

Abstract
The ability of calcium-bound calmodulin (CaM) to recognize most of its target peptides is caused by its binding to two hydrophobic residues (‘anchors’). In most of the CaM complexes, the anchors pack against the hydrophobic pockets of the CaM domains and are surrounded by fully conserved Met side chains. Here, by using metadynamics simulations, we investigate quantitatively the energetics of the final step of this process using the M13 peptide, which has a high affinity and spans the sequence of the skeletal myosin light chain kinase, an important natural CaM target. We established the accuracy of our calculations by a comparison between calculated and NMR-derived structural and dynamical properties. Our calculations provide novel insights into the mechanism of protein/peptide recognition: we show that the process is associated with a free energy gain similar to that experimentally measured for the CaM complex with the homologous smooth muscle MLCK peptide (Ehrhardt et al., 1995, Biochemistry 34, 2731). We suggest that binding is dominated by the entropic effect, in agreement with previous proposals. Furthermore, we explain the role of conserved methionines by showing that the large flexibility of these side chains is a key feature of the binding mechanism. Finally, we provide a rationale for the experimental observation that in all CaM complexes the C-terminal domain seems to be hierarchically more important in establishing the interaction.
INTRODUCTION
A large variety of proteins adapt their shape to recognize their molecular partners. Among these, calmodulin (CaM) is probably the most prototypical example (1): this protein, which acts as a Ca2+ messenger in all eukaryotic cells, recognizes more than 100 different protein partners involved in various fundamental biological mechanisms (2) with a tight dissociation constant in the range of nanomolars.
CaM is a small acidic protein, consisting of two mostly helical globular domains, connected by a flexible linker: each domain contains two calcium-binding EF-hand motifs (3). Upon calcium binding, the two globular domains undergo a structural transition from a closed to an open conformation, characterized by a rearrangement of the interhelix axes. As a consequence, the hydrophobic core of each domain becomes exposed and able to accommodate the target into its cavity. Typically, peptide segments of ∼20 amino acids (4) are sufficient for tight binding (Fig. 1).
FIGURE 1.

(a) Structure of CaM bound to its highest affinity target peptide (M13), as determined by NMR spectroscopy (8): CaM (red) and M13 (blue) Cα chains are drawn as ribbons; the side chains of the two anchors (W-4 and F-17), which bind to the C- and N-domains, are represented as yellow and green sticks, respectively. (b) PDB entries and peptide sequences of the 15 CaM/peptide complex structures which have been determined so far (anthrax edema factor, which is a CaM/protein complex (58), is not included here). The helical regions (as defined by the program DSSP (42)) are boxed. The two anchors (10) are colored as in panel a. For convenience, sequences are printed with the first of the two anchors aligned.
The structural determinants of a variety of CaM/peptide complexes have been established by NMR spectroscopy and x-ray crystallography. These structures reveal that, despite their very low sequence identity (Fig. 1), some key features are common to the vast majority of the CaM-binding peptides (4–7). They are all amphiphilic and able to adopt upon binding an α-helical structure, independent of whether they are unstructured in their unbound states (4,8,9). Two hydrophobic residues play a pivotal role in binding (10), acting as “anchors” to the protein (Fig. 1). Some of the shortest peptides contain only one anchor. The CaM sequence, on the other hand, is highly conserved throughout species, probably having evolved to provide the optimal compromise to achieve a high binding affinity with an elevated number of quite different target sequences (11). Its domains accommodate the peptide by burying the anchors into two specific hydrophobic pockets, each contained in one of the globular domains (8). This process is thought to be assisted by an unusually large number (eight in the two domains, and one at the linker) of highly conserved (12,13) methionines, which have been proposed to play a key role in the target recognition (4,8,10,11,14). This suggestion is supported by the observation that in CaM the methionine side chains experience the largest loss of conformational freedom upon peptide binding (15–17). Additional contributions to the binding are provided by electrostatic interactions between basic residues of the target peptide with negatively charged residues suitably positioned along the CaM sequence. It is intriguing to notice that, despite the significant sequence identity (∼45%), the N- and C-terminal domains have a distinct hierarchical role in target recognition—although some of the peptides bind only the C-terminal domain, no structure is known in which the interaction is established only with the N-terminal domain (18). The affinity of tightly binding peptides to the isolated C-terminal domain is comparable to the one measured for full-length CaM (19). Moreover, when only one anchor is present, it binds preferentially to the C-terminal domain (20).
Despite the large interest shown in the field and the plethora of both experimental and theoretical studies, several questions remain unsolved before we can say we have understood in detail the mechanisms which allow CaM to recognize both in vitro and in vivo so many different sequences. It would for instance be very important to have a quantitative and reliable description of the thermodynamics of the binding process. Another important question is to understand theoretically the role of key residues—i.e., the anchors and the methionines—in the recognition process. To do so, it is necessary to study in detail the effect of dehydration of the binding pockets. It would also be very important to find a satisfactory theoretical explanation for the observed higher importance of the C-terminal domain.
To find an answer to these questions, we have investigated the final steps of CaM-peptide complex formation using metadynamics simulations (21), an approach successfully used to simulate rare events and reconstruct free energy profiles (22–25). Compared to other free energy methods, metadynamics does not require a priori atomic resolution structures of the transition endpoints and mechanisms, and its accuracy can be estimated by well-established guidelines for the choice of its parameters (26). To the best of our knowledge, this is the first of such calculations in the context of peptide- or protein-protein interactions.
We focus on the complex between CaM and M13 (8), a peptide which is part of the skeletal muscle myosin light chain kinase (skMLCK) (27,28). This complex has the highest affinity known for CaM natural targets (dissociation constant Kd ∼0.22 nM (29)) and involves an important biological CaM partner in the muscle tissue. Based on the NMR structure of the complex (8), we calculate here the free energy profile as a function of the coordination numbers of each anchor with its pocket. The calculated value is in excellent agreement with that observed for the CaM complex with the highly homologous smooth muscle MLCK peptide (30,31).
Our results, which are consistent with the NMR experiments within experimental error (8,15), i), provide additional insights into the role of the conserved methionines in the substrate recognition; ii), suggest that peptide binding is structurally and energetically different in the two sites, consistent with the hierarchical more important role of the C-terminal anchor relative to the N-terminal one (32); and iii), suggest that substrate binding might be dominated by the entropic effect, as previously postulated (16,33), with a free energy gain similar to that measured for the homologous smooth muscle MLCK system (31).
COMPUTATIONAL DETAILS
Structure equilibration
The NMR structure of the CaM/M13 complex (2BBM entry in the Protein Data Bank (PDB)) (8) was inserted in a water cubic box with an 80-Å long edge. The ionic strength of the NMR structure (8) was reproduced by adding in random positions 33 K+ ions and 25 Cl− ions. The total number of atoms was ∼51,000. The AMBER03 force field was used for the solute molecules and counterions (34). The TIP3P water model (35) was adopted. The electrostatic effect was taken into account by the particle mesh Ewald (PME) method (36), with a 12-Å cutoff and a 0.75-Å-spaced Fourier grid; a dielectric constant of 1 was assumed. Twelve angstroms was also the cutoff applied to the van der Waals forces. The time step was 1.5 fs. The nonbonded atoms pair list was updated every 30 fs. The SHAKE algorithm (37) was used to fix all bond lengths. Constant temperature (300 K) and constant pressure (1 atm) simulations were achieved with a Langevin thermostat (38) with a damping coefficient of 1 ps−1 and Nosè-Hoover Langevin barostat (39,40) with oscillation period 200 fs and decay coefficient 100 fs.
Before starting the metadynamics, the system was equilibrated using the following computational procedure: i), energy minimization of the solvent, using the conjugate gradient algorithm up to a convergence of 10−4 kcal/mol (the total energy being 2 × 105 kcal/mol); ii), 1 ns of 300 K molecular dynamics (MD) of the solvent and of the counterions: this simulation was meant to equilibrate the solvent and the ions' spatial distribution around the solute; iii), energy minimization of the entire system, using the same procedure as in i); and iv), 3.5 ns of MD at 300 K. The last 2 ns were used to collect statistics. In all simulations, trajectory frames were stored every 0.6 ps for analysis and calculation of system properties.
Metadynamics
History-dependent metadynamics (21,22,24,26) was performed based on the last MD snapshot, using the same setup as above. We used one collective variable describing the interactions between anchor W-4 and CaM and one describing the interactions between F-17 and CaM (Fig. 1). These are the coordination numbers CW-4 and CF-17, calculated by means of a continuous function (22) of all pairs of nonpolar carbons in each anchor and its pocket:
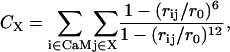 |
(1) |
where X = W-14 or F-17, rij is the distance between selected carbon atoms i of the protein and j of either anchor (see Fig. 1 of Supplementary Material), and r0 = 6 Å is a parameter that takes into consideration their typical carbon-carbon distance (4/4.5 Å) and the thermal motions' amplitude (1.5/2 Å). The sum involves specific nonpolar carbon atoms, namely those belonging to the side chains (starting from the Cγ) forming the two binding sites: F92, I100, L105, M109, M124, L125, V136, F141, M144, and M145 for W-4 and F19, I27, L32, M36, M51, L52, V55, I63, F68, M71, and M72 for F-17 binding sites. Metadynamics parameters followed the suggestions of Laio et al. (26): the Gaussian width δS = 5, the Gaussian weight W = 0.05 kcal/mol, and the insertion time τG = 300 fs.
The intrinsic error of the metadynamics approach was calculated as in Laio et al. (26) by assuming that the calculation does not depend on the starting structure and on the particular sequence of visited configurations but only on the sum of the added Gaussians. Then, the error ε depends only on the ratio between i), the width of each Gaussian (δS), the total size (S), of the configuration space (in this case it turns out to be ∼70 for both CW-4 and CF-17) and the insertion frequency 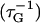 , and ii), the intrinsic diffusion coefficients (D) of collective variables:
, and ii), the intrinsic diffusion coefficients (D) of collective variables:
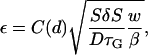 |
(2) |
where C(d) is a coefficient which depends on the number d of collective variables (here, C(2) ≃ 0.3; see Laio et al. (26) for details), w is the Gaussian weight, and β is the inverse temperature. Diffusion coefficients D were calculated as in Carte and Hynes (41) over a 1-ns unbiased MD trajectory, with the same equilibrated starting structure as the metadynamics run, and turn out to be 0.0015 fs−1 and 0.0012 fs−1, for CW-4 and CF-17, respectively.
Properties
The following properties were calculated:
Hydration numbers of the two anchors, calculated as the number of water oxygens within an ∼8-Å radius from them.
The degree of α-helical character of the peptide during simulation, calculated as from Kabsch and Sander (42).
The NMR contact restraints' cost function, defined as from Schwieters et al. (38): the cost function was averaged over MD trajectory frames, and in particular for the metadynamics trajectories each frame was weighted by the Boltzmann factor calculated with its free energy.
- Van der Waals (EvdW) and Coulomb (ECoul) energies between anchors and protein as well as between the protein-binding pocket and the rest of the system. Test calculations showed that, as expected, the fluctuations of the interaction energies between the solvent and the protein are much larger than the differences calculated here; therefore they are not presented in this work:
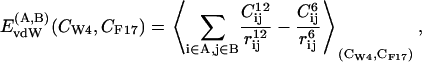
(3)
where
(4) 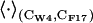 denotes an average over trajectory snapshots with values of the collective variables equal to (CW-4, CF-17) within a tolerance parameter, which was chosen as 2δS; rij is the distance between atoms i and j; qi, qj are their partial charges; and
denotes an average over trajectory snapshots with values of the collective variables equal to (CW-4, CF-17) within a tolerance parameter, which was chosen as 2δS; rij is the distance between atoms i and j; qi, qj are their partial charges; and 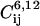 are the Lennard-Jones parameters for the atom pair (i, j). Atom i and j belong to A and B, where A or B includes the anchor, the binding pocket, the rest of the protein, and the solvent with the counterions. Each bin in the grid of CW-4, CF-17 values has on average ∼100 snapshots. The dispersion of each energy term was calculated as the average standard deviation from the binned energies of Eqs. 3 and 4:
are the Lennard-Jones parameters for the atom pair (i, j). Atom i and j belong to A and B, where A or B includes the anchor, the binding pocket, the rest of the protein, and the solvent with the counterions. Each bin in the grid of CW-4, CF-17 values has on average ∼100 snapshots. The dispersion of each energy term was calculated as the average standard deviation from the binned energies of Eqs. 3 and 4:
where [CW-4, CF-17] indicates a bin on the collective variables' grid,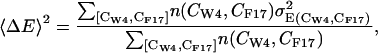
(5) 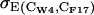 is the standard deviation within the bin of the energy E(CW-4, CF-17) calculated as from Eqs. 3 and 4, and n(CW-4, CF-17) is the number of trajectory snapshots in the bin.
is the standard deviation within the bin of the energy E(CW-4, CF-17) calculated as from Eqs. 3 and 4, and n(CW-4, CF-17) is the number of trajectory snapshots in the bin. - S2 order parameters (43–45) of the protein were calculated in terms of bond vector autocorrelation functions (ACFs) (17):
where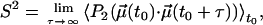
(6) 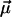 is the normalized bond vector (fitted to the NMR structure of the complex, (8)) and P2 is the second-order Legendre polynomial. We averaged each ACF for 600 ps and calculated its average limiting value after 150 ps, which is a reasonable upper bound for “microscopic” correlation times (43); the standard deviation was taken as an error estimate. The calculation was performed for the backbone N-H bonds and for side-chain X-C bonds, where X is a side-chain heavy atom and C is a methyl carbon.
is the normalized bond vector (fitted to the NMR structure of the complex, (8)) and P2 is the second-order Legendre polynomial. We averaged each ACF for 600 ps and calculated its average limiting value after 150 ps, which is a reasonable upper bound for “microscopic” correlation times (43); the standard deviation was taken as an error estimate. The calculation was performed for the backbone N-H bonds and for side-chain X-C bonds, where X is a side-chain heavy atom and C is a methyl carbon.
All MD simulations were performed using the NAMD code (46), locally modified to incorporate the changes necessary to perform metadynamics. Interaction energies were calculated from the obtained trajectories using GROMACS (47). The cost function was calculated with VMD-XPLOR (48). Pictures were produced with VMD (49).
RESULTS AND DISCUSSION
In the following section, we first briefly outline some results from the MD simulation carried out on the CaM/M13 complex to equilibrate the system and from the successive metadynamics. Then, we provide a detailed description of the energetics of the final hydration process as followed by metadynamics. Comparison is made between our results and NMR data.
Structural features
During the 3.5 ns of MD equilibration period, the structure of the complex fluctuates around an average conformation within a 3-Å root mean square deviation (RMSD) from the initial NMR (8) structure (Fig. 2 in Supplementary Material). The secondary structure elements (42) are fully preserved. The two sites that host the M13 peptide anchors maintain the structural differences observed in the NMR structure (RMSD 3.3 Å). In particular, the W-4-binding pocket is roughly 2 Å narrower and 2 Å deeper than that of F-17 both in the NMR structure and in the last MD snapshot (Fig. 2 of Supplementary Material).
The coordination numbers of the peptide's anchors, here defined as CW-4 and CF-17 (see Computational Details), have adimensional units, and their values are roughly equal to the number of carbon-carbon internuclear distances between the two anchors and their pockets, which are within 6 Å or less (see Computational Details). CW-4 and CF-17 fluctuate in the simulation around the average values 90 ± 6 and 67 ± 7 (Fig. 3 of Supplementary Material).
Validation of the molecular dynamics trajectory by NMR restraints
We checked the accuracy of our MD calculations by measuring the fraction of the CaM/M13 interatomic distances that satisfy the experimental NMR restraints (8). We also compared the calculated experimental NMR order parameters (15,43,45), thus comparing static and dynamical features.
The number of distance violations during the MD converges to 7% ± 0.4% of the total after 3.5 ns. Of these, a very small fraction of violations (1% of the total) exceeds the RMSD of the complex (3.0 Å) (Fig. 2); these correspond to nuclear Overhauser effect (NOE) restraints involving atoms in the two inter-EF-hand loops (six restraints) and intramolecular ones within the peptide (10 restraints), which are intrinsically very flexible regions anyway (15,50). Another fraction of restraints (10%) experiences a small violation for almost the whole run. Although this implies that some permanent rearrangement of the experimental structure occurs, which can be detected at the end of the MD (see Fig. 2), the overall extent of the conformational change is relatively small. This is well within the experimental error of the NMR structure determination as suggested by comparison of the 2BBM structure with that of the highly homologous CaM/smooth muscle MLCK complex (31).
FIGURE 2.

MD simulation of the CaM/M13 complex. I. Comparison of the Cα traces of the initial structure (PDB entry 2BBM, shown in blue) and the final MD structure (red). II. Top: number of violated NMR restraints as a function of time. Middle: violation distance for each violated restraint (averaged over the frames in which violations occur). Bottom: total time for which each restraint is violated, if any violation occurs. In the last two, intramolecular restraints range from 1 to 1,486 for CaM and from 1,487 to 1,650 for M13; intermolecular restraints range from 1,651 to 1,782. III. Values of CW-4 (black) and CF-17 (red) order parameters (see definition in Computational Details) along the MD trajectory (top), and RMSD of backbone atoms with respect to the experimental structure (bottom) are plotted as a function of time. IV. S2 order parameters of CaM NH and methionines' methyl groups: experimental (green solid circles) and calculated (red empty circles).
The calculated S2 order parameters are also in good agreement with those obtained experimentally (Fig. 2 and Fig. 6 of Supplementary Material) (15), and in a previous MD simulation (17). In particular, the methionine methyl group S2 parameters of the complex, which are very different from those of the protein in the free state (15–17), agree well with the experimental values in both states (Fig. 2 and Table 1).
TABLE 1.
MD simulations of CaM and of the CaM/M13 complex. S2 methyl order parameters and RMSDs of Met side chains (heavy atoms) on each MD trajectory, taking as a reference the two starting experimental structures, respectively
| W-4 site free CaM | ||||
|---|---|---|---|---|
| RMSD (Å)
|
2.3 ± 0.4
|
|||
| M109 | M124 | M144 | M145 | |
| S2 (calc) | 0.51 ± 0.10 | 0.30 ± 0.08 | 0.21 ± 0.10 | 0.24 ± 0.05 |
| S2 (exp) | 0.15 ± 0.02 | 0.19 ± 0.02 | 0.12 ± 0.02 | 0.22 ± 0.02 |
| complex
| ||||
| RMSD (Å)
|
1.8 ± 0.2
|
|||
| M109 | M124 | M144 | M145 | |
| S2 (calc) | 0.41 ± 0.10 | 0.65 ± 0.16 | 0.44 ± 0.12 | 0.46 ± 0.09 |
| S2 (exp) | 0.36 ± 0.03 | 0.84 ± 0.06 | 0.36 ± 0.03 | 0.29 ± 0.02 |
| F-17 site free CaM
| ||||
| RMSD (Å)
|
2.8 ± 0.6
|
|||
| M36 | M51 | M71 | M72 | |
| S2 (calc) | 0.25 ± 0.07 | 0.23 ± 0.07 | 0.14 ± 0.08 | 0.38 ± 0.09 |
| S2 (exp) | 0.26 ± 0.02 | 0.18 ± 0.02 | 0.16 ± 0.02 | 0.26 ± 0.02 |
| complex
| ||||
| RMSD (Å)
|
2.0 ± 0.2
|
|||
| M36 | M51 | M71 | M72 | |
| S2 (calc) | 0.45 ± 0.10 | 0.52 ± 0.10 | 0.27 ± 0.12 | 0.76 ± 0.14 |
| S2 (exp) | 0.31 ± 0.02 | 0.28 ± 0.02 | 0.40 ± 0.02 | 0.76 ± 0.05 |
Methionine mobility during the dynamics of the CaM/M13 complex. Comparison of RMSDs of methionine residues (heavy atoms) for the W-4 and F-17 binding sites. M109, M124, M144, and M145 are present in the W-4 site, M36, M51, M71, and M72 in the F-17 site.
Overall, this comparison provides a rough estimate of the quality of this MD simulation, both in terms of force field used and sampling convergence and allows us to conclude that both the static and the dynamical features of the complex are sufficiently well described in our MD simulation.
Free energy landscape
The free energy surface as a function of the coordination numbers CW-4 and CF-17 was calculated using 8 ns of metadynamics (Fig. 3 a). The estimated error associated to this procedure is 2.3 kcal/mol (26).
FIGURE 3.

Metadynamics calculations. Free energy (kcal/mol) associated to the final step of dehydration of the anchors' binding pockets in the CaM/M13 complex. I. Two-dimensional and II. Three-dimensional plots of the free energy as a function of the anchor coordination numbers CW-4 and CF-17, as defined in the Computational Details. The coloring scheme in I. provides the energies, in II. the values of the cost function based on the NMR restraints (8,38). In II., the 21 experimental structures deposited in the PDB are represented as black dots. III. Contribution of the methionines in the W-4 binding site, i.e., CW-4(Met)/CW-4(total). IV. Same for the F-17 binding site.
Around the equilibrium configuration, the free energy surface is nearly flat, with minima separated by 1.5 kcal/mol. The two basins Ga and Gb are to be related to the occupation by the F-17 anchor of the two CaM pockets that are delimited by the two isoleucines I-27 and I-63 (Fig. 4).
FIGURE 4.

Metadynamics calculations. Molecular recognition of the W-4 (top) and F-17 (bottom) anchors from the partially hydrated states (D1 and D2 in Fig. 3) to the dehydrated complex (Ga and Gb). (a) Schematic representation of the anchors (blue) conformations: the light blue cartoon indicates the full hydrophobic pocket; Methionine side chains (yellow) and Ile side chains (green) are also shown; arrows indicate transitions explicitly observed in the simulation. (b) Corresponding three-dimensional structures of the binding sites. The peptide Cα traces are shown in blue. CaM's solvent accessible surface is colored according to atom types (sulfur, oxygen, nitrogen, and carbon in yellow, red, blue, and gray, respectively).
As in the initial NMR structure, the CaM methionines of the binding pockets closely interact with the two peptide anchors. In the W-4 binding site, M109, M124, M144, and M145 interact with the substrate, whereas at the F-17 site, conformations Ga and Gb differ for the conformations of M36, M51, M71, and M72; of the two, Gb is the conformation of the F-17 site most similar to that of the W-4 pocket, in that it has a symmetrical arrangement of the methionines around the anchor (Fig. 4). Thus, the relative orientation of the F-17 binding site with respect to the two isoleucines is a key distinguishing feature of the two minima (see also Fig. 8 of Supplementary Material).
A shallow minimum D1 is present above Ga and Gb, 5.5 kcal/mol higher in free energy. This local minimum corresponds to a state in which the W-4 anchor is partially hydrated; the number of coordinated waters within 8 Å increases from ∼11 in G to 20 in D1. The G → D1 transition is achieved by orienting this residue in a direction orthogonal to that assumed in G. The conformation of the other anchor, F-17, is instead practically the same as in the NMR structures, and CF-17 assumes values similar to those in Ga,b. In the W-4 binding site, methionines undergo a tiny rearrangement, to interact more closely with themselves and with the rest of the hydrophobic pocket, with the exception of M124, which moves apart to open the way to W-4 (Fig. 4).
The even higher D2 metastable conformation at 8 kcal/mol corresponds to the exit of F-17 from the pocket. In this conformation, F-17 is fully exposed to the solvent: its water coordination number is 60, to be compared to ∼10 in Ga,b. The peptide becomes locally unwound. The methionine residues retract toward the cavity, interacting mostly with other intramolecular residues. Also in this case, only one anchor, F-17, rearranges; the value of CW-4 is the same as that of Ga,b. Thus, the dissociation of each anchor from its binding pocket does not involve significant rearrangements of the other.
Finally, in the minimum D3 both anchors are partially dehydrated similarly to D1 and D2. 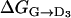 is 11 kcal/mol, which is slightly lower than
is 11 kcal/mol, which is slightly lower than 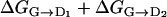 = 13.5 kcal/mol. This fact and the fact that D3 is not perfectly aligned to D1 and D2 (Fig. 3) are suggestive of a small cooperative interaction between the two sites, although ΔΔG is as small as 2.5 kcal/mol, that is, of the same order of the metadynamics estimated error.
= 13.5 kcal/mol. This fact and the fact that D3 is not perfectly aligned to D1 and D2 (Fig. 3) are suggestive of a small cooperative interaction between the two sites, although ΔΔG is as small as 2.5 kcal/mol, that is, of the same order of the metadynamics estimated error.
Thus, we conclude that 5.5 ± 3.2 kcal/mol and 8.0 ± 3.2 kcal/mol are required to partially solvate the peptide at the W-4 and at the F-17 sites, respectively (error propagation is considered on the free energy differences because no systematic error is assumed (26)). These values are compatible with the 5.5 kcal/mol value measured for the highly homologous and structurally similar smooth muscle MLCK peptide (30,31) (Fig. 1). They are also in excellent agreement with several other protein/ligand complexes (51).
Comparison of the metadynamics with NMR data
The average number of violated NMR restraints is 7% ± 0.9%, that is, the same as that of the MD simulation (7% ± 0.4%). Notice that its dispersion is slightly larger because of a larger conformational space explored in metadynamics. Indeed, the average energy cost function is 126 ± 21 kcal/mol (85 ± 23 kcal/mol in the unbiased MD).
The cost energy function, plotted versus CW-4, F-17, correlates well with our free energy (Fig. 3). However, as expected, the minimum of the cost function G*, at about CW-4, F-17 = (80, 80), does not exactly coincide with the minima of the free energy, Ga,b. Also, the CW-4, F-17 values calculated for the NMR bundle (21 structures, PDB entry 2BBN) (8) are different both from G and G*: this reflects the fact that for given CW-4, F-17 values the structure ensemble explored during the simulation is slightly different from the experimental ensemble. The effect is particularly notable at the F-17 site where the two experimental conformations, related to the free energy minima Ga and Gb, give rise to a narrower interval of CF-17 values than in the metadynamics simulation. Also considering the structural rearrangement already observed in the unbiased MD, these relatively small differences are to be attributed to i), the use of a necessarily approximate force field (in fact, in the NMR structural determination (8) the weight of the force field was very small compared to the experimentally derived cost function); and ii), the errors associated to the metadynamics setup.
Recognition mechanism of the M13 anchors
Because of the structural differences between the two anchors, in one case (F-17) hydration is assisted by local unwinding of the M13 helix, whereas for W-4 there is only a smaller rearrangement that leaves the helix unchanged. Our calculations also show that F-17 dissociates completely (D2 minimum), whereas W-4 only rearranges and partly binds to the protein (D1 minimum). This is consistent with the experimentally proven hierarchical features of peptide binding in that the dissociation of the N-terminal domain of CaM (F-17 anchor) is known indeed to occur first (19), whereas the C-terminal domain (W-4 anchor) follows.
The role of the methionines in the CaM/M13 complex is well established (4,10,11,14). Here we provide novel information about the response of these residues upon binding by observing their conformation during the dehydration process (Fig. 4). We observe that seven out of eight of the methionine residues have the same conformation in the partially dehydrated states D1, D2, and D3 as in the nonligated state, as observed by comparing the results here with both the x-ray structure (52) and our MD simulations (53) (RMSD < 2 Å). Instead, as already discussed above, M124 changes its conformation relative to the free state significantly to let W-4 move toward the solvent (RMSD 3.4 Å). This conformation also differs from that of the bound state. These findings are fully consistent with the proposal, based on systematic mutagenesis of the methionines (12,13), that M124 is the most important methionine implicated in CaM target binding (15).
We further notice that the contribution of the eight methionines is ∼60%/70% of the total coordination of the anchors in the barrier between the two minima, whereas it is only 20% in both the final complexed and in the partially hydrated states. It is therefore clear that the methionines assist the dehydration process.
Role of the nonbonded interactions and of the hydrophobic effect
To investigate the role of protein/peptide interactions in the process upon partial dehydration (from D1, 2 to G), we calculated the AMBER (54) van der Waals and Coulomb interaction energies between the anchors, the pockets, and the rest of the system. Such calculations are expected to be highly approximated because fluctuations around average values are usually of the same order of magnitude as the difference between fully and partially dehydrated states. They can, however, still provide useful qualitative insights.
The variations of both van der Waals (Fig. 5) and Coulomb (Fig. 10 of Supplementary Material) energies between the anchors and the system (protein + solvent) or the pockets and the system (∼2 kcal/mol) in passing from G to D1 and from G to D2 are significantly smaller than the free energy changes (5.5 and 8 kcal/mol); interestingly, these energies are similar not only for the dehydrated and partially hydrated states, but practically during the entire dehydration process (Fig. 5). These rather small changes arise from a compensation of several contributions, for which the changes with CW-4, F-17 do exceed the energy dispersions. A detailed analysis is presented as Supplementary Material, where all individual terms are reported. However, these differences are smaller than the energy fluctuations during the simulation, which are 5 kcal/mol for the interactions between the anchors and the system and 10 kcal/mol for those between the pockets and the rest of the system.
FIGURE 5.

Metadynamics calculations. Van der Waals interactions (kcal/mol) of W-4, F-17, and their pockets with the whole system (including solvent), calculated with the AMBER force field (34) on the metadynamics trajectory. The four components are defined including both (hydrophobic) side-chain and (polar) backbone atoms. Values at the free energy minimum Ga (CW-4, F-17 = 90, 45) are taken as the reference. Color scale equal to that of the free energy (Fig. 3).
We can therefore explain the role that the entropic effect plays in the recognition of CaM and M13, although the large fluctuations of the calculated energies cannot firmly establish this point. Such an entropy gain associated to dehydration had already been suggested for a variety of CaM/peptide complexes (55) and, more generally, for a plethora of protein/peptide complexes (56).
Calorimetric studies have shown that formation of CaM complexes is associated either to enthalpy or to entropy-driven processes. The total entropy change for the overall complexation process has, for instance, been measured for two CaM complexes with peptides whose sequence and structure are very similar to that of M13 (Fig. 1), namely, smooth muscle MLCK and CaM-dependent protein kinase I CaM complexes; in both cases, TΔS is positive (14,57).
This can be explained by considering that the calorimetric data reflect the overall process of the interaction, whereas we describe only the final stage. In our calculations, we observe no significant changes either in the relative orientation of the protein domains, or in the linker conformation when going from D1 and D2 to Ga,b (Fig. 7 of Supplementary Material). It is quite possible that, in some CaM peptides, the entropic effect of the overall process is more than counterbalanced by loss of the protein conformational entropy (15,16,33), which is expected to occur in the first steps of the recognition.
CONCLUSION
In conclusion, we have performed metadynamics simulations on the CaM/M13 complex (8). We have validated our computational results obtained both during the preliminary equilibration period and during the metadynamics against the NMR data; we showed that the number of violations of the experimental distance restraints, as well as their magnitude, converge to stable values in the unbiased MD simulation and that these values are maintained in the metadynamics. The calculated S2 order parameters, especially those of the methionines, also agree with the experimental values (15). Thus, we can conclude that our simulations reproduce at least the overall structural and dynamical properties of the CaM complex in water solution.
Our metadynamics data provide new insights into the final stage of peptide dehydration (31) as they show that the two sites have different dissociation mechanisms. Both exploit the flexibility of methionine side chains, which have been shown experimentally to play a key role in binding (12,13,15). However, the W-4 anchor of M13 is overall more tightly bound to its pocket, so that its degree of hydration is only partial and requires (besides the increase of conformational flexibility of the methionines observed at both sites) the relocation of the M124 side chain. This process explains what makes this CaM residue particularly important for binding, as observed experimentally by mutagenesis data (12,13). On the contrary, the F-17 anchor does not need structural rearrangements, being already at the equilibrium characterized by two distinct conformations. This feature, which is also observed experimentally in the NMR bundle (8), is likely to explain the different importance in molecular recognition played by the N- and the C-terminal domains.
The dehydration process leads to a free energy loss similar to that observed for the CaM complex with the homologous smooth muscle MLCK peptide (31). A simple estimate of the nonbonded interaction energies suggests that the process might be mostly entropy driven as previously suggested (16,33).
Several approaches had been previously presented to predict a priori the stability of protein/protein complexes. Here, we have presented a metadynamics simulation that described the final events that lead to the interaction, thus providing a first step toward predicting the complete energetics of the molecular recognition between proteins and their target peptides or proteins. The challenge is now to design metadynamics-based approaches that could allow the treatment of more than a few reaction coordinates, thus making it possible to describe quantitatively the complete process.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
The authors thank Dr. Alessandro Laio for helpful discussions on the choosing criteria of metadynamics coordinates, and Dr. Rosa Bulo for providing the source code of the modified version of NAMD. The structures of the four minima in Fig. 4 are available at http://www.sissa.it/∼fiorin/cam+m13/.
References
- 1.Ikura, M., and J. B. Ames. 2006. Genetic polymorphism and protein conformational plasticity in the calmodulin superfamily: two ways to promote multifunctionality. Proc. Natl. Acad. Sci. USA. 103:1159–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Carafoli, E. 2002. Calcium signaling: a tale for all seasons. Proc. Natl. Acad. Sci. USA. 99:1115–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kawasaki, H., and R. H. Kretsinger. 1995. Calcium-binding proteins 1: EF-hands. Protein Profile. 2:297–490. [PubMed] [Google Scholar]
- 4.O'Neil, K. T., and W. F. DeGrado. 1990. How calmodulin binds its targets: sequence independent recognition of amphiphilic α-helices. Trends Biochem. Sci. 15:59–64. [DOI] [PubMed] [Google Scholar]
- 5.Crivici, A., and M. Ikura. 1995. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 24:85–116. [DOI] [PubMed] [Google Scholar]
- 6.Yap, K. L., J. Kim, K. Truong, M. Sherman, T. Yuan, and M. Ikura. 2000. Calmodulin target database. J. Struct. Funct. Genomics. 1:8–14. [DOI] [PubMed] [Google Scholar]
- 7.Hoeflich, K. P., and M. Ikura. 2002. Calmodulin in action: diversity in target recognition and activation mechanisms. Cell. 108:739–742. [DOI] [PubMed] [Google Scholar]
- 8.Ikura, M., G. M. Clore, A. M. Gronenborn, G. Zhu, C. B. Klee, and A. Bax. 1992. Solution structure of a calmodulin-target peptide complex by multidimensional NMR. Science. 256:632–638. [DOI] [PubMed] [Google Scholar]
- 9.Zhang, M., M. Li, J. H. Wang, and H. J. Vogel. 1994. The effect of met→leu mutations on calmodulin's ability to activate cyclic nucleotide phosphodiesterase. J. Biol. Chem. 269:15546–15552. [PubMed] [Google Scholar]
- 10.Hultschig, C., H. J. Hecht, and R. Frank. 2004. Systematic delineation of a calmodulin peptide interaction. J. Mol. Biol. 343:559–568. [DOI] [PubMed] [Google Scholar]
- 11.Shifman, J. M., and S. L. Mayo. 2003. Exploring the origins of binding specificity through the computational redesign of calmodulin. Proc. Natl. Acad. Sci. USA. 100:13274–13279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chin, D., and A. R. Means. 1996. Methionine to glutamine substitutions in the C-terminal domain of calmodulin impair the activation of three protein kinases. J. Biol. Chem. 271:30465–30471. [DOI] [PubMed] [Google Scholar]
- 13.Edwards, R. A., M. P. Walsh, C. Sutherland, and H. J. Vogel. 1998. Activation of calcineurin and smooth muscle myosin light chain kinase by Met-to-Leu mutants of calmodulin. Biochem. J. 331:149–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wintrode, P. L., and P. L. Privalov. 1997. Energetics of target peptide recognition by calmodulin: a calorimetric study. J. Mol. Biol. 266:1050–1062. [DOI] [PubMed] [Google Scholar]
- 15.Lee, A. L., S. A. Kinnear, and A. J. Wand. 2000. Redistribution and loss of side chain entropy upon formation of a calmodulin-peptide complex. Nat. Struct. Biol. 7:72–77. [DOI] [PubMed] [Google Scholar]
- 16.Cavanagh, J., and M. Akke. 2000. May the driving force be with you—whatever it is. Nat. Struct. Biol. 7:11–13. [DOI] [PubMed] [Google Scholar]
- 17.Prabhu, N. V., A. L. Lee, A. J. Wand, and K. A. Sharp. 2003. Dynamics and entropy of a calmodulin-peptide complex studied by NMR and molecular dynamics. Biochemistry. 42:562–570. [DOI] [PubMed] [Google Scholar]
- 18.Atkinson, R. A., C. Joseph, G. Kelly, F. W. Muskett, T. A. Frenkiel, D. Nietlispach, and A. Pastore. 2001. Ca2+-independent binding of an EF-hand domain to a novel motif in the α-actinin-titin complex. Nat. Struct. Biol. 8:853–857. [DOI] [PubMed] [Google Scholar]
- 19.Persechini, A., K. McMillan, and P. Leakey. 1994. Activation of myosin light chain kinase and nitric oxide synthase activities by calmodulin fragments. J. Biol. Chem. 269:16148–16154. [PubMed] [Google Scholar]
- 20.Shuman, C. F., R. Jiji, K. S. Kerfeldt, and S. Linse. 2006. Reconstitution of calmodulin from domains and subdomains: influence of target peptide. J. Mol. Biol. 358:870–881. [DOI] [PubMed] [Google Scholar]
- 21.Laio, A., and M. Parrinello. 2002. Escaping free-energy minima. Proc. Natl. Acad. Sci. USA. 99:12562–12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Iannuzzi, M., A. Laio, and M. Parrinello. 2003. Efficient exploration of reactive potential energy surfaces using Car-Parrinello molecular dynamics. Phys. Rev. Lett. 90:238–302. [DOI] [PubMed] [Google Scholar]
- 23.Ensing, B., A. Laio, F. L. Gervasio, M. Parrinello, and M. L. Klein. 2004. A minimum free energy reaction path for the e2 reaction between fluoro ethane and a fluoride ion. J. Am. Chem. Soc. 126:9492–9493. [DOI] [PubMed] [Google Scholar]
- 24.Gervasio, F. L., A. Laio, and M. Parrinello. 2005. Flexible docking in solution using metadynamics. J. Am. Chem. Soc. 127:2600–2607. [DOI] [PubMed] [Google Scholar]
- 25.Branduardi, D., F. L. Gervasio, A. Cavalli, M. Recanatini, and M. Parrinello. 2005. The role of the peripheral anionic site and cation-pi interactions in the ligand penetration of the human AChE gorge. J. Am. Chem. Soc. 127:9147–9155. [DOI] [PubMed] [Google Scholar]
- 26.Laio, A., A. Rodriguez-Fortea, F. L. Gervasio, M. Ceccarelli, and M. Parrinello. 2005. Assessing the accuracy of metadynamics. J. Phys. Chem. B. 109:6714–6721. [DOI] [PubMed] [Google Scholar]
- 27.Blumenthal, D. K., and J. T. Stull. 1980. Activation of skeletal muscle myosin light chain kinase by calcium(2+) and calmodulin. Biochemistry. 19:5608–5614. [DOI] [PubMed] [Google Scholar]
- 28.Blumenthal, D. K., and J. T. Stull. 1982. Effects of pH, ionic strength, and temperature on activation by calmodulin and catalytic activity of myosin light chain kinase. Biochemistry. 21:2386–2391. [DOI] [PubMed] [Google Scholar]
- 29.Findlay, W. A., S. R. Martin, K. Beckingham, and P. M. Bayley. 1995. Recovery of native structure by calcium binding site mutants of calmodulin upon binding of sk-MLCK target peptides. Biochemistry. 34:2087–2094. [DOI] [PubMed] [Google Scholar]
- 30.Meador, W. E., A. R. Means, and F. A. Quiocho. 1992. Target enzyme recognition by calmodulin: 2.4 Å structure of a calmodulin-peptide complex. Science. 257:1251–1255. [DOI] [PubMed] [Google Scholar]
- 31.Ehrhardt, M. R., J. L. Urbauer, and A. J. Wand. 1995. The energetics and dynamics of molecular recognition by calmodulin. Biochemistry. 34:2731–2738. [DOI] [PubMed] [Google Scholar]
- 32.Persechini, A., K. Kano, and P. L. Stemmer. 2000. Ca2+ binding and energy coupling in the calmodulin-myosin light chain kinase complex. J. Biol. Chem. 275:4199–4204. [DOI] [PubMed] [Google Scholar]
- 33.Yang, C., G. S. Jas, and K. Kuczera. 2004. Structure, dynamics and interaction with kinase targets: computer simulations of calmodulin. Biochim. Biophys. Acta. 1697:289–300. [DOI] [PubMed] [Google Scholar]
- 34.Ponder, J. W., and D. A. Case. 2003. Force fields for protein simulations. Adv. Prot. Chem. 66:27–85. [DOI] [PubMed] [Google Scholar]
- 35.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 36.Essman, U., L. Perela, M. L. Berkowitz, T. Darden, H. Lee, and L. G. Pedersen. 1995. A smooth particle mesh Ewald method. J. Chem. Phys. 103:8577–8592. [Google Scholar]
- 37.Ryckaert, J. P., G. Ciccotti, and H. J. C. Berendsen. 1977. Numerical integration of the Cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J. Comp. Phys. 23:327–341. [Google Scholar]
- 38.Schwieters, C. D., J. J. Kuszewski, N. Tjandra, and G. M. Clore. 2003. The Xplor-NIH NMR molecular structure determination package. J. Magn. Res. 160:66–74. [DOI] [PubMed] [Google Scholar]
- 39.Martyna, G., D. Tobias, and M. Klein. 1994. Constant pressure molecular dynamics algorithms. J. Chem. Phys. 101:4177–4189. [Google Scholar]
- 40.Feller, S. E., Y. Zhang, R. W. Pastor, and B. R. Brooks. 1995. Constant pressure molecular dynamics simulation: the Langevin piston method. J. Chem. Phys. 103:4613–4621. [Google Scholar]
- 41.Carter, E. A., and J. T. Hynes. 1991. Solvation dynamics for an ion pair in a polar solvent: time-dependent fluorescence and photochemical charge transfer. J. Chem. Phys. 94:5961–5979. [Google Scholar]
- 42.Kabsch, W., and C. Sander. 1983. Dictionary of protein secondary structure: pattern recognition of hydrogen-bonded and geometrical features. Biopolymers. 22:2577–2637. [DOI] [PubMed] [Google Scholar]
- 43.Lipari, G., and A. Szabo. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 1. Theory and range of validity. J. Am. Chem. Soc. 104:4546–4559. [Google Scholar]
- 44.Lipari, G., and A. Szabo. 1982. Model-free approach to the interpretation of nuclear magnetic resonance relaxation in macromolecules. 2. Analysis of experimental results. J. Am. Chem. Soc. 104:4559–4570. [Google Scholar]
- 45.Lee, A., P. Flynn, and A. Wand. 1999. Comparison of 2H and 13C NMR relaxation techniques for the study of protein methyl group dynamics in solution. J. Am. Chem. Soc. 121:2891–2902. [Google Scholar]
- 46.Kalé, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Phillips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. NAMD2: greater scalability for parallel molecular dynamics. J. Comp. Phys. 151:283–312. [Google Scholar]
- 47.Lindahl, E., B. Hess, and D. Van der Spoel. 2001. GROMACS 3.0: a package for molecular simulation and trajectory analysis. J. Mol. Mod. 7:306–317. [Google Scholar]
- 48.Schwieters, C. D., and G. M. Clore. 2001. The VMD-XPLOR visualization package for NMR structure refinement. J. Magn. Res. 149:239–244. [DOI] [PubMed] [Google Scholar]
- 49.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD—visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 50.Barbato, G., M. Ikura, L. E. Kay, R. W. Pastor, and A. Bax. 1992. Backbone dynamics of calmodulin studied by 15N relaxation using inverse detected two-dimensional NMR spectroscopy: the central helix is flexible? Biochemistry. 31:5269–5278. [DOI] [PubMed] [Google Scholar]
- 51.Puvanendrampillai, D., and J. B. O. Mitchell. 2003. Protein Ligand Database (PLD): additional understanding of the nature and specificity of protein-ligand complexes. Bioinformatics. 19:1856–1857. [DOI] [PubMed] [Google Scholar]
- 52.Chattopadhyaya, R., W. E. Meador, A. R. Means, and F. A. Quiocho. 1992. Calmodulin structure refined at 1.7 Å resolution. J. Mol. Biol. 228:1177–1192. [DOI] [PubMed] [Google Scholar]
- 53.Fiorin, G., R. R. Biekofsky, A. Pastore, and P. Carloni. 2005. Unwinding the helical linker of calcium-loaded calmodulin: a molecular dynamics study. Proteins. 61:829–839. [DOI] [PubMed] [Google Scholar]
- 54.Cheatham, T. E., P. Cieplak III, and P. A. Kollman. 1999. A modified version of the Cornell et al. force field with improved sugar pucker phases and helical repeat. J. Biomol. Struct. Dyn. 16:845–862. [DOI] [PubMed] [Google Scholar]
- 55.Yang, C., and K. Kuczera. 2002. Molecular dynamics simulations of calcium-free calmodulin in solution. J. Biomol. Struct. Dyn. 19:801–819. [DOI] [PubMed] [Google Scholar]
- 56.Vidossich, P., and P. Carloni. 2006. Binding of phosphinate and phosphonate inhibitors to aspartic proteases: a first-principles study. J. Phys. Chem. B. 110:1437–1442. [DOI] [PubMed] [Google Scholar]
- 57.Brokx, R. D., M. M. Lopez, H. J. Vogel, and G. I. Makhatadze. 2001. Energetics of target peptide binding by calmodulin reveals different modes of binding. J. Biol. Chem. 276:14083–14091. [DOI] [PubMed] [Google Scholar]
- 58.Drum, C. L., S. Z. Yan, J. Bard, Y. Q. Shen, D. Lu, S. Soelaiman, Z. Grabarek, A. Bohm, and W. J. Tang. 2002. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature. 415:396–402. [DOI] [PubMed] [Google Scholar]