

Abstract
Molecular dynamics simulations have been performed to investigate the partitioning of the volatile anesthetic halothane from an aqueous phase into a coexisting hydrated bilayer, composed of 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipids, with embedded α-helical peptide bundles based on the membrane-bound portions of the α- and δ-subunits, respectively, of nicotinic acetylcholine receptor. In the molecular dynamics simulations halothane molecules spontaneously partitioned into the DOPC bilayer and then preferentially occupied regions close to lipid headgroups. A single halothane molecule was observed to bind to tyrosine (Tyr-277) residue in the α-subunit, an experimentally identified specific binding site. The binding of halothane attenuated the local loop dynamics of α-subunit and significantly influenced global concerted motions suggesting anesthetic action in modulating protein function. Steered molecular dynamics calculations on a single halothane molecule partitioned into a DOPC lipid bilayer were performed to probe the free energy profile of halothane across the lipid-water interface and rationalize the observed spontaneous partitioning. Partitioned halothane molecules affect the hydrocarbon chains of the DOPC lipid, by lowering of the hydrocarbon tilt angles. The anesthetic molecules also caused a decrease in the number of peptide-lipid contacts. The observed local and global effects of anesthetic binding on protein motions demonstrated in this study may underlie the mechanism of action of anesthetics at a molecular level.
INTRODUCTION
Despite the use of general anesthetics (GA) in medicine for more than 150 years, their mechanism of action on the central nervous system remains a matter of debate. Two contenders for the primary site of action in this controversy are the lipid membrane and the proteins embedded in it. Starting with the early experiments by Meyer and Overton (1,2), many studies (3,4) have suggested that the lipid bilayers act as primary targets for anesthetics, because of their hydrophobicity. The GA molecules achieve a high concentration in the lipid bilayer, and were suggested to perturb its structure and dynamics, only indirectly affecting membrane protein function through nonspecific mechanisms. On the other hand, proteins have hydrophobic domains, which make them equally plausible targets, as demonstrated in the case of firefly luciferase (5) or apoferritin (6). Further support for protein-centered hypotheses came from work with a series of chemical compounds known as nonimmobilizers, which partition readily into lipid membranes, but do not produce anesthesia (7–9). However, recent theoretical studies (10,11) have renewed interest in lipid effects of GA molecules.
Interest in the specific interactions of the GA molecules with membrane proteins emerged from experiments with the ligand-gated ion channels (LGIC) (12–14). The gene superfamily of LGIC includes nicotinic acetylcholine (nACh), GABAA, glycine, and serotonin (5-HT3) receptors (15). Each has a pentameric arrangement of different subunits about a central, ion-conducting axis (Fig. 1) (16). Each subunit in turn consists of a large N-terminal extracellular domain, and four helical transmembrane (TM) regions (M1–M4). The amino-acid residues in M2 subunit line the ion channel and the residues in M3 and M4 interact with the lipid layers (12,17). The atomic structure of the closed form of nACh receptor at 4 Å resolution has been published recently (18,19). The structure was obtained by electron microscopy from the muscle-derived electric organ of Torpedo marmarota membranes. In general, anesthetics potentiate the effect of agonist on GABAA and glycine receptors, and strongly inhibit the nACh receptors (20). In searching for the anesthetic binding site underlying these effects, photolabeling experiments have been conducted on the nACh receptors (21), in particular α- and δ-subunits. Halothane was found to label residues in the TM regions and an agonist-dependent, tyrosine-containing site was found between TM-1 and -3 in the δ-subunit (20).
FIGURE 1.

Crystal structure of acetylcholine receptor from the Protein DataBank (PDB ID: 1OED) showing the pentameric nature of the assembly and the transmembrane region within each subunit. M2 is the channel-lining helix of each subunit.
Molecular dynamics (MD) simulations have been used to investigate the distribution of small molecules such as cholesterol (22,23) and halothane (24–27) within model membranes. However, in these studies, the molecules were either grown in the bilayer or substituted for some lipid molecules in the initial configuration. Simulation studies have also been conducted to investigate effects of halothane on proteins in water, such as synthetic α-helix bundles (28), and on gramicidin A in lipid membranes (29). Simulations of the LGIC have been limited because of the lack of high-resolution structural data to set reliable starting conditions. Because of the recent publication of the high-resolution cryoEM structure of the nicotinic receptor (18,19), combined with the experimental evidence of possible binding sites in these proteins (20) and the availability of tetrascale computing platforms, it is now feasible to characterize the interactions between anesthetic molecules and membrane proteins in their native environment using MD simulations (30–33).
Accordingly, MD simulations have been carried out to investigate membrane-bound α-helical peptide bundles based on the α- and δ-subunits of nAChR in a di-oleoyl-phosphatidyl-choline (DOPC) lipid bilayer with the aim to study the interaction and possible binding of the inhalational anesthetic halothane (CF3CBrClH) with these membrane-bound species.
METHODS
Description of the systems
The initial coordinates of the two subunits were taken from the PDB structure 1OED. A small preequilibrated patch of 72 DOPC lipids was replicated to generate a 288-lipid system. The system was then solvated using the solvate plug-in of VMD (34) ensuring adequate hydration of the DOPC lipid bilayer (number of water molecules/lipid nw/l = 32.8) (35). The DOPC system was equilibrated for 2 ns and the area per lipid and lamellar spacing were monitored. From this equilibrated system, a patch of 147 DOPC lipid molecules was extracted that would be sufficient to wrap the target four-helix peptide bundles, and this patch was further equilibrated for 4 ns. The peptide bundles were then inserted into this preequilibrated DOPC lipid bilayer system with the initial orientation being nearly parallel to the bilayer normal. To maintain charge neutrality, two sodium and two chlorine counterions were added to the α- and δ-nAChR-DOPC systems, respectively, using the Autoionize plug-in of VMD. Both simulation systems were subjected to energy minimization, followed by a short run of 500 ps in which the peptide bundles were held fixed to allow the adjustment of lipid and water. The minimized systems were then subjected to 3 ns of constant pressure and temperature (NPT) ensemble molecular dynamics runs before the anesthetic molecules were introduced. Next, 10 halothane molecules were placed in the aqueous phase on both sides of the equilibrated DOPC-bound α- and δ-nAChR peptide bundles, by removing overlapping water molecules. To improve the statistics and efficient anesthetic sampling in the bilayer, the concentration of halothane molecules used in the simulation was ∼8.5% (nhal/nDOPC), which is somewhat higher than that achieved at typical clinical concentrations (∼3%). Previous simulations studying the effects of halothane on pure lipids compared high and low concentration simulations, and found qualitative agreement between the resulting anesthetic distributions and effects (24,27). For comparison, simulations were also performed on halothane-free systems. Each system was then subjected to additional energy minimization runs using conjugate gradient method, followed by equilibration runs.
Molecular dynamics simulations
MD simulations were performed using the NAMD2 (36) software package, on the Intel Xeon 3.2 GHz processor machine Tungsten at the National Center for Supercomputing Applications. The Nosé-Hoover method with Langevin dynamics and Langevin piston were applied to maintain a pressure of 1 bar and temperature of 305 K. A multiple time step (37,38) was used and all the hydrogen atoms were constrained using the RATTLE algorithm to allow a time step of 1.5 fs. The CHARMM 22 (39) and CHARMM 27 (40) force fields were used for the peptide bundles and lipids, respectively, and water was modeled by TIP3P (41). Parameters developed by Scharf and Laasonen (42) were employed for halothane molecules. A cutoff distance of 12 Å and a pair-list distance of 15 Å were used to compute all nonbonded interactions and periodic boundary conditions were imposed. The full electrostatics interactions were computed with the particle-mesh Ewald method with a tolerance of 10−6 and updated every two time steps. All the analysis was performed within the software environment of VMD.
Free energy calculations
Steered molecular dynamics (SMD) simulations (43,44), using NAMD2, were also carried out on a system consisting of a single halothane molecule dragged from the DOPC lipid into the water phase. SMD calculations involve the application of external forces to accelerate the processes to overcome energy barriers. SMD calculations allow construction of a potential of mean force (PMF) or free energy difference estimated from the work (W) values using Jarzynski's equality (44,45) as
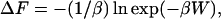 |
where β ≡ 1/kBT and W is the external work. SMD simulations have been used in various applications including study of transport of glycerol molecules through the aquaporin ion channel (46), binding and unbinding of adhesion proteins (47), and unfolding linkers between helical repeats in spectrinlike proteins (48,49). For the calculation of free energy profile, a single halothane molecule was placed in bulk water region outside of a preequilibrated patch of 72 DOPC molecules. The halothane molecule was spontaneously inserted into the bilayer in <2 ns and was located just below the headgroup region of the lipid bilayers similar to the other systems described in this study. The system was further equilibrated for another eight nanoseconds before starting SMD simulations to better equilibrate the halothane in the bilayer. Sixteen random snapshots were taken from the last two nanoseconds of the equilibration run as starting configurations for SMD runs. These 16 configurations take into account different orientations of the halothane molecule inside the lipid bilayer and are likely to give improved statistics for free energy profile calculations. In the SMD simulations reported here, external forces were applied to the center of mass of the halothane molecule, moving it in a direction parallel to the normal of the bilayer (z) with a constant velocity of 3.33 × 10−3Å/ps into the aqueous phase with a force constant of 10 Kcal/mol Å2, which is large enough to allow for stiff spring approximation (43). The pulling velocity used in this study is considerably lower than values used in other SMD simulations such as acetylcholine unbinding from ligand-binding domain (50), unbinding of retinoic acid from its receptor (51), and extraction of lipids from phospholipid membranes (52), justifying our construction of the PMF from the number of trajectories used in this study.
For the analysis of SMD runs, two extrapolation methods developed by Zuckerman and co-workers, which estimate the free energy values from a relatively short set of work values (45,53), were used. Both methods rely on the blocked averages of free energies obtained through Jarzynski's equality by extrapolating to infinite data limits. In linear extrapolation, an estimate of free energy is obtained by exploiting the monotonically changing nature of the blocked averages. The cumulative integral extrapolation method improves on the linear extrapolation method by using an integration scheme to obtain free energy estimates using 5–40 times less data than traditional Jarzynski's approach.
RESULTS AND DISCUSSION
Characterization of membrane-protein system
The α- and δ-nAChR peptide bundles were equilibrated for 3 ns in the DOPC lipid system before introduction of the halothane molecules. Root mean-squared deviation (RMSD) is used to monitor the structural similarity of the subunits in the lipid environment with the cryo-EM “parent.” The RMSD was calculated by superimposing all frames of the simulation on to the relevant 1OED subunit coordinates. After the initial increase in the RMSD values due to the optimization of the subunits in the lipid environment (and with the absence of other subunits), the RMSD values leveled to a value of ∼3.3 Å and ∼3.0 Å for the α- and δ-nAChR peptide bundles, respectively. We also monitored the individual helix RMSD values and found that helices M1 and M3 had lower values compared to M2 and M4 in both α- and δ-nAChR peptide bundles. The M2 helix of the subunits forms the pore of the nACh receptor (as shown in Fig. 1) so the absence of other subunits in our simulations may be responsible for the higher RMSD values for M2 helix in both the subunits. The intracellular connecting loop between M3 and M4 was not resolved in the crystal structure, so these residues were not included in our simulations. The lack of connection between M4 and M3 may have resulted in the higher RMSD for the M4 helix. Root mean-square fluctuation (RMSF) provides information about the residue mobility relative to the average structure, analogous to crystallographic B-factors. The two-peptide bundles structures are stable with RMSF values <1 Å for most of the residues except for the terminal and loop residues. The M2 helix adopts a more kinked conformation in the vicinity of Leu-251 compared to the crystal structure, as was observed in simulations involving only M2δ peptides (54). The tilt angle of the subunit was measured as the angle between the smallest component of the moment of inertia tensor and the bilayer normal. The asymptotic tilt angle for both the subunits in the lipid bilayer was ∼24°, consistent with the mean tilt angle of 21° reported from the analysis of 45 transmembrane helix x-ray structures (55). The interhelix distance, which reflects the stability of the bundle itself, was also measured as an average of the Cα-Cα distance of helices and remained more or less constant during the equilibration process.
Partition of halothane in lipid bilayers
In previous simulations (24–27,29) of the distribution of halothane molecules in bilayers in saturated and polysaturated lipid bilayers, the halothane molecules were initially placed inside the bilayer, either at locations consistent with experimental predictions, or were distributed evenly. In this MD study a different approach was adopted, in which the halothane molecules were initially placed in the water, close to one or another of the lipid bilayer surfaces. Snapshots of α- and δ-nAChR-DOPC simulation systems at time t = 0 and 16 ns, respectively, are shown in Fig. 2. The initial placement of the halothane molecules in both systems was identical, with five halothane molecules on each side of the bilayer. At the end of 16 ns, all of the halothane molecules had partitioned into the bilayers, most of the partitioning occurring in the first 5 ns of simulation. In the α-nAChR-DOPC system, eight and two halothane molecules partitioned into top and bottom layers, respectively, and in the δ-nAChR-DOPC system, six and four halothane molecules partitioned into top and bottom layers, respectively. This difference is not statistically significant. Fig. 3 shows the trajectories of the 10-halothane molecules in the case of the α- and δ-nAChR-DOPC systems, in which all the halothane molecules were spontaneously inserted into the bilayer.
FIGURE 2.

Snapshots of bilayer-subunit-halothane systems at (a) t = 0, (b) t = 16 ns. The subunits are shown in ribbon representation. Shown also are the tyrosine residues and lipids in licorice and line representation, respectively. Lipid hydrogen atoms and water molecules are not shown for clarity. The halothane molecules are represented as van der Waals spheres. The cluster formed by halothane molecules 4, 5, and 9 is also highlighted.
FIGURE 3.

The trajectories of center-of-mass positions of individual halothane molecules over the entire simulation with the starting configuration as in Fig. 2 in the z direction (bilayer normal). The center-of-mass position of lipid headgroups and carbon atom C5 of alkyl chains are also shown. The halothane molecule 3 shown in panel a migrates from bottom leaflet to the top leaflet and 6 entered the α-subunit and remained stable. The initial periodic wrapping of trajectories for bilayer crossing is removed for clarity.
The distribution of halothane molecules in δ-nAChR-DOPC is more symmetrical with respect to the two leaflets of the bilayer than in the α-nAChR-DOPC system (Figs. 2 and 3). In both cases, halothane molecules were predominantly located around the carbon atoms C2–C5 of the hydrocarbon chains. This is consistent with the previous simulations on pure lipid systems (24–27) and NMR findings (56) that anesthetic molecules are preferentially located near the complex lipid-water interface. In the α-nAChR-DOPC system, the distribution of halothane molecules at the center of the bilayer is not zero, due to the migration of a single halothane molecule from bottom layer to top layer in the lipid membrane. In the Fig. 4, the density profiles of various components of the systems averaged over last the 10 ns are shown. The peak positions of halothane distribution and C5 carbon atoms of the alkyl chains overlap in α-nAChR-DOPC and δ-nAChR-DOPC systems. The atom distributions of C5 and C9 (representing the double-bond) appear to be different for the two systems, which is likely due to the difference in halothane distribution in the two systems. Also in the α-nAChR-DOPC, significant thinning (∼5 Å) of the bilayer was observed. We also observed migration of halothane molecule 3 along the bilayer normal from the bottom leaflet to the top, via the hydrocarbon core in the α-nAChR-DOPC system, in agreement with the results for a coarse-grain model at low anesthetic concentrations (27). In the δ-nAChR-DOPC system, halothane molecules 4, 5, and 9 formed a cluster and stabilized above the headgroup region in the hydrocarbon alkyl chain region of the bottom leaflet (see Fig. 2). Similar behavior of pairing of anesthetics was observed in previous simulations of Tang et al. (29). The cluster essentially behaves like a nonanesthetic molecule (26) due to partial cancellation of the dipole moment, and favors the apolar region of hydrophobic chains compared to the polar lipid-water interface. In the α-nAChR-DOPC system, halothane molecule 6 stays just above the headgroup region, due to the binding of 6 in the vicinity of Tyr-277, which will be described in more detail below.
FIGURE 4.

The density profiles along the bilayer normal, z, averaged over last 10-ns of the MD trajectory for α-nAChR and δ-nAChR subunits. The z = 0 corresponds to the bilayer normal. The density of the headgroups, alkyl chains, and C9 carbon represent the location of double-bond in the alkyl chains; the C5 carbon, representing the most probable location of halothane molecules along with protein and halothane molecules, is also shown.
Free energy profile of halothane across DOPC bilayer
Several small, uncharged molecules such as carbon dioxide, oxygen, and anesthetic molecules diffuse into cell membranes without the help of any membrane proteins. Encouraged by the observed spontaneous insertion of the halothane molecules into the lipid bilayer, the free energy profile of the halothane molecules was computed. Excess chemical potentials of small solute molecules, both polar and nonpolar, have been calculated in previous computational studies (57,58). Free energy calculations of transport of small neutral molecules such as water, oxygen, and chloroform across dimyristoylphosphatidylcholine lipid bilayers using cavity-insertion Widom calculations (59) have been performed before (60). We report here the results of steered molecular dynamics (SMD) simulations of halothane molecule extraction from the DOPC lipid into aqueous phase. From the 16 different SMD runs, it was observed that the deviation in work values increased with the distance of the halothane molecule from its equilibrium position, especially when the halothane molecule reaches the aqueous phase. To estimate the PMF, two extrapolation methods (53) described earlier were used, in addition to the exponential average method. The linear and cumulative extrapolation methods were developed to address the issues of improving the PMF estimation in case of limited sampling. The extrapolation methods have also been used recently to estimate the binding energy of acetylcholine (50) in which similar deviation in work values was observed. The PMF constructed from the time analysis of the 16 SMD runs using exponential and these extrapolation methods is shown in Fig. 5. The distance is measured from the equilibration location of the halothane molecules, which is just below the headgroup of the DOPC bilayer.
FIGURE 5.

The PMF profiles of a single halothane molecule extracted from its equilibrium position in DOPC lipid bilayer into the solution computed from 16 individual steered molecular dynamics simulations using Jarzynski's equality of exponential average (black solid), block exponential average (gray dashed), linear (light gray dashed), and cumulative integral (black dashed) methods. The origin represents the equilibrated position of the halothane molecule, which is below the headgroup of the DOPC lipid bilayer. Inset is the demonstration of stiff spring approximation showing the trajectories of constraint (gray) and center of mass of halothane (black).
From Fig. 5 it can be seen that the PMF estimated from the extrapolation methods is more rugged in nature and is lower than the exponential average estimate using Jarzynski's equality. Similar findings were observed in a recent work of estimation of binding energy of a ligand (50). The cumulative integral extrapolation PMF profile reveals that external work of several Kcal/mol is required to extract an equilibrated halothane molecule from the lipid bilayer into the solution phase. The shape of the PMF calculated here corresponds well with the previous free energy calculations of 1,1,2-trifluoroethane across the water-hexane interface (57). The PMF profile has a minima just near the interface at the starting position, confirming the preference of anesthetic molecules toward the complex interfacial region consisting of both lipid and water phases. The plot also reveals that the halothane molecules experience a considerably higher free energy barrier upon leaving the membrane. The linear regime, however, should be considered with caution since this is the region of the membrane with the slowest relaxation times. On the other hand, the free energy barrier that the halothane molecule has to cross to enter the headgroup region of the bilayer from the aqueous phase, is small at ∼1 Kcal/mol, which is consistent with the observed spontaneous insertion.
Halothane effects on the lipids
Lipid-protein interactions may play an important functional role in processes such as gating and desensitization of membrane channel proteins. Indeed, lipid-protein complexes have been shown to affect the properties of lipid bilayers with embedded peptide bundles (33). The effects of inhalational anesthetics such as halothane on the lipid-protein interface takes on a special significance in view of the most probable location of these molecules, which is close to the headgroup region of the lipids as shown in this study and many others. Experiments have demonstrated an association between the lipid-disordering activity of general anesthetics and desensitization of acetylcholine receptors (61). In this study, the number of lipid-protein contacts was measured as a function of time in the presence and absence of halothane molecules and were defined as the number of lipids within a distance of 3.5 Å of the protein. A decrease of 17% and 8% in the lipid-protein contacts was observed in the α-nAChR-DOPC and δ-nAChR-DOPC, respectively. These results indicate that low-affinity ligands like halothane might exert significant effects on the protein function by affecting lipid-protein interactions.
Anesthetic molecules are also considered to affect different membrane properties, which in turn can affect the membrane protein functions (11). As mentioned previously, the halothane molecules in α-nAChR and δ-nAChR DOPC distribute preferentially just below the headgroup region in both leaflets. The dipole moment of halothane molecules was observed to align in a direction perpendicular to the bilayer normal and along the lipid headgroups. The charges used in the present simulation were taken from Scharf et al. (42) and the dipole moment of the halothane molecule is ∼2 Debye. Previous NMR experiments (62,63) have shown that the anesthetic halocarbons such as halothane with higher dipole moments aggregate preferentially near the complex headgroup region compared to nonanesthetic halocarbons with lower dipole moment. However, in the case of α-nAChR DOPC system, the distribution is broader and more uneven with 80% of halothane molecules in the top layer. The effect of halothane distribution on the alkyl chain order parameter and alkyl chain tilt angles for DOPC systems with α- and δ-subunits was measured and is shown in Fig. 6. The order parameters are averaged over both sn-1 and sn-2 chains of DOPC lipid and the characteristic dip at carbon C9 due to the presence of a double bond. The partitioning of the halothane molecules decreases the order parameters, similar to the results obtained in pure lipid systems (25). The decrease is larger in the case of α-nAChR-DOPC, where the order of carbon atoms close to the headgroup is affected more due to the predominant partitioning of halothane molecules in the upper leaflet (see Fig. 3 a), which is not the case in the lower leaflet, or in the δ-nAChR-DOPC system. The average tilt angle of both hydrocarbon chains is defined as the angle between the vector joining C2–C18 carbons and the membrane normal. The tilt angles were averaged over the two leaflets and for both the sn-1 and sn-2 chains of DOPC alkyl chains. As can be seen from the Fig. 6 b, the presence of halothane molecules in both systems shifts the average orientation of the alkyl chains and also makes the distribution broader, although the shapes of the distribution do not differ much. The most probable values of the tilt angles increase in the presence of halothane molecules, which is likely because of the disordering of the chains. The density profiles of the carbon atoms C2 to C18 are plotted in Fig. 7 to illustrate the loss of order in the alkyl chains with the partitioning of halothane molecules.
FIGURE 6.

The effect of the partitioning of the halothane molecules on the lipid alkyl chains for α-subunit (a, b) and δ-subunit (c, d). (a, c) Comparison of the deuterium order parameter (SCD) of the alkyl chains in DOPC in the presence and absence of halothane molecules averaged over the last 10-ns of the simulations. (b, d) Comparison of the distribution of the tilt angles of the alkyl chains in both the leaflets in the presence and absence of halothane molecules.
FIGURE 7.

The density profiles of carbon atoms C2–C18 of the alkyl chains for the α-subunit-DOPC system in the absence (a, b) and presence of halothane (c, d). The profiles are calculated for the last 10-ns of both simulations.
Halothane effects on α- and δ-subunit structure
The structural stability of the α-and δ- subunit structures was evaluated by the RMSD and RMSF calculations. The presence of halothane molecules has no detectable effect on the secondary structure of either subunit, as seen in Fig. 8, which is similar to the previous simulation results on gramicidin A channels (29). To understand the effect of halothane molecules on the local dynamics, RMSF profiles of both subunits in the presence and absence of the halothane molecules were calculated and is shown in Fig. 9. To compute the RMSF values, all the frames of the simulation run were fit to a common reference point (the initial configuration). This removes the effects of any translational and rotational motions in the molecule. As expected, the terminal residues (beginning of M1, end of M3, beginning and end of M4) have higher RMSF values (were more mobile) as compared to the central residues in both subunits. The presence of halothane molecules has significant effects on the local dynamics of both subunits. As described in the previous section, a single halothane molecule 6 enters the α-subunit and stays in the groove between helices M3 and M4, near Tyr-277 and the M2-M3 loop (Figs. 2 and 3). This halothane molecule appears to reduce the flexibility of the M2-M3 loop, as well as the mobility of residues in close proximity. For example, residues in the M3 and M4 helices have reduced RMSF values in the presence of 6 (Fig. 6 a). Most striking, however, is the difference in the M2-M3 loop structure in the presence and absence of halothane 6, as shown in Fig. 10, which also shows the proximity of the halothane molecule to Tyr-277, analogous to the photolabeled site (20).
FIGURE 8.

The root mean-squared deviation (RMSD) of the backbone Cα atoms in subunits in the presence and absence of halothane molecules: panels a and c for M1, M2, M3 helices in α- and δ-, respectively; and panels b and d M4 helix in α- and δ-, respectively.
FIGURE 9.

Comparison of root mean-squared fluctuations (RMSF) of the backbone Cα atoms in (a) α-subunit and (b) δ-subunit in the presence and absence of halothane molecules.
FIGURE 10.

Molecular representation of the effect of halothane molecule 6 on the dynamics of the loop connecting helices M2 and M3 in the α-subunit. The helices of the subunit are color-coded: no halothane (red) and after binding of halothane 6 (16-ns snapshot) (cyan). The hydrophobic residues in the binding cavity are shown in green. The halothane 6 reduces the mobility of the loop and the aromatic residue Tyr-277.
Although exhibiting high mobility and a lack of preferred orientation, the halothane molecule 6 enters the α-subunit and occupies a position, which is consistently closer to the Tyr-277 residue throughout the simulation run. The evolution of distance between center of mass of Tyr-277 residue and center of mass of halothane 6 in α-subunit is shown in Fig. 11 a. Two angles were calculated to characterize the orientation of the Tyr-277 residue in the presence and absence of halothane molecule 6. The notation SN is the angle between the axis normal to the bilayer and a vector perpendicular to the plane of the aromatic ring, while SL is the angle between the bilayer normal and the vector from Cβ to Cα in Tyr. The presence of the halothane molecule is associated with smaller fluctuations in both angles. This quenching of the Tyr-277 residue motion is further confirmed by the significant lowering of the RMSF value from 1.4 to 0.4 in the presence of halothane 6. The anesthetic binding modulation of protein motion is consistent with our previous simulations on four helix bundle/ halothane complexes (28), and with simulations of the gramicidin receptor (29).
FIGURE 11.

The binding of halothane molecule 6 near the aromatic residue Tyr-277 and its effects on the motion of the aromatic ring. The time-evolution of (a) the distance between the center of mass of the Tyr-277 residue and the halothane molecule 6 in α-subunit; (b) SN, the angle between the vector perpendicular to the aromatic plane of Tyr-277 and the bilayer normal (c) SL; and the angle between the vector connecting Cβ and Cα of Tyr-277 and the bilayer normal.
Protein flexibility is a vital aspect of the relationship between protein structure and function, as it facilitates the ability to explore energy landscapes, sampling different conformations, and binding and unbinding of different ligands (64). It was previously shown in experiments on mammalian protein bovine serum albumin that the anesthetic (halothane and isoflurane) binding causes attenuation of local side-chain dynamics and stabilization of folded protein conformation (65,66). In this study, similar observations were made regarding the effect of halothane on the protein structure. In the case of the δ-subunit, where no binding of halothane molecules occurred in the simulation time frame, the effects of halothane on the mobility, and consequently the flexibility, of different parts of the subunit were negligible. However, in the case of the α-subunit, a single binding event significantly alters the flexibility of the loop connecting the two transmembrane helices and the residues in respective helices.
To get a better picture of the interactions between different residues of the protein in the presence and absence of ligands, the cross-correlation matrices (67) were calculated. The cross-correlation (normalized) matrix can help identify regions of the proteins that move in concert and also can reflect the effect of ligands on the dynamical nature of the protein. It can be obtained from the covariance matrix, and for any two atoms i and j, the corresponding covariance matrix element is obtained from the expression
 |
where
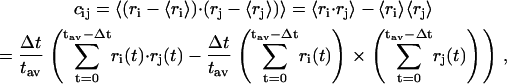 |
and tav is the averaging time, Δt is the time step of the simulation, and angular brackets denote the time averaging. The values range from −1 to +1 representing completely anticorrelated and correlated motions, respectively, and the plots in Fig. 12 show the effect of the presence of the halothane on the correlated motion of different helices. The cross-correlation plots averaged over last 6-ns of simulation are shown for the α-bundle without (Fig. 12, a and b) halothane. It is very clear from the plot that the presence of halothane molecules in the system has enhanced the correlations between the helices. The significant correlations that appear after the halothane binding are highlighted as 1 and 2 in the Fig. 12 b. The halothane molecule 6 binding to the α-bundle to Tyr-277 on helix M3 has favorable van der Waals interactions with helix M4, increasing the correlation between the helices as seen in 2 of Fig. 12 b, and which helps make the α-nAChR system more compact. The compact nature of the bundle with halothane binding is also reflected in reduction of radius of gyration Rg, calculated in the plane of the subunits normal to the pore axis monitored for α-nAChR subunit in the presence and absence of halothane molecules, which gives an estimate of the “breathing” motion of the protein. The compactness of the bundle introduces new correlations between helices M1 and M4, as also seen in Fig. 12 b. The cross-correlation analysis clearly demonstrates the effect of anesthetic binding on the global protein dynamics, which may play a role in altering the protein functions and suggests new mechanisms of molecular nature of anesthetic action on proteins.
FIGURE 12.

The cross-correlation plots for the α-subunit (a) without and (b) with binding of halothane. The plot is constructed from time average over the last 6-ns of a total of 16 ns of MD simulation for each system. The color scale is also shown. Significant correlations upon halothane binding are shown as 1 and 2.
CONCLUDING REMARKS
In this MD study, the inhalational anesthetic, halothane, has been observed to spontaneously partition from the aqueous phase into a DOPC lipid bilayer containing a membrane protein. In agreement with early work, the equilibrium distribution of the halothane molecules in the bilayer at the end of a 16-ns MD run shows a preference for the acyl chain/glycerophosphate headgroup interface. The partitioned halothane molecules introduced disorder in the acyl chains, and reduced the lipid-protein contacts. In recently published MD simulations studying the gating mechanism of nicotinic receptor (31), essential dynamics analysis revealed the dynamic nature of the M2-M3 loop and it was proposed in the study that the loop may play a role in gating mechanism by allosterically transmitting the effects to channel gate. The existence of a hydrophobic cavity in the region close to the M2-M3 loop makes it very amenable to binding of anesthetics, which was in fact observed in photolabeling studies of nAChR subunits (20). Mutational studies on neuronal nicotinic receptors in the M2-M3 loop in the α-subunit resulted in significant reduction in halothane sensitivity demonstrating the critical role played by the M2-M3 loop in transduction of anesthetic binding to channel inhibition (68). A major finding of this study is the effect of halothane binding to the α-nAChR peptide bundle on the dynamics of M2-M3 loop. One of the halothane molecules was observed to partition spontaneously from the solution into the hydrophobic cavity close to the M2-M3 loop in the vicinity of αTyr-277 between the helices M3 and M4, which was one of the experimentally photolabeled sites. In addition, the fluctuation of the aromatic plane of the αTyr-277 residue was also significantly affected. Cross-correlation analysis in these simulations revealed that the binding of halothane molecule also significantly affects the correlated motions between the helices. The present observations suggest that the binding of anesthetics can significantly affect the protein function. In the specific case of nicotinic receptors, it can be suggested that halothane binding plays a role in channel inhibition by altering the dynamics of M2-M3 loop, which is implicated in transmitting the effects to the channel gate. This work provides new insight into the interactions between anesthetics, lipids, and membrane proteins. Further computational work with fully assembled nAChR in the presence of cholesterol will be valuable for further validation of these observations.
Acknowledgments
This research was supported in part by National Institutes of Health. Computer time from National Center for Supercomputing Applications under the aegis of National Resource Allocation Committee is greatly appreciated.
References
- 1.Meyer, K. H. 1937. Contribution to the theory of narcosis. Trans. Faraday Soc. 33:1062–1068. [Google Scholar]
- 2.Overton, C. E. 1901. Studien uber die narkose, zugleich ein beitrag zur allegemeiner pharmakologie. Gustav Fischer, Jena, Switzerland. [Studies of Narcosis (translation). 1990. Chapman and Hall, London.]
- 3.Pringle, M. J., and K. W. Miller. 1979. Differential effects on phospholipid phase-transitions produced by structurally related long-chain alcohols. Biochemistry. 18:3314–3320. [DOI] [PubMed] [Google Scholar]
- 4.Ueda, I., T. Tatara, J. S. Chiou, P. R. Krishna, and H. Kamaya. 1994. Structure-selective anesthetic action of steroids—anesthetic potency and effects on lipid and protein. Anesth. Analg. 78:718–725. [DOI] [PubMed] [Google Scholar]
- 5.Franks, N. P., A. Jenkins, E. Conti, W. R. Lieb, and P. Brick. 1998. Structural basis for the inhibition of firefly luciferase by a general anesthetic. Biophys. J. 75:2205–2211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu, R., P. J. Loll, and R. G. Eckenhoff. 2005. Structural basis for high affinity volatile anesthetic binding in a natural 4-helix bundle protein. FASEB J. 19:567–576. [DOI] [PubMed] [Google Scholar]
- 7.Fang, Z., P. Ionescu, B. S. Chortkoff, L. Kandel, J. Sonner, M. J. Laster, and E. I. Eger. 1997. Anesthetic potencies of n-alkanols: results of additivity and solubility studies suggest a mechanism of action similar to that for conventional inhaled anesthetics. Anesth. Analg. 84:1042–1048. [DOI] [PubMed] [Google Scholar]
- 8.Kandel, L., B. S. Chortkoff, J. Sonner, M. J. Laster, and E. I. Eger. 1996. Nonanesthetics can suppress learning. Anesth. Analg. 82:321–326. [DOI] [PubMed] [Google Scholar]
- 9.Koblin, D. D., B. S. Chortkoff, M. J. Laster, E. I. Eger, M. Halsey, and P. Ionescu. 1994. Polyhalogenated and perfluorinated compounds that disobey the Meyer-Overton hypothesis. Anesth. Analg. 79:1043–1048. [DOI] [PubMed] [Google Scholar]
- 10.Cantor, R. S. 1997. The lateral pressure profile in membranes: a physical mechanism of general anesthesia. Biochemistry. 36:2339–2344. [DOI] [PubMed] [Google Scholar]
- 11.Cantor, R. S. 1999. Lipid composition and the lateral pressure profile in bilayers. Biophys. J. 76:2625–2639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Krasowski, M. D., and N. L. Harrison. 1999. General anaesthetic actions on ligand-gated ion channels. Cell. Mol. Life Sci. 55:1278–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mihic, S. J., Q. Ye, M. J. Wick, V. V. Koltchine, M. A. Krasowski, S. E. Finn, M. P. Mascia, C. F. Valenzuela, K. K. Hanson, E. P. Greenblatt, R. A. Harris, and N. L. Harrison. 1997. Sites of alcohol and volatile anaesthetic action on GABAA and glycine receptors. Nature. 389:385–389. [DOI] [PubMed] [Google Scholar]
- 14.Yamakura, T., E. Bertaccini, J. R. Trudell, and R. A. Harris. 2001. Anesthetics and ion channels: molecular models and sites of action. Annu. Rev. Pharmacol. Toxicol. 41:23–51. [DOI] [PubMed] [Google Scholar]
- 15.Ortells, M. O., and G. G. Lunt. 1995. Evolutionary history of the ligand-gated ion channel superfamily of receptors. Trends Neurosci. 18:121–127. [DOI] [PubMed] [Google Scholar]
- 16.Karlin, A. 2002. Emerging structure of the nicotinic acetylcholine receptors. Nature. 3:102–114. [DOI] [PubMed] [Google Scholar]
- 17.Corringer, P.-J., N. L. Novere, and J.-P. Changeux. 2000. Nicotinic receptors at the amino acid level. Annu. Rev. Pharmacol. Toxicol. 40:431–438. [DOI] [PubMed] [Google Scholar]
- 18.Miyazawa, A., Y. Fujiyoshi, and N. Unwin. 2003. Structure and gating mechanism of the acetylcholine receptor pore. Nature. 423:949–955. [DOI] [PubMed] [Google Scholar]
- 19.Unwin, N. 2005. Refined structure of the nicotinic acetylcholine receptor at 4 Å resolution. J. Mol. Biol. 346:967–989. [DOI] [PubMed] [Google Scholar]
- 20.Chiara, D. C., L. J. Dangott, R. G. Eckenhoff, and J. B. Cohen. 2003. Identification of nicotinic acetylcholine receptor amino acids photolabeled by the volatile anesthetic halothane. Biochemistry. 42:13457–13467. [DOI] [PubMed] [Google Scholar]
- 21.Eckenhoff, R. G. 1996. An inhalational anesthetic binding domain in the nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 93:2807–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Chiu, S. W., E. Jakobsson, R. J. Mashl, and L. H. Scott. 2002. Cholesterol-induced modifications in lipid bilayers: a simulation study. Biophys. J. 83:1842–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tu, K. C., M. L. Klein, and D. J. Tobias. 1998. Constant-pressure molecular dynamics investigation of cholesterol effects in a dipalmitoylphosphatidylcholine bilayer. Biophys. J. 75:2147–2156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu, K. C., M. Tarek, M. L. Klein, and D. Scharf. 1998. Effects of anesthetics on the structure of a phospholipid bilayer: molecular dynamics investigation of halothane in the hydrated liquid crystal phase of dipalmitoylphosphatidylcholine. Biophys. J. 75:2123–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Koubi, L., M. Tarek, M. L. Klein, and D. Scharf. 2000. Distribution of halothane in a dipalmitoylphosphatidylcholine bilayer from molecular dynamics calculations. Biophys. J. 78:800–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koubi, L., L. Saiz, M. Tarek, D. Scharf, and M. L. Klein. 2003. Influence of anesthetic and nonimmobilizer molecules on the physical properties of a polyunsaturated lipid bilayer. J. Phys. Chem. B. 107:14500–14508. [Google Scholar]
- 27.Pickholz, M., L. Saiz, and M. L. Klein. 2005. Concentration effects of volatile anesthetics on the properties of model membranes: a coarse-grain approach. Biophys. J. 88:1524–1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Davies, L. A., Q. F. Zhong, M. L. Klein, and D. Scharf. 2000. Molecular dynamics simulation of four-α-helix bundles that bind the anesthetic halothane. FEBS Lett. 478:61–66. [DOI] [PubMed] [Google Scholar]
- 29.Tang, P., and Y. Xu. 2002. Large-scale molecular dynamics simulations of general anesthetic effects on the ion channel in the fully hydrated membrane: the implication of molecular mechanisms of general anesthesia. Proc. Natl. Acad. Sci. USA. 99:16035–16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Henchman, R. H., H.-L. Wang, S. M. Sine, and P. Taylor. 2005. Ligand-induced conformational change in the seven nicotinic receptor ligand binding domain. Biophys. J. 88:2564–2576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Law, R. J., R. H. Henchman, and J. A. McCammon. 2005. A gating mechanism proposed from a simulation of a human seven-nicotinic acetylcholine receptor. Proc. Natl. Acad. Sci. USA. 102:6813–6818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Saiz, L., and M. L. Klein. 2005. The transmembrane domain of the acetylcholine receptor: insights from simulations on synthetic peptide models. Biophys. J. 88:959–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Saiz, L., S. Bandyopadhyay, and M. L. Klein. 2004. Effect of the pore region of a transmembrane ion-channel n the physical properties of a simple membrane. J. Phys. Chem. B. 108:2608–2613. [Google Scholar]
- 34.Humphrey, W., A. Dalke, and K. Schulten. 1996. VMD: visual molecular dynamics. J. Mol. Graph. 14:33–38. [DOI] [PubMed] [Google Scholar]
- 35.Nagle, J. F., and S. Tristram-Nagle. 2000. Structure of lipid bilayers. Biochim. Biophys. Acta. 1469:159–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kale, L., R. Skeel, M. Bhandarkar, R. Brunner, A. Gursoy, N. Krawetz, J. Philips, A. Shinozaki, K. Varadarajan, and K. Schulten. 1999. Molecular dynamics programs design. NAMD2: greater scalability for parallel molecular dynamics. J. Comput. Phys. 151:283–312. [Google Scholar]
- 37.Martyna, G. J., M. E. Tuckerman, D. J. Tobias, and M. L. Klein. 1996. Explicit reversible integrators for extended systems. Mol. Phys. 87:1117–1157. [Google Scholar]
- 38.Tuckerman, M. E., and G. J. Martyna. 2000. Understanding modern molecular dynamics: techniques and applications. J. Phys. Chem. B. 104:159–178. [Google Scholar]
- 39.MacKerell, A. D., D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiorkiewicz-Kuczera, D. Yin, and M. Karplus. 1998. All-atom empirical potential for molecular modeling and dynamics studies of proteins. J. Phys. Chem. B. 102:3586–3616. [DOI] [PubMed] [Google Scholar]
- 40.Feller, S. E., and A. D. MacKerell. 2000. An improved empirical potential energy function for molecular simulations of phospholipids. J. Phys. Chem. B. 104:7510–7515. [Google Scholar]
- 41.Jorgensen, W. L., J. Chandrasekhar, J. D. Madura, R. W. Impey, and M. L. Klein. 1983. Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79:926–935. [Google Scholar]
- 42.Scharf, D., and K. Laasonen. 1996. Structure, effective pair potential and properties of halothane. Chem. Phys. Lett. 258:276–282. [Google Scholar]
- 43.Park, S., F. Khalili-Araghi, E. Tajkhorshid, and K. Schulten. 2003. Free energy calculation from steered molecular dynamics simulations using Jarzynski's equality. J. Chem. Phys. 119:3559–3566. [Google Scholar]
- 44.Park, S., and K. Schulten. 2004. calculating potentials of mean force from steered molecular dynamics simulations. J. Chem. Phys. 120:5946–5961. [DOI] [PubMed] [Google Scholar]
- 45.Jarzynski, C. 1997. Nonequilibrium equality for free energy differences. Phys. Rev. Lett. 78:2690–2693. [Google Scholar]
- 46.Jensen, M., S. Park, E. Tajkhorshid, and K. Schulten. 2002. Energetics of glycerol conduction through aquaglyceroporin GlpF. Proc. Natl. Acad. Sci. USA. 99:6731–6736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bayas, M. V., K. Schulten, and D. Leckband. 2003. Forced detachment of the CD2–CD58 complex. Biophys. J. 84:2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ortiz, V., S. O. Nielsen, M. L. Klein, and D. E. Discher. 2005. Unfolding a linker between helical repeats. J. Mol. Biol. 349:638–647. [DOI] [PubMed] [Google Scholar]
- 49.Grubmüller, H., B. Heymann, and P. Tavan. 1996. Ligand binding and molecular mechanics calculation of the streptavidin-biotin rupture force. Science. 271:997–999. [DOI] [PubMed] [Google Scholar]
- 50.Zhang, D., J. Gullingsrud, and J. A. McCammon. 2006. Potentials of mean force for acetylcholine unbinding from the α7 nicotinic acetylcholine receptor ligand-binding domain. J. Am. Chem. Soc. 128:3019–3026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kosztin, D., S. Izrailev, and K. Schulten. 1997. Unbinding of retinoic acid from its receptor studied by steered molecular dynamics. Biophys. J. 76:188–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Stepaniants, S., S. Izrailev, and K. Schulten. 1997. Extraction of lipids from phospholipid membranes by steered molecular dynamics. J. Mol. Model. (Online). 2:473–475. [Google Scholar]
- 53.Ytreberg, M. F., and D. M. Zuckerman. 2004. Efficient use of nonequilibrium measurement to estimate free energy differences for molecular systems. J. Comput. Chem. 25:1749–1759. [DOI] [PubMed] [Google Scholar]
- 54.Law, R. J., D. P. Tieleman, and M. S. P. Sansom. 2003. Pores formed by the nicotinic receptor M2 peptide: a molecular dynamics simulation study. Biophys. J. 84:14–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bowie, J. U. 1997. Helix packing in membrane proteins. J. Mol. Biol. 272:780–789. [DOI] [PubMed] [Google Scholar]
- 56.Tang, P., B. Yan, and Y. Xu. 1997. Different distribution of fluorinated anesthetics and non-anesthetics in model membranes: an 19FNMR study. Biophys. J. 72:1676–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pohorille, A., and M. A. Wilson. 1996. Excess chemical potential of small solutes across water-membrane and water-hexane interfaces. J. Phys. Chem. 104:3760–3773. [DOI] [PubMed] [Google Scholar]
- 58.Marrink, S.-J., and J. C. Berendsen. 1994. Simulation of water transport through a lipid membrane. J. Phys. Chem. 98:4155–4168. [Google Scholar]
- 59.Widom, B. 1963. Some topics in the theory of fluids. J. Chem. Phys. 39:2808–2812. [Google Scholar]
- 60.Jedlovsky, P., and M. Mezei. 2000. Calculation of the free energy profile of H2O, O2, CO, CO2, NO, CHCl3 in a lipid bilayer with a cavity insertion variant of the Widom method. J. Am. Chem. Soc. 122:5125–5131. [Google Scholar]
- 61.Firestone, L. L., J. K. Alifimoff, and K. W. Miller. 1994. Does general anesthetic-induced desensitization of the torpedo acetylcholine receptor correlate with lipid disordering? Mol. Pharmacol. 46:508–515. [PubMed] [Google Scholar]
- 62.Baber, J., J. F. Ellena, and D. Cafiso. 1995. Distribution of general anesthetics in phospholipid bilayers determined using 2H NMR and 1H-1H NOE spectroscopy. Biochemistry. 34:6533–6539. [DOI] [PubMed] [Google Scholar]
- 63.North, C., and D. Cafiso. 1997. Contrasting membrane localization and behavior of halogenated cyclobutanes that follow or violate the Meyer-Overton hypothesis of general anesthetic potency. Biophys. J. 72:1754–1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Frauenfelder, H., S. G. Sligar, and P. G. Wolynes. 1991. The energy landscapes and motions of proteins. Science. 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 65.Johansson, J. S., H. Zou, and J. W. Tanner. 1999. Bound volatile general anesthetics alter both local protein dynamics and global protein stability. Anesthesiology. 90:235–245. [DOI] [PubMed] [Google Scholar]
- 66.Eckenhoff, R. G. 1998. Do specific or nonspecific interactions with proteins underlie inhalational anesthetic action? Mol. Pharmacol. 54:610–615. [PubMed] [Google Scholar]
- 67.Hunenberger, P. H., A. E. Mark, and W. F. van Gunsteren. 1995. Fluctuations and cross-correlation analysis of protein motions observed in nanosecond molecular dynamics simulations. J. Mol. Biol. 252:492–503. [DOI] [PubMed] [Google Scholar]
- 68.Downie, D. L., F. Vincente-Agullo, A. Campos-Caro, T. J. Bushell, W. R. Lieb, and N. P. Franks. 2002. Determinants of the anesthetic sensitivity of neuronal nicotinic acetylcholine receptors. J. Biol. Chem. 277:10367–10373. [DOI] [PubMed] [Google Scholar]