

Abstract
The three-dimensional backbone structure of the transmembrane domain of Vpu from HIV-1 was determined by solid-state NMR spectroscopy in two magnetically-aligned phospholipid bilayer environments (bicelles) that differed in their hydrophobic thickness. Isotopically labeled samples of Vpu2–30+, a 36-residue polypeptide containing residues 2–30 from the N-terminus of Vpu, were incorporated into large (q = 3.2 or 3.0) phospholipid bicelles composed of long-chain ether-linked lipids (14-O-PC or 16-O-PC) and short-chain lipids (6-O-PC). The protein-containing bicelles are aligned in the static magnetic field of the NMR spectrometer. Wheel-like patterns of resonances characteristic of tilted transmembrane helices were observed in two-dimensional 1H/15N PISEMA spectra of uniformly 15N-labeled Vpu2–30+ obtained on bicelle samples with their bilayer normals aligned perpendicular or parallel to the direction of the magnetic field. The NMR experiments were performed at a 1H resonance frequency of 900 MHz, and this resulted in improved data compared to lower-resonance frequencies. Analysis of the polarity-index slant-angle wheels and dipolar waves demonstrates the presence of a transmembrane α-helix spanning residues 8–25 in both 14-O-PC and 16-O-PC bicelles, which is consistent with results obtained previously in micelles by solution NMR and mechanically aligned lipid bilayers by solid-state NMR. The three-dimensional backbone structures were obtained by structural fitting to the orientation-dependent 15N chemical shift and 1H-15N dipolar coupling frequencies. Tilt angles of 30° and 21° are observed in 14-O-PC and 16-O-PC bicelles, respectively, which are consistent with the values previously determined for the same polypeptide in mechanically-aligned DMPC and DOPC bilayers. The difference in tilt angle in C14 and C16 bilayer environments is also consistent with previous results indicating that the transmembrane helix of Vpu responds to hydrophobic mismatch by changing its tilt angle. The kink found in the middle of the helix in the longer-chain C18 bilayers aligned on glass plates was not found in either of these shorter-chain (C14 or C16) bilayers.
INTRODUCTION
Although the vast majority of membrane-associated proteins are helical (1), structures of members of this class of proteins are significantly underrepresented in the Protein Data Bank because of the difficulties in sample preparation. This is largely because the hydrated phospholipid bilayer environment that is essential for maintaining the native, active conformations of these proteins interferes with crystallization, required for x-ray diffraction, and rapid isotropic reorientation, required for solution NMR spectroscopy.
Solid-state NMR spectroscopy has the potential to determine atomic resolution structures of proteins with membrane-spanning domains in phospholipid bilayers. There are two basic approaches to high-resolution solid-state NMR, since sample manipulations are essential supplements to radiofrequency irradiations to obtain high-resolution NMR spectra of immobile molecules. The measurements of spectral parameters associated with individual resonances provide distance or angular constraints as input for the calculation of protein structures. Unoriented “powder” samples of proteins can be investigated with magic angle sample spinning methods, including membrane proteins in phospholipid bilayers (2–4). In contrast, the aligned sample method utilized in this study is effective with stationary samples where the proteins are immobilized and uniaxially aligned by their environment (5–7). Proteins associated with phospholipid bilayers can be aligned mechanically on glass plates or magnetically through the formation of bicelles that result from the addition of short-chain phospholipids. We have obtained solid-state NMR spectra of a number of membrane proteins in magnetically aligned bicelles, and have shown that these samples are suitable for structure determination by solid-state NMR spectroscopy (8,9). Large bicelles that align with their normals perpendicular to the direction of the applied magnetic field (“perpendicular” or “unflipped” bicelles) can be used to determine the structures of membrane proteins because the protein molecules undergo rotational diffusion about the bilayer normal at a rate that is rapid compared to the relevant NMR timescales (104 Hz) determined by the heteronuclear dipolar coupling and chemical shift interactions. Among the advantages of studying membrane peptides in bicelles are that the proteins and the lipids are fully hydrated, it is feasible to use a sealed sample tube that ensures sample stability for long periods of time, and the cylindrical sample tube can be placed inside a solenoid coil for optimal radiofrequency response.
In many cases, protein-containing bicelle samples can be prepared by simply adding an appropriate mixture of long-chain (e.g., C14 or C16) and short-chain (e.g., C6) phospholipids and water to the purified polypeptide. The components self-assemble into bilayers that serve to immobilize the protein and align it magnetically, fulfilling both prerequisites of structure determination by solid-state NMR of aligned samples. The molecular architecture of bicelles has been described with a variety of models (10–15). Significantly, all of the properties of solid-state NMR spectra of both the protein and lipid components are consistent with the long-chain phospholipids forming planar bilayers and the short-chain phospholipids “capping” either edges of disks or internal voids. The size and other properties of bicelles are affected by the molar ratio of long-chain to short-chain phospholipids (q). In addition, bicelles have the useful property that they can be “flipped” from perpendicular to parallel alignments of their bilayer normals relative to the direction of the applied magnetic field (11). Bicelle samples prepared from ether-linked phospholipids (16,17) are stable for many months in sealed glass tubes (8).
Vpu is a small, 81-residue membrane protein whose sequence is encoded in the genome of HIV-1 (18,19); in addition to a hydrophobic transmembrane helix in the N-terminal domain, there are two shorter amphipathic helices that reside in the plane of the membrane and comprise its cytoplasmic C-terminal domain (20,21). The two principal biological activities of Vpu are known to be associated with the two different domains (20–23). The degradation of the CD4/gp160 complexes is affected by the cytoplasmic domain in the C-terminal half of the protein. Vpu also affects the budding of new virus particles, and this may be associated with the channel activity found in the N-terminal domain (7). We have determined the three-dimensional structure of the transmembrane domain of Vpu by NMR spectroscopy in several lipid environments, including weakly aligned micelles, and mechanically-aligned bilayers on glass plates (7). In this article, we describe the structures of the same transmembrane domain in magnetically aligned bicelles with two different hydrophobic thicknesses (14-O-PC/6-O-PC bicelles and 16-O-PC/6-O-PC bicelles). We compare the three-dimensional structures determined in bicelles with that previously determined in mechanically aligned bilayers on glass plates and show that the transmembrane α-helix of Vpu encompasses residues 8–25 in all environments, but there are substantial differences in tilt angles and the properties of a kink in the middle of the helix that reflect hydrophobic mismatch. This expands upon our earlier comparisons, which were limited to the tilt and rotation of the ideal helical segments in phospholipid bilayers on glass plates (24).
MATERIALS AND METHODS
Sample preparation
We have previously described the expression and purification of the transmembrane domain of Vpu (7). Vpu2–30+, the polypeptide that represents the transmembrane domain of Vpu, has the sequence QPIQIAIVALVVAIIIAIVVWSIVIIEGRGGKKKK, where the underlined residues are from native Vpu of BH10 isolate, and six additional residues GGKKKK were added to facilitate the isolation, purification, and sample preparation of the hydrophobic minimal transmembrane domain of the protein (7). Uniformly 15N-labeled samples were obtained by growing bacteria in M9 minimal medium with (15NH4)2SO4 as the sole nitrogen source. Selectively 15N-labeled protein was obtained by growing bacteria in minimal medium with unlabeled ammonium sulfate supplemented with 19 unlabeled amino acids and the selected 15N-labeled amino acid. After purification of the inclusion bodies by nickel chelate chromatography and cleavage with cyanogen bromide, the peptide was purified by HPLC. It was stored as a lyophilized powder.
Samples of Vpu2–30+ in bicelles were prepared by dissolving the purified polypeptide in an aqueous solution containing the short-chain lipid and then adding this solution to a dispersion of the long-chain lipids in water (25); ∼3 mg of the lyophilized peptide was first dissolved in 1 mL trifluoroethanol, and sonicated for 5 min in a bath sonicator. The peptide concentration was determined by measuring the absorbance at 280 nm. The organic solvent was then evaporated under a stream of nitrogen gas to obtain a thin, transparent protein film, which was placed under high vacuum overnight. The lipids 6-O-PC and 14-O-PC were obtained dissolved in chloroform (Avanti Polar Lipids, Alabaster, AL). Volumes corresponding to 46.5 mg of 14-O-PC and to 9.5 mg of 6-O-PC were aliquoted separately in glass tubes. The solvent was evaporated under a stream of nitrogen gas and the lipids were placed under high vacuum overnight. An aqueous solution of 6-O-PC, obtained by adding 100 μL of water to the short-chain lipid, was added to the dry protein film. A dispersion of 14-O-PC in water was prepared by adding 80 μL of water to the long-chain lipid, followed by extensive vortexing and three freeze/heating cycles (liquid nitrogen/45°C). The protein/6-O-PC solution was transferred to the 14-O-PC dispersion, previously warmed to >40°C. The resulting solution was vortexed and freeze-heated several times and then allowed to equilibrate to room temperature. Upon bicelle formation, the previously white dispersion of 14-O-PC becomes a clear, nonviscous solution between 0°C and 10°C. The bicelle sample for q = 3.2 and a lipid concentration of 28% (w/v), was ∼300 mM in 14-O-PC in a 200-μL volume, at pH 5.0. A small, flat-bottomed NMR tube with a 4-mm outer diameter (New Era Enterprises, Vineland, NJ) was filled with 90 μL of the bicelle solution, using a precooled glass pipette, at 4°C. The NMR tube was sealed with a tight-fitting rubber cap, pierced with a thin syringe to remove residual air from the sample and create a tight seal. Samples of thicker bicelles, composed of 16-O-PC (Avanti Polar Lipids, Alabaster, AL) and 6-O-PC with q = 3.0 and cL = 28% (w/v) were prepared similarly.
To monitor the bicelle alignment and measure the order parameter by 2H NMR, 14-O-PC/6-O-PC bicelle samples were prepared by adding a small amount of DMPC lipid deuterated in the fatty acid chains (DMPC-d54) to the 14-O-PC ([DMPC-d54]/[14-O-PC] = 1:50). Similarly, samples of 16-O-PC/6-O-PC bicelles were added to deuterated DPPC (DPPC-d62). All deuterated lipids were purchased from Avanti Polar Lipids. Deuterium-depleted H2O (Cambridge Isotope Laboratories, Andover, MA) was used for the deuterated samples.
Parallel bicelles were prepared by adding YbCl3.6H2O (Sigma, St. Louis, MO) to the samples directly in the NMR tube. The tube was cooled to 4°C, the rubber cap was quickly removed, and 5 μL of freshly prepared 100 mM YbCl3.6H2O in water were added to the sample. The lanthanide concentration, 3 mM, i.e., 1 mol %, of the long-chain lipid, was chosen to be slightly above the minimum needed to induce parallel bicelle alignment, as verified by 31P NMR.
To prepare 2H2O-exchanged bicelle samples, the water in the sample was evaporated slowly under a stream of nitrogen gas at room temperature. When the volume decreased by ∼50%, the sample was chilled at 4°C and an equal amount of 2H2O was added to it and mixed thoroughly. The process was then repeated until the desired concentration of 2H2O was obtained.
Solid-state NMR spectroscopy
The NMR experiments were performed on a spectrometer with a 1H resonance frequency of 900 MHz consisting of a Bruker Avance console and Magnex magnet and room temperature shims. All samples of 14-O-PC/6-O-PC and 16-O-PC/6-O-PC bicelles were equilibrated in the magnetic field at 40°C and 50°C, respectively, for at least 30 min before the NMR measurements. Bicelle alignments were checked and order parameters were measured separately with a single-channel homebuilt 2H NMR probe with a 5-mm ID solenoid coil. The double-resonance 1H/15N probe used in the experiments is described below.
A radio-frequency strength B1 = 50 kHz was used for 1H irradiation during acquisition and Lee-Goldburg irradiation in PISEMA, and 1 ms cross-polarization mixing time was applied for both one-dimensional 1H-15N cross-polarization and separated local field experiments. CP-MOIST (26) was used for cross-polarization to compensate for power mismatch. Composite-pulse decoupling by SPINAL-16 (27,28) proved particularly effective for heteronuclear 1H decoupling during acquisition and was used in all cases. The frequency jumps to satisfy the Lee-Goldburg off-resonance irradiation for PISEMA experiments were 35.4 kHz. Typical parameters were 6 s recycle delay, 1 K scans with 5- to 10-ms acquisition time for one-dimensional experiments, and 128–256 scans with 5-ms acquisition time for two-dimensional experiments. The optimal 1H carrier position was determined experimentally to be ∼9 ppm, which agrees with the prediction for transmembrane helices based on the typical position chosen for mechanically oriented bilayers with the normal parallel to the static field (∼3 ppm), when taking into account the 90° orientation and the bicelle order parameter.
The two-dimensional PISEMA spectra of uniformly 15N-labeled samples resulted from a total of 64 t1 increments and 512 t2 complex points with 128 scans for each t1 increment. Selectively labeled spectra for 15N alanine, isoleucine, leucine, tryptophan and valine Vpu2–30+ were obtained with 32 or 64 t1 increments, depending on the sensitivity of each sample. The spectra of the selectively 15N-labeled samples were used qualitatively to assign the spectrum of the uniformly 15N-labeled protein, which was used for all of the quantitative frequency measurements. This eliminates variability due to slight differences in magnetic susceptibility or other factors from sample to sample. The data were zero-filled in both the t2 and t1 dimension, yielding a 1024 × 1024 real matrix. A suitable phase-shifted sine bell multiplication followed by 50–100 Hz of exponential multiplication was applied in both dimensions before Fourier transformation. The NMR data were processed using the program NMRPipe/NMRDraw (29). All chemical shifts were externally referenced to 15N-labeled solid ammonium sulfate, set to 26.8 ppm, corresponding to the signal from liquid ammonia at 0 ppm.
The bicelle order parameter was measured using the 2H quadrupolar splitting of the methylene deuterons in the fatty acid chains located near the top of the phospholipid headgroups in the 2H one-dimensional NMR spectrum of aligned bicelles (30). The splitting was compared to that for a full powder pattern obtained from a liposome sample of the long-chain lipid. All spectra were acquired at a deuterium frequency of 107.47 MHz, using a standard quadrupole echo pulse sequence with a 3-μs 90° pulse, 300-ms recycle delay, 250-kHz spectral width, and 1 K scans.
900 MHz NMR probe for PISEMA experiments on bicelle samples
As described above, the NMR experiments were performed on a spectrometer that consisted of a commercial console and magnet. The probe is the key technological link between the sample and the radiofrequency irradiations and responses, and we built this specifically for experiments on bicelle samples. There are many challenges in the design and construction of double-resonance probes for solid-state NMR experiments at high fields in narrow-bore magnets because of the high-power irradiations that are involved. The resolution in the experimental spectra, especially in the dipolar-coupling frequency dimension, benefits from higher-power irradiation. In practice, the limiting factor in experiments is the ability of the probes to withstand prolonged radiofrequency irradiations. Sound mechanical design and construction is paramount to have stable tuning of the radiofrequency circuits and to avoid arcing under high-power irradiation, since stray capacitances and inductances are unavoidable at high frequencies. The principal tuning components in the probe are Voltronics (www.voltronicsorp.com) variable capacitors (model NMNT6-10ENL). The small diameter of this type of capacitor is essential in this application because of limited space available within the confines of a probe with an outer diameter of 48 mm; however, their reduced power handling capability compared to physically larger capacitors means that that American Technical Ceramics (www.atceramics.com) fixed capacitors (series 700C) must be placed in series. As shown in Fig. 1, the electronic components are located on three separate ground planes. The solenoid sample coil, with a 4-mm inner diameter, is located on the top ground plane, which is constructed of copper-clad Teflon. The coil leads are soldered to pads located at the bottom of the ground plane. The two lower ground planes are constructed from solid brass with screw holes for securing the variable capacitors and open holes for the home-built ferrules. The ferrules are torch-soldered to the ground planes, and are used to attach coaxial cables. The grounded variable capacitors are screwed directly into the silver-plated ground planes, and the ungrounded variable capacitors are screwed into plastic standoffs, which are then screwed into the ground plane. The coaxial cables are secured using small cable ties after they are inserted into the ferrules. The ground planes are layered by sliding onto three threaded tubes and held in place with screws and washers. The threaded tubes are attached to a base made of brass, which is not silver-plated. The net result is that the components are securely positioned and grounded, which means that the stray capacitances are fixed and can be absorbed into the tuning circuit. Samples are changed by lowering the probe out of the magnet and removing the cap to gain access to the sample coil. Since the removable gold-plated cap is in close proximity to the electrical components, gold-plated beryllium copper fingers are attached to the ground planes with screws and used to ground the cap when it is placed on the probe to avoid mechanical and tuning instabilities.
FIGURE 1.

The 900-MHz double-resonance probe used to obtain the experimental results. (A) Photograph of the probe with the outer casing and cap removed. (B) Circuit diagram. The bicelle sample in the sealed cylindrical tube is placed in the 4-mm solenoid sample coil located on the top ground plane.
With a nonlossy test sample (15N ammonium sulfate powder), B1 fields of 60 kHz can be generated with 600 Watts of 15N power and 50 Watts of 1H power. With bicelle samples, there is a loss of efficiency at 900 MHz due to effects of their high dielectric constant. We typically obtain 53 kHz B1 for the 1H resonances with 86 Watts.
Dipolar waves and structure calculations
Once the uniformly 15N-labeled spectrum was assigned, the 1H-15Ndipolar couplings were plotted as a function of the residue number, and fit to a sinusoid of period 3.6, corresponding to the pitch of an α-helix (31). The dipolar waves identify the α-helical transmembrane region of the peptide and can be used to characterize the length, curvature, and relative orientation of the helix. The fit to the data was carried out by nonlinear fitting of the phase and amplitude of a sinusoid as previously described (32).
A set of structures was obtained from the orientationally dependent experimental 15N-1H dipolar coupling and 15N chemical shift frequencies in bicelles. The structural fitting algorithm (33) was adapted to fit the data in bicelles assuming the following relation of the NMR frequencies in bicelles versus those in oriented bilayers, valid at sufficiently fast uniaxial averaging (9):
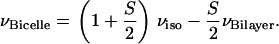 |
This equation takes into account the perpendicular orientation of the bicelle disks (the factor −1/2) and the presence of a fast wobbling motion which gives rise to an additional scaling by the order parameter S. Here νiso is the isotropic frequency for the chemical shift and the dipolar interactions (which is zero in the latter case). The order parameter S, measured by 2H NMR, was set to 0.75 for 14-O-PC/6-O-PC and 0.80 for 16-O-PC/6-O-PC bicelles. The Ramachandran angles φ and ψ between consecutive residues were obtained by direct calculation from the dipolar and chemical shift frequencies, assuming constant peptide plane geometry. Explicit analytic relations linking NMR anisotropic frequencies for adjacent residues as functions of φ and ψ have been given (33). The structural fitting was carried out in a sequential manner from N-terminus to C-terminus. Only the solutions that satisfied the predefined deviation (in Hz) from the experimental data for all the residues were carried out all the way until the last residue; whereas if the maximum allowed error was reached before the C-terminus, a solution was rejected. The 15N chemical shift tensor values were assigned constant values (σ11 = 64 ppm, σ22 = 77 ppm, σ33 = 222 ppm), assuming an experimental accuracy of ±200 Hz in each dimension for the spectral frequencies, which presumably includes the variations in the tensor values and the scaling factor in the dipolar dimension. For every calculation, the torsion angles were allowed to vary within ±15° for φ and ψ relative to the average values for an α-helix, φ = −61°, ψ = −45°. The convergence of the structural fit (in Å) is evaluated by comparing the multiple structural solutions from repeated calculations within these defined limits for φ and ψ. To be able to pick up multiple solutions (when they exist), the starting values for the torsion angles were randomized at each sequential step. The ensemble of 100 structural solutions gives rise to an RMS deviation of ∼0.3 Å, which gives an estimate of the precision of the calculated structures. Structure calculations were carried out in Matlab on a Windows Laptop PC.
The coordinates have been deposited in the Protein Data Bank (PDB codes 2GOF and 2GOH for structures determined in C14 and C16 bicelles, respectively), for release upon publication.
RESULTS AND DISCUSSION
Alignment of the transmembrane domain of Vpu in phospholipid bicelles
Protein-containing bicelles align spontaneously in a strong magnetic field at temperatures above the gel-to-liquid crystalline phase transition temperature of the long-chain phospholipids (30). The direction and extent of the alignment of the bicelles were verified with 31P and 2H NMR spectra obtained after adding small amounts of deuterated lipids to the samples (data not shown). More importantly, the partial resolution of the overlapped single-line 15N resonances in the spectra in Fig. 2 clearly indicate that the protein-containing bicelle samples are well aligned in the magnetic field of the NMR spectrometer. Significantly, there is no evidence of underlying “powder pattern” intensity, which would be indicative of unoriented material, in any of the spectra.
FIGURE 2.

Schematic diagrams of parallel and perpendicular bicelles containing a transmembrane helix, and one-dimensional 15N chemical shift NMR spectra of uniformly 15N-labeled transmembrane domain of Vpu in aligned bicelles. Bicelles are aligned magnetically with their normals (n) either parallel (A and B) by addition of lanthanide ions shown in black dots, or perpendicular (C and D) with respect to the applied magnetic field (Bo). One-dimensional spectra of the transmembrane domain of Vpu aligned in 14-O-PC/6-O-PC (q = 3.2) bicelles are shown in A and C. One-dimensional spectra in 16-O-PC/6-O-PC (q = 3.0) bicelles are shown in B and D. The spectra of parallel bicelle samples (A and B) were obtained in 90% 2H2O/10% H2O and those in perpendicular bicelle samples (C and D) were obtained in H2O.
The uniaxial alignment of the protein-containing bicelles is a consequence of the interaction of the magnetic field and the magnetic susceptibility anisotropy of the liquid crystalline phospholipid bilayers. The negative sign of the anisotropy tensor results in the bicelles aligning with their normals perpendicular to the direction of the magnetic field (30,34,35) (Fig. 2, C and D). Paramagnetic lanthanide ions (e.g., Tm3+ and Yb3+) affect the direction of alignment (11,36), since the binding of paramagnetic lanthanide ions to the lipids changes the sign of the magnetic susceptibility anisotropy tensor of the bicelles (35). In our experiments, the addition of Yb3+, with its large positive magnetic susceptibility anisotropy tensor, causes the bicelles to change orientation by 90° and align with their normals parallel to the direction of the applied magnetic field (Fig. 2, A and B).
1H decoupled one-dimensional 15N NMR spectra of the transmembrane domain of Vpu in parallel and perpendicular bicelles are compared in Fig. 2. Each amide site in the protein contributes a single-line 15N resonance whose frequency is determined by the orientation of the peptide plane relative to the direction of the magnetic field. Many of the resonances from these uniformly 15N-labeled samples overlap because the amide sites in a tilted membrane-spanning α-helix have similar orientations and, hence, chemical shift frequencies. The appearance of these spectra is consistent with the resonances having line widths of 1–2 ppm, as measured from fully resolved resonances in selectively 15N-labeled samples (8,9).
The dramatic differences in the 15N chemical shift frequencies for proteins in parallel (Fig. 2, A and B) and perpendicular bicelles (Fig. 2, C and D) demonstrate that the orientations of individual peptide planes relative to the magnetic field are reflected in the spectra. Notably, no evidence of line broadening is observed in the 15N NMR spectra due to the addition of the paramagnetic lanthanide ions. Since the spectra in parallel bicelles (Fig. 2, A and B) were obtained in aqueous solution containing ∼90% 2H2O, resonances from residues in the nonhelical N- and C-terminal regions of the polypeptide are not present, due to the presence of a deuteron rather than a proton at those amide sites. Only resonances from residues in the transmembrane helix, which are highly resistant to hydrogen exchange, are present in the spectra obtained by cross-polarization. For a polypeptide containing a single helix, such as Vpu2–30+, comparisons between one-dimensional experimental spectra and those simulated for a range of tilt angles yields a reasonable approximation of the overall tilt angle despite the resonance overlap observed in the spectra of uniformly 15N-labeled samples. The data shown in Fig. 2 indicate that the tilt angles for the transmembrane helix domain of Vpu are 30° in 14-O-PC/6-O-PC bicelles and 21° in 16-O-PC/6-O-PC bicelles. The transmembrane helix clearly responds to hydrophobic mismatch of the bilayers in magnetically aligned bicelle samples by changing its tilt angle, just as it does for bilayer samples aligned on glass plates (24).
PISEMA spectra of the transmembrane domain of Vpu in phospholipid bicelles
Two-dimensional PISEMA spectra of the uniformly 15N-labeled transmembrane domain of Vpu in 14-O-PC/6-O-PC and 16-O-PC/6-O-PC bicelles are shown in Fig. 3, A and B, respectively. The spectrum with resonances <120 ppm was obtained from perpendicular bicelles, whereas that with resonances >120 ppm was obtained from parallel bicelles with added Yb3+ ions. The wheel-like patterns of resonances observed in the spectra of uniformly labeled samples are characteristic of a tilted transmembrane helix (37–39). PISA-wheel patterns correspond to helical wheel projections and serve as direct indices of secondary structure and topology (38,40). In the two-dimensional PISEMA spectra, each amide resonance is characterized by 1H-15N dipolar coupling and 15N chemical shift frequencies that serve as orientation constraints for structure calculations that are independent of the analysis of spectral patterns.
FIGURE 3.

Two-dimensional 15N chemical shift/1H-15N dipolar coupling PISEMA spectra of uniformly 15N-labeled transmembrane domain of Vpu in (A) 14-O-PC/6-O-PC (q = 3.2) bicelles and (B) 16-O-PC/6-O-PC (q = 3.0) bicelles. The displayed two-dimensional spectra are composites of spectra obtained with bicelles aligned with their bilayer normals parallel (left) and perpendicular (right) to the magnetic field. Superimposed on the experimental data are ellipses corresponding to the ideal PISA wheels for an α-helix with uniform dihedral angles (φ = −61° and ψ = −45°) tilted at 30° (A) and 21° (B) with respect to the bilayer normal. The line is drawn at the average isotropic chemical shift frequency of 120 ppm. The sequential assignments of the amide resonances are indicated by residue numbers. The spectra of parallel bicelle samples were obtained in 90% 2H2O/10% H2O and those of perpendicular bicelle samples were obtained in H2O.
In parallel 14-O-PC/6-O-PC bicelles, a total of 15 individual backbone amide resonances are resolved in the PISEMA spectrum in the spectral region 150–190 ppm associated with a transmembrane helix. The resonance assignments were obtained using the “shotgun” NMR approach, which takes advantage of the scattering of each type of residue throughout the amino acid sequence and the symmetry inherent in helical structures (39). All of the resonances in the spectrum were assigned to individual residues in the transmembrane helix based on spectra from Ile, Val, Ala, Leu, and Trp selectively 15N-labeled samples and were confirmed by the results of 2H/1H exchange experiment on a selectively 15N Ile-labeled sample (data not shown). The frequencies are listed in Table 1.
TABLE 1.
Orientation frequencies of the transmembrane helix of Vpu in various lipid environments
| DHPC*15N shift (ppm) | 14C bicelle
|
14C bicelle + Yb†
|
16C bicelle
|
16C bicelle + Yb
|
|||||
|---|---|---|---|---|---|---|---|---|---|
| Residue | 15N shift (ppm) | Coupling (kHz) | 15N shift (ppm) | Coupling (kHz) | 15N shift (ppm) | Coupling (kHz) | 15N shift (ppm) | Coupling (kHz) | |
| I8 | 114.7 | 88.3 | 3.50 | 183.3 | 6.42 | 83.9 | 4.06 | 195.9 | 7.45 |
| V9 | 118.4 | 111.9 | 2.51 | 146.4 | 4.94 | 103.0 | 3.25 | 165.3 | 6.27 |
| A10 | 119.5 | 91.2 | 1.71 | 177.6 | 3.42 | 88.3 | 2.41 | 190.6 | 4.76 |
| L11 | 116.3 | 84.0 | 2.77 | 189.5 | 5.00 | 79.1 | 3.43 | 200.5 | 6.32 |
| V12 | 117.3 | 101.0 | 3.64 | 157.3 | 6.84 | 93.3 | 4.20 | 172.5 | 7.86 |
| V13 | 117.8 | 105.5 | 1.97 | 153.1 | 4.14 | 96.3 | 2.85 | 170.6 | 5.92 |
| A14 | 120.3 | 87.8 | 2.13 | 188.8 | 3.57 | 82.4 | 2.73 | 201.1 | 4.82 |
| I15 | 116.0 | 86.9 | 3.54 | 172.2 | 7.03 | 86.6 | 4.21 | 186.3 | 8.24 |
| I16 | 118.7 | 107.1 | 3.16 | 152.7 | 4.84 | 99.0 | 3.77 | 172.1 | 6.45 |
| I17 | 117.5 | 99.0 | 1.72 | 165.3 | 3.66 | 92.6 | 2.27 | 179.2 | 5.04 |
| A18 | 120.6 | 87.8 | 2.13 | 197.0 | 4.14 | 80.8 | 2.91 | 208.4 | 5.48 |
| I19 | 115.7 | 92.2 | 3.74 | 155.3 | 6.80 | 86.1 | 4.28 | 172.5 | 7.86 |
| V20 | 121.9 | 108.7 | 2.50 | 152.7 | 4.84 | 99.5 | 3.18 | 172.1 | 6.45 |
| V21 | 118.1 | 91.2 | 1.71 | 177.6 | 3.42 | 85.8 | 2.54 | 190.6 | 4.76 |
| W22 | 119.0 | 84.0 | 2.77 | 196.7 | 5.33 | 79.1 | 3.43 | 206.4 | 6.43 |
| S23 | 112.8 | 107.1 | 3.16 | 151.0 | 6.16 | 99.0 | 3.77 | 169.0 | 7.23 |
| I24 | 120.5 | 105.5 | 1.97 | 158.3 | 3.76 | 96.3 | 2.85 | 169.1 | 5.51 |
| V25 | 119.0 | 89.5 | 1.87 | 188.8 | 3.57 | 82.4 | 2.73 | 201.1 | 4.82 |
15N isotropic chemical shift values of Vpu2–30+ in 100 mM DHPC-d40 at 50°C are from Park et al. (7).
A 3-mM quantity of YbCl3 was added to flip the bicelle sample parallel to the magnetic field.
In perpendicular bicelles, the resolution is also excellent even though the spectral range is half that obtained from parallel bicelles. The narrow line widths (<2 ppm for the 15N chemical shift and ∼250 Hz for the 1H-15N dipolar couplings) observed in both dimensions require fast axial diffusion of the proteins (Drot ≥ 105 s−1) about the direction of the bilayer normal in perpendicular bicelles (8,9). Although this motion is compatible, quantitatively and qualitatively, with the description of bicelles as spinning disks, the spectra of Vpu2–30+ also display single line resonances in planar bilayers oriented on glass plates with their normals tilted at various angles, including perpendicular to the direction of the applied magnetic field (9,41), indicating that the polypeptide itself undergoes rapid rotational diffusion within the bilayers. Narrow single-line resonances have also been observed in perpendicular bicelles for substantially larger polypeptides, including MerFt, a 60-residue membrane protein with two transmembrane segments connected by a structured loop (8), and CXCR1, a 350-residue G-protein-coupled receptor with seven transmembrane helices (42). Whether the averaging in perpendicular bicelles is due solely to rotational diffusion of the protein within the bilayer or to overall rotational diffusion of the bicelles is a subject for investigation, but does not affect the interpretation of the experimental results obtained on these samples.
The bicelle samples were “flipped” from perpendicular to parallel alignments by addition of YbCl3, as described in Materials and Methods. The PISEMA spectra of the uniformly and selectively labeled samples in parallel bicelles display the same PISA wheel pattern of resonances previously observed in lipid bilayers on glass plates (7). Spectra from the two orientations of the bicelles are complementary, and can be used to separate overlapped resonances and to resolve ambiguities in spectral assignments.
Dipolar wave analysis
The magnitudes of the heteronuclear dipolar couplings measured from the assigned PISEMA spectrum of uniformly 15N-labeled samples of Vpu2–30+ (Fig. 3) are plotted as a function of residue number in Fig. 4. Structural distortions of helices, including kinks and curvature, are readily detected using dipolar waves (31,32,43), since the resonance patterns from ideal helices are well fit by simple mathematical functions with the 3.6 period of the secondary structure. Dipolar waves derived from solid-state NMR data give absolute measurements of helix orientations because the polypeptides are immobile and the bilayers have a known alignment in the magnetic field. The automated fitting of a dipolar wave to the experimental data clearly identifies those residues that contribute to the transmembrane helix of the polypeptide. It also serves as a fast, visual verification of the assignments, since misassigned resonances generally fall off the wave. The dipolar waves for the residues in the transmembrane helix of Vpu are compared in Fig. 4 for two different thicknesses of bilayers. The range and average value of the dipolar couplings give precise measures of the helix tilt, and the correspondence of the phase of the two plots indicates that the helix rotation is the same in the two samples. The transmembrane α-helical segment of Vpu2–30+ has 18 residues, from 8 to 25 in the sequence, in all of the bicelle samples investigated, in agreement with the structures previously determined in micelles and mechanically oriented bilayers on glass plates (7). Recently, it has been reported that the segment of Vpu comprising residues 1–40 forms a highly ordered α-helical segment from residues 7 through 25 in DOPC/DOPG (9:1) liposomes at 40°C, although the peptide forms a longer α-helix spanning residues 7–27 (4).
FIGURE 4.

Dipolar waves of 1H-15N dipolar couplings of the transmembrane helix of Vpu in 14-O-PC/6-O-PC bicelles (A) and in 16-O-PC/6-O-PC bicelles (B). The data points of solid and open circles are from the parallel and perpendicular bicelles, respectively. (C) Comparison between the dipolar waves of normalized dipolar couplings (Δ) with S = 0.8 obtained from parallel 14-O-PC/6-O-PC bicelles and those of dipolar couplings (X) obtained from 14-O-PC bilayers on glass plates (24).
The experimental dipolar couplings are well fit by a single dipolar wave. The RMS errors for the C14 and C16 perpendicular bicelles are 0.22 and 0.17 kHz, respectively, which are equivalent to the experimental line widths in the heteronuclear dipolar coupling frequency dimension of the PISEMA spectra. Similarly, the RMS errors for the fits of the C14 and C16 parallel bicelles are 0.51 and 0.49 kHz, respectively. These results indicate that the transmembrane helix of Vpu in both C14 and C16 bicelles is well described as an 18-residue undistorted α-helix. Previously, we showed that this same helix has very similar properties in C14 phospholipid bilayers mechanically aligned on glass plates (24). In contrast, a kink was found in the middle of the helix C18:1 phospholipid bilayers mechanically aligned on glass plates, as well as in weakly aligned DHPC micelles (7,24).
Comparison of magnitudes of the dipolar couplings in C14 bilayers aligned mechanically on glass plates (24) and C14 parallel (flipped) bicelles provides a measurement of the order parameter S of the bicelle sample. The average values of dipolar couplings in C14 phospholipids are 6.12 kHz and 4.88 kHz in bilayers and bicelles, which give a value for the order parameter of the bicelles of ∼0.8. This value is similar to that obtained by measuring the 2H quadrupolar splittings of the methylene deuterons in the fatty acyl chains (30). The dipolar waves of parallel 14-O-PC/6-O-PC bicelles whose dipolar couplings are normalized with the order parameter S = 0.8, are consistent with those of 14-O-PC bilayers on glass plates (Fig. 4 C). These results indicate that the tilt and rotation angles of the transmembrane helix of Vpu are similar in mechanically aligned bilayers and magnetically aligned bicelles.
Structure calculations
Structural fitting (33) enables the calculation of protein structures from the chemical shift and heteronuclear dipolar coupling frequencies associated with individual resonances in two-dimensional PISEMA spectra. In addition to being able to determine the three-dimensional backbone structures of proteins, the structural fitting algorithm is sensitive enough to detect minor deviations from ideality in helices (7,33,44,45). A structural fit to a fully assigned spectrum is equivalent to a direct calculation of the protein structure. The results of structural fitting for the two types of perpendicular bicelle samples are shown in Fig. 5 for the transmembrane helix of Vpu. The structures were calculated sequentially from the N-terminus to the C-terminus, which provides the torsion angles (φ and ψ) between each pair of residues. As a result, there are no ambiguities with respect to the relative orientations of the helical segments.
FIGURE 5.

Superimposition of 100 calculated backbone structures of the transmembrane helix of Vpu in (A) 14-O-PC/6-O-PC bicelles and (B) 16-O-PC/6-O-PC bicelles. The structures were obtained by the method of structural fitting of the experimental solid-state NMR data and are aligned for the best overlap of residues Ile8–Val25. An average tilt angle of the helix is represented. (C and D) Helical wheel diagrams of the transmembrane helix of Vpu in (C) 14-O-PC/6-O-PC and (D) 16-O-PC/6-O-PC perpendicular bicelles. Residue L11 is marked for comparison of the rotation angles of the helices in the two different bicelle samples.
For structural fitting of the experimental data, the experimentally measured values of the order parameter (S = 0.75 for C14 bicelles and S = 0.80 for C16 bicelles) were used in the calculations. However, we find that the order parameter cannot be varied in the calculations by more than a few percent without the quality of the fit to the data degrading significantly. This suggests a simplified approach for future studies where an estimate of S based on the q of the bicelles is used as a starting point, and then its value is varied within ±0.05 as a fitting parameter in the structure calculations. By performing repeated structural fits to the assigned PISEMA spectra in Fig. 3, 100 structural solutions were generated for the protein in perpendicular bicelles of two thicknesses, and superimposed in Fig. 5, A and C. The mean value of the RMS deviation of ∼0.3 Å relative to the average structure provides a measure of the precision of the structure calculations. Average backbone torsion angles of each residue in the transmembrane helix of Vpu in bicelles are summarized in Table 2. The torsion angles are consistent with an α-helix: φ = −61° and ψ = −45°, with a deviation of ±15°.
TABLE 2.
Average backbone torsion angles of the transmembrane helix of Vpu in bicelles
| 14C bicelle
|
16C bicelle
|
|||
|---|---|---|---|---|
| – | φ (dev) | ψ (dev) | φ (dev) | ψ (dev) |
| I8 | −72.5 (0.5) | −39.4 (1.2) | −72.3 (0.9) | −39.5 (1.4) |
| V9 | −55.0 (1.0) | −59.4 (0.7) | −55.4 (1.9) | −58.9 (1.3) |
| A10 | −56.4 (1.6) | −30.0 (0.2) | −56.7 (2.6) | −30.7 (1.2) |
| L11 | −70.2 (2.7) | −44.1 (2.4) | −69.1 (2.4) | −45.1 (0.5) |
| V12 | −61.4 (2.0) | −44.0 (0.8) | −60.6 (0.9) | −43.9 (0.9) |
| V13 | −60.5 (1.9) | −59.9 (0.7) | −59.8 (0.9) | −58.9 (1.9) |
| A14 | −61.4 (3.4) | −36.5 (3.6) | −62.8 (3.2) | −35.6 (4.0) |
| I15 | −73.8 (2.0) | −55.7 (2.0) | −73.7 (2.8) | −56.2 (2.6) |
| I16 | −52.7 (2.9) | −48.0 (2.3) | −54.3 (3.3) | −47.0 (2.3) |
| I17 | −59.1 (1.3) | −37.1 (8.1) | −59.4 (1.7) | −35.5 (3.2) |
| A18 | −53.4 (3.5) | −37.1 (2.6) | −55.6 (1.1) | −35.5 (0.6) |
| I19 | −71.2 (1.2) | −41.1 (0.9) | −71.6 (1.0) | −41.0 (1.1) |
| V20 | −56.8 (1.0) | −57.7 (1.7) | −57.2 (1.4) | −56.6 (2.3) |
| V21 | −46.1 (0.3) | −32.1 (1.9) | −46.6 (1.2) | −31.8 (2.4) |
| W22 | −71.1 (1.9) | −46.4 (7.7) | −71.4 (3.0) | −46.4 (9.1) |
| S23 | −59.4 (7.5) | −51.9 (0.6) | −59.2 (9.0) | −51.8 (0.9) |
| I24 | −60.2 (1.3) | −58.9 (2.8) | −59.6 (0.9) | −59.6 (1.6) |
| V25 | ND | ND | ND | ND |
Data were obtained from 20 structures of the transmembrane helix of Vpu in 14C bicelles (PDB code 2GOF) and 16C bicelles (PDB code 2GOH). Values are presented in degrees, with the standard deviation of the torsion angle indicated in parentheses.
The structure of the transmembrane helix in 14-O-PC bicelles shows an average tilt angle of 30°, similar to the value of 27° obtained for DMPC bilayers on glass plates (24). This finding suggests that the thickness of the hydrophobic region of phospholipid bilayers is not significantly different in mechanically aligned bilayers or magnetically aligned bicelles, or in ether-linked or ester-linked phospholipids. Our finding of a tilt angle of 27–30° for the transmembrane helix of Vpu in C14 phospholipid bilayers by solid-state NMR experiments on an expressed polypeptide is significantly different from the finding of a 6.5 ± 1.7° tilt angle based on infrared dichroism of synthetic Vpu1–31 reconstituted in C14 phospholipid vesicles (47). Our finding of a tilt angle of 21° in C16 ether-linked phospholipids in bicelles is similar to that obtained from C18:1 ether-linked phospholipid bilayers (18°) on glass plates (24) and 18:1 lipid vesicles (≤20°) investigated with magic angle spinning methods (4). These results are consistent in showing that the hydrophobic thickness of C16 phospholipid bilayers is similar to that of C18:1 phospholipid bilayers, which have been found to have a hydrophobic thickness of 27 ± 1 Å (48).
The change in tilt angle appears to be the principal compensation mechanism for hydrophobic mismatch of the Vpu transmembrane helix, since the rotation angle of the helix is very similar in the various samples (24). As the bilayer thickness decreases from ∼27 Å to 15.5 Å, the helix tilt increases from 18° to 51°. Therefore, the difference in helix tilt between C14 and C16 bicelles can be explained as a hydrophobic mismatch effect due to the different length of the fatty acyl chains of the long-chain lipids. The peptide assumes a transmembrane α-helical conformation in both cases and the tilt is increased in 14-O-PC bicelles to compensate for a shorter hydrophobic length of the lipid bilayer.
The rotation angle of the helix at a specific residue (ω) is mapped onto the dipolar waves, where ω is given by the angle about the helix axis relative to the most tilted NH bond. (31,32,43). Helical wheel diagrams of perpendicular bicelles show how the phase of the sinusoids maps to the periodicity of the helix (Fig. 5, B and D). Residue L11 is marked to enable comparison of the rotation angles of the helix in 14-O-PC/6-O-PC and 16-O-PC/6-O-PC bicelles. The similar phases of all dipolar waves indicate that the helix rotation is the same in all samples (Fig. 4, A and B); the rotation angle of the L11 residue in 14:0 and 16:0 perpendicular bicelles is 288° (Fig. 5, B and D).
CONCLUSIONS
Comparisons of the three-dimensional structures of the transmembrane helix of Vpu in bicelles with different hydrophobic thicknesses with those determined previously in micelles and phospholipid bilayers aligned mechanically on glass plates show that the hydrophobic thickness of the bilayers is the dominant influence on the properties of a transmembrane helix (Fig. 6). In all membrane environments examined, the transmembrane helix of Vpu includes residues Ile8-Val25 and has the same rotation angle. However, the tilt angles of the helices were different due to the hydrophobic mismatch between the length of the transmembrane helix and the hydrophobic region of the phospholipids.
FIGURE 6.

Backbone structures of the transmembrane helix of Vpu determined in the following lipid environments: (A) 100 mM DHPC micelles by solution NMR; (B) DOPC/DOPG (9:1, w/w) bilayers aligned on glass plates (PDB code 1PJE); (C) 16-O-PC/6-O-PC (3:1, w/w) bicelles (PDB code 2GOH); and (D) 14-O-PC/6-O-PC (3.2:1, w/w) bicelles (PDB code 2GOF). The Cα of Ile17 is marked to indicate the rotation of each helix. The shaded boxes indicate the thickness of hydrophobic region of lipid bilayers. The images were created using the program MOLMOL (46).
In addition to providing more details than earlier studies, including the coordinates for the three-dimensional backbone structures in two different phospholipid environments, these results demonstrate that a membrane protein has the same features in ether-linked phospholipid bilayers and in ether-linked phospholipid bicelles. This provides assurance that magnetically aligned bicelles provide a phospholipid bilayer environment that is well suited for structure determination of membrane proteins by solid-state NMR spectroscopy.
Acknowledgments
This research was supported by grant RO1GM066978 from the National Institutes of Health. It utilized the Biomedical Technology Resource for NMR Molecular Imaging of Proteins supported by grant P41EB002031. A.A.D. was supported by postdoctoral fellowship F32GM065833. The 900 MHz NMR spectrometer was obtained with grants from the National Institutes of Health, the National Science Foundation, and the Department of Energy.
Abbreviations used: 6-O-PC, 1,2-di-O-hexyl-sn-glycero-3-phosphocholine; 14-O-PC, 1,2-di-O-tetradecyl-sn-glycero-3-phosphocholine; 16-O-PC, 1,2-di-O-hexadecyl-sn-glycero-3-phosphocholine; DHPC, 1,2-dihexanoyl-sn-glycero-3-phosphocholine; DMPC, 1,2-dimyristoyl-sn-glycero-3-phosphocholine; DOPC, 1,2-dioleoyl-sn-glycero-3-phosphocholine; DOPG, 1,2-dioleoyl-sn-glycero-3-[phospho-rac-(1-glycerol)]; DPPC, 1,2-dipalmitoyl-sn-glycero-3-phosphocholine; PISA wheel, polarity-index slant-angle wheel; PISEMA, polarization inversion spin exchange at the magic angle; RMS, root mean-square.
References
- 1.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 2.Eilers, M., W. Ying, P. J. Reeves, H. G. Khorana, and S. O. Smith. 2002. Magic angle spinning nuclear magnetic resonance of isotopically labeled rhodopsin. Methods Enzymol. 343:212–222. [DOI] [PubMed] [Google Scholar]
- 3.Andronesi, O. C., S. Becker, K. Seidel, H. Heise, H. S. Young, and M. Baldus. 2005. Determination of membrane protein structure and dynamics by magic-angle-spinning solid-state NMR spectroscopy. J. Am. Chem. Soc. 127:12965–12974. [DOI] [PubMed] [Google Scholar]
- 4.Sharpe, S., W. M. Yau, and R. Tycko. 2006. Structure and dynamics of the HIV-1 Vpu transmembrane domain revealed by solid-state NMR with magic-angle spinning. Biochemistry. 45:918–933. [DOI] [PubMed] [Google Scholar]
- 5.De Angelis, A. A., D. H. Jones, C. V. Grant, S. H. Park, M. F. Mesleh, and S. J. Opella. 2005. NMR experiments on aligned samples of membrane proteins. Methods Enzymol. 394:350–382. [DOI] [PubMed] [Google Scholar]
- 6.Ketchem, R. R., W. Hu, and T. A. Cross. 1993. High-resolution conformation of gramicidin A in a lipid bilayer by solid-state NMR. Science. 261:1457–1460. [DOI] [PubMed] [Google Scholar]
- 7.Park, S. H., A. A. Mrse, A. A. Nevzorov, M. F. Mesleh, M. Oblatt-Montal, M. Montal, and S. J. Opella. 2003. Three-dimensional structure of the channel-forming trans-membrane domain of virus protein “u” (Vpu) from HIV-1. J. Mol. Biol. 333:409–424. [DOI] [PubMed] [Google Scholar]
- 8.De Angelis, A. A., A. A. Nevzorov, S. H. Park, S. C. Howell, A. A. Mrse, and S. J. Opella. 2004. High-resolution NMR spectroscopy of membrane proteins in aligned bicelles. J. Am. Chem. Soc. 126:15340–15341. [DOI] [PubMed] [Google Scholar]
- 9.Nevzorov, A. A., A. A. De Angelis, S. H. Park, and S. J. Opella. 2005. Uniaxial motional averaging of the chemical shift anisotropy of membrane proteins in bilayer environments. In NMR Spectroscopy of Biological Solids. M. Williams, editor. Marcel Dekker, New York.
- 10.Sanders 2nd, C. R., and J. P. Schwonek. 1992. Characterization of magnetically orientable bilayers in mixtures of dihexanoylphosphatidylcholine and dimyristoylphosphatidylcholine by solid-state NMR. Biochemistry. 31:8898–8905. [DOI] [PubMed] [Google Scholar]
- 11.Prosser, R. S., S. A. Hunt, J. A. DiNatale, and R. R. Vold. 1996. Magnetically aligned membrane model systems with positive order parameter: switching the sign of Szz with paramagnetic ions. J. Am. Chem. Soc. 118:269–270. [Google Scholar]
- 12.Struppe, J., and R. R. Vold. 1998. Dilute bicellar solutions for structural NMR work. J. Magn. Reson. 135:541–546. [DOI] [PubMed] [Google Scholar]
- 13.Glover, K. J., J. A. Whiles, G. Wu, N. Yu, R. Deems, J. O. Struppe, R. E. Stark, E. A. Komives, and R. R. Vold. 2001. Structural evaluation of phospholipid bicelles for solution-state studies of membrane-associated biomolecules. Biophys. J. 81:2163–2171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gaemers, S., and A. Bax. 2001. Morphology of three lyotropic liquid crystalline biological NMR media studied by translational diffusion anisotropy. J. Am. Chem. Soc. 123:12343–12352. [DOI] [PubMed] [Google Scholar]
- 15.Nieh, M. P., V. A. Raghunathan, C. J. Glinka, T. A. Harroun, G. Pabst, and J. Katsaras. 2004. Magnetically alignable phase of phospholipid “bicelle” mixtures is a chiral nematic made up of wormlike micelles. Langmuir. 20:7893–7897. [DOI] [PubMed] [Google Scholar]
- 16.Cavagnero, S., H. J. Dyson, and P. E. Wright. 1999. Improved low pH bicelle system for orienting macromolecules over a wide temperature range. J. Biomol. NMR. 13:387–391. [DOI] [PubMed] [Google Scholar]
- 17.Ottiger, M., and A. Bax. 1999. Bicelle-based liquid crystals for NMR-measurement of dipolar couplings at acidic and basic pH values. J. Biomol. NMR. 13:187–191. [DOI] [PubMed] [Google Scholar]
- 18.Cohen, E. A., E. F. Terwilliger, J. G. Sodroski, and W. A. Haseltine. 1988. Identification of a protein encoded by the vpu gene of HIV-1. Nature. 334:532–534. [DOI] [PubMed] [Google Scholar]
- 19.Strebel, K., T. Klimkait, and M. A. Martin. 1988. A novel gene of HIV-1, vpu, and its 16-kilodalton product. Science. 241:1221–1223. [DOI] [PubMed] [Google Scholar]
- 20.Marassi, F. M., C. Ma, H. Gratkowski, S. K. Straus, K. Strebel, M. Oblatt-Montal, M. Montal, and S. J. Opella. 1999. Correlation of the structural and functional domains in the membrane protein Vpu from HIV-1. Proc. Natl. Acad. Sci. USA. 96:14336–14341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ma, C., F. M. Marassi, D. H. Jones, S. K. Straus, S. Bour, K. Strebel, U. Schubert, M. Oblatt-Montal, M. Montal, and S. J. Opella. 2002. Expression, purification, and activities of full-length and truncated versions of the integral membrane protein Vpu from HIV-1. Protein Sci. 11:546–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schubert, U., S. Bour, A. V. Ferrer-Montiel, M. Montal, F. Maldarell, and K. Strebel. 1996. The two biological activities of human immunodeficiency virus type 1 Vpu protein involve two separable structural domains. J. Virol. 70:809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Opella, S. J., S. H. Park, S. Lee, D. Jones, A. Nevzorov, M. Mesleh, A. Mrse, F. M. Marassi, M. Oblatt-Montal, M. Montal, K. Strebel, and S. Bour. 2005. Structure and Function of Vpu from HIV-1. In Viral Membrane Proteins: Structure, Function, and Drug Design. W. Fisher, editor. Kluwer Academic, New York.
- 24.Park, S. H., and S. J. Opella. 2005. Tilt angle of a trans-membrane helix is determined by hydrophobic mismatch. J. Mol. Biol. 350:310–318. [DOI] [PubMed] [Google Scholar]
- 25.Howard, K. P., and S. J. Opella. 1996. High-resolution solid-state NMR spectra of integral membrane proteins reconstituted into magnetically oriented phospholipid bilayers. J. Magn. Reson. B. 112:91–94. [DOI] [PubMed] [Google Scholar]
- 26.Levitt, M. H., D. Suter, and R. R. Ernst. 1986. Spin dynamics and thermodynamics in solid-state NMR cross polarization. J. Chem. Phys. 84:4243–4255. [Google Scholar]
- 27.Fung, B. M., A. K. Khitrin, and K. Ermolaev. 2000. An improved broadband decoupling sequence for liquid crystals and solids. J. Magn. Reson. 142:97–101. [DOI] [PubMed] [Google Scholar]
- 28.Sinha, N., C. V. Grant, C. H. Wu, A. A. De Angelis, S. C. Howell, and S. J. Opella. 2005. SPINAL modulated decoupling in high field double- and triple-resonance solid-state NMR experiments on stationary samples. J. Magn. Reson. 177:197–202. [DOI] [PubMed] [Google Scholar]
- 29.Delaglio, F., S. Grzesiek, G. W. Vuister, G. Zhu, J. Pfeifer, and A. Bax. 1995. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR. 6:277–293. [DOI] [PubMed] [Google Scholar]
- 30.Prosser, R. S., J. S. Hwang, and R. R. Vold. 1998. Magnetically aligned phospholipid bilayers with positive ordering: a new model membrane system. Biophys. J. 74:2405–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mesleh, M. F., S. Lee, G. Veglia, D. S. Thiriot, F. M. Marassi, and S. J. Opella. 2003. Dipolar waves map the structure and topology of helices in membrane proteins. J. Am. Chem. Soc. 125:8928–8935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mesleh, M. F., and S. J. Opella. 2003. Dipolar Waves as NMR maps of helices in proteins. J. Magn. Reson. 163:288–299. [DOI] [PubMed] [Google Scholar]
- 33.Nevzorov, A. A., and S. J. Opella. 2003. Structural fitting of PISEMA spectra of aligned proteins. J. Magn. Reson. 160:33–39. [DOI] [PubMed] [Google Scholar]
- 34.Sanders 2nd, C. R. 1994. Qualitative comparison of the bilayer-associated structures of diacylglycerol and a fluorinated analog based upon oriented sample NMR data. Chem. Phys. Lipids. 72:41–57. [DOI] [PubMed] [Google Scholar]
- 35.Cardon, T. B., E. K. Tiburu, and G. A. Lorigan. 2003. Magnetically aligned phospholipid bilayers in weak magnetic fields: optimization, mechanism, and advantages for X-band EPR studies. J. Magn. Reson. 161:77–90. [DOI] [PubMed] [Google Scholar]
- 36.Caporini, M. A., A. Padmanabhan, T. B. Cardon, and G. A. Lorigan. 2003. Investigating magnetically aligned phospholipid bilayers with various lanthanide ions for X-band spin-label EPR studies. Biochim. Biophys. Acta. 1612:52–58. [DOI] [PubMed] [Google Scholar]
- 37.Opella, S. J., F. M. Marassi, J. J. Gesell, A. P. Valente, Y. Kim, M. Oblatt-Montal, and M. Montal. 1999. Structures of the M2 channel-lining segments from nicotinic acetylcholine and NMDA receptors by NMR spectroscopy. Nat. Struct. Biol. 6:374–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marassi, F. M., and S. J. Opella. 2000. A solid-state NMR index of helical membrane protein structure and topology. J. Magn. Reson. 144:150–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Marassi, F. M., and S. J. Opella. 2003. Simultaneous assignment and structure determination of a membrane protein from NMR orientational restraints. Protein Sci. 12:403–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wang, J., J. Denny, C. Tian, S. Kim, Y. Mo, F. Kovacs, Z. Song, K. Nishimura, Z. Gan, R. Fu, J. R. Quine, and T. A. Cross. 2000. Imaging membrane protein helical wheels. J. Magn. Reson. 144:162–167. [DOI] [PubMed] [Google Scholar]
- 41.Park, S. H., A. A. Mrse, A. A. Nevzorov, A. A. De Angelis, and S. J. Opella. 2006. Rotational diffusion of membrane proteins in aligned phospholipid bilayers by solid-state NMR spectroscopy. J. Magn. Reson. 178:162–165. [DOI] [PubMed] [Google Scholar]
- 42.Park, S. H., S. Prytulla, A. A. De Angelis, J. M. Brown, H. Kiefer, and S. J. Opella. 2006. High-resolution NMR spectroscopy of a GPCR in aligned bicelles. J. Am. Chem. Soc. 128:7402–7403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mesleh, M. F., G. Veglia, T. M. DeSilva, F. M. Marassi, and S. J. Opella. 2002. Dipolar waves as NMR maps of protein structure. J. Am. Chem. Soc. 124:4206–4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zeri, A. C., M. F. Mesleh, A. A. Nevzorov, and S. J. Opella. 2003. Structure of the coat protein in fd filamentous bacteriophage particles determined by solid-state NMR spectroscopy. Proc. Natl. Acad. Sci. USA. 100:6458–6463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thiriot, D. S., A. A. Nevzorov, L. Zagyanskiy, C. H. Wu, and S. J. Opella. 2004. Structure of the coat protein in Pf1 bacteriophage determined by solid-state NMR spectroscopy. J. Mol. Biol. 341:869–879. [DOI] [PubMed] [Google Scholar]
- 46.Koradi, R., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:51–55, 29–32. [DOI] [PubMed] [Google Scholar]
- 47.Kukol, A., and I. T. Arkin. 1999. vpu transmembrane peptide structure obtained by site-specific fourier transform infrared dichroism and global molecular dynamics searching. Biophys. J. 77:1594–1601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wiener, M. C., and S. H. White. 1992. Structure of a fluid dioleoylphosphatidylcholine bilayer determined by joint refinement of x-ray and neutron diffraction data. III. Complete structure. Biophys. J. 61:434–447. [DOI] [PMC free article] [PubMed] [Google Scholar]