [PubMed] [Google Scholar]
- 52.Adelman, J. P., K. Z. Shen, M. P. Kavanaugh, R. A. Warren, Y. N. Wu, A. Lagrutta, C. T. Bond, and R. A. North. 1992. Calcium-activated potassium channels expressed from cloned complementary DNAs. Neuron. 9:209–216. [DOI] [PubMed] [Google Scholar]
- 53.Butler, A., S. Tsunoda, D. P. McCobb, A. Wei, and L. Salkoff. 1993. mSlo, a complex mouse gene encoding “maxi” calcium-activated potassium channels. Science. 261:221–224. [DOI] [PubMed] [Google Scholar]
- 54.Dworetzky, S. I., J. T. Trojnacki, and V. K. Gribkoff. 1994. Cloning and expression of a human large-conductance calcium-activated potassium channel. Brain Res. Mol. Brain Res. 27:189–193. [DOI] [PubMed] [Google Scholar]
- 55.Tseng-Crank, J., C. D. Foster, J. D. Krause, R. Mertz, N. Godinot, T. J. DiChiara, and P. H. Reinhart. 1994. Cloning, expression, and distribution of functionally distinct Ca2+-activated K+ channel isoforms from human brain. Neuron. 13:1315–1330. [DOI] [PubMed] [Google Scholar]
- 56.Pallanck, L., and B. Ganetzky. 1994. Cloning and characterization of human and mouse homologs of the Drosophila calcium-activated potassium channel gene, slowpoke. Hum. Mol. Genet. 3:1239–1243. [DOI] [PubMed] [Google Scholar]
- 57.Magleby, K. L. 2003. Gating mechanism of BK (Slo1) channels: so near, yet so far. J. Gen. Physiol. 121:81–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang, Y., A. Lee, J. Chen, M. Cadene, B. T. Chait, and R. MacKinnon. 2002. Crystal structure and mechanism of a calcium-gated potassium channel. Nature. 417:515–522. [DOI] [PubMed] [Google Scholar]
- 59.Jiang, Y., A. Pico, M. Cadene, B. T. Chait, and R. MacKinnon. 2001. Structure of the RCK domain from the E. coli K+ channel and demonstration of its presence in the human BK channel. Neuron. 29:593–601. [DOI] [PubMed] [Google Scholar]
- 60.Cui, J., D. H. Cox, and R. W. Aldrich. 1997. Intrinsic voltage dependence and Ca2+ regulation of mslo large conductance Ca-activated K+ channels. J. Gen. Physiol. 109:647–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Delano, W. L. 2002. The Pymol molecular graphics system. Delano Scientific, San Carlos, CA.. http://www.pymol.org. [Online].
- 62.Guex, N., and M. C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- 63.Hu, L., H. Yang, J. Shi, and J. Cui. 2006. Effects of multiple metal binding sites on calcium and magnesium-dependent activation of BK channels. J. Gen. Physiol. 127:35–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cox, D. H., J. Cui, and R. W. Aldrich. 1997. Allosteric gating of a large conductance Ca-activated K+ channel. J. Gen. Physiol. 110:257–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang, G., and F. T. Horrigan. 2005. Cysteine modification alters voltage- and Ca2+-dependent gating of large conductance (BK) potassium channels. J. Gen. Physiol. 125:213–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stock, J. B., M. G. Surette, W. R. McCleary, and A. M. Stock. 1992. Signal transduction in bacterial chemotaxis. J. Biol. Chem. 267:19753–19756. [PubMed] [Google Scholar]
- 67.Bellsolell, L., J. Prieto, L. Serrano, and M. Coll. 1994. Magnesium binding to the bacterial chemotaxis protein CheY results in large conformational changes involving its functional surface. J. Mol. Biol. 238:489–495. [DOI] [PubMed] [Google Scholar]
- 68.Stock, A. M., E. Martinez-Hackert, B. F. Rasmussen, A. H. West, J. B. Stock, D. Ringe, and G. A. Petsko. 1993. Structure of the Mg2+-bound form of CheY and mechanism of phosphoryl transfer in bacterial chemotaxis. Biochemistry. 32:13375–13380. [DOI] [PubMed] [Google Scholar]
- 69.Lukat, G. S., A. M. Stock, and J. B. Stock. 1990. Divalent metal ion binding to the CheY protein and its significance to phosphotransfer in bacterial chemotaxis. Biochemistry. 29:5436–5442. [DOI] [PubMed] [Google Scholar]
- 70.Needham, J. V., T. Y. Chen, and J. J. Falke. 1993. Novel ion specificity of a carboxylate cluster Mg(II) binding site: strong charge selectivity and weak size selectivity. Biochemistry. 32:3363–3367. [DOI] [PubMed] [Google Scholar]
- 71.Horrigan, F. T., and R. W. Aldrich. 2002. Coupling between voltage sensor activation, Ca2+ binding and channel opening in large conductance (BK) potassium channels. J. Gen. Physiol. 120:267–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dudev, T., and C. Lim. 2003. Principles governing Mg, Ca, and Zn binding and selectivity in proteins. Chem. Rev. 103:773–788. [DOI] [PubMed] [Google Scholar]
- 73.Falke, J. J., R. B. Bass, S. L. Butler, S. A. Chervitz, and M. A. Danielson. 1997. The two-component signaling pathway of bacterial chemotaxis: a molecular view of signal transduction by receptors, kinases, and adaptation enzymes. Annu. Rev. Cell Dev. Biol. 13:457–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Babu, C. S., T. Dudev, R. Casareno, J. A. Cowan, and C. Lim. 2003. A combined experimental and theoretical study of divalent metal ion selectivity and function in proteins: application to E. coli ribonuclease H1. J. Am. Chem. Soc. 125:9318–9328. [DOI] [PubMed] [Google Scholar]
- 75.Lewit-Bentley, A., and S. Rety. 2000. EF-hand calcium-binding proteins. Curr. Opin. Struct. Biol. 10:637–643. [DOI] [PubMed] [Google Scholar]
- 76.Ikura, M. 1996. Calcium binding and conformational response in EF-hand proteins. Trends Biochem. Sci. 21:14–17. [PubMed] [Google Scholar]
- 77.Nelson, M. R., and W. J. Chazin. 1998. An interaction-based analysis of calcium-induced conformational changes in Ca2+ sensor proteins. Protein Sci. 7:270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Creighton, T. E. 1992. Proteins: Structures and Molecular Principles. New York: W.H. Freeman.
- 79.Linse, S., P. Brodin, C. Johansson, E. Thulin, T. Grundstrom, and S. Forsen. 1988. The role of protein surface charges in ion binding. Nature. 335:651–652. [DOI] [PubMed] [Google Scholar]
- 80.Linse, S., C. Johansson, P. Brodin, T. Grundstrom, T. Drakenberg, and S. Forsen. 1991. Electrostatic contributions to the binding of Ca2+ in calbindin D9k. Biochemistry. 30:154–162. [DOI] [PubMed] [Google Scholar]
- 81.Sheng, J.-Z., A. Weljie, L. Sy, S. Ling, H. J. Vogel, and A. P. Braun. 2005. Homology modeling identifies C-terminal residues that contribute to the Ca2+ sensitivity of a BKCa channel. Biophys. J. 89:3079–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bao, L., C. Kaldany, E. C. Holmstrand, and D. H. Cox. 2004. Mapping the BKCa channel's “Ca2+ bowl”: side-chains essential for Ca2+ sensing. J. Gen. Physiol. 123:475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]