

Abstract
Verapamil and amlodipine are calcium ion influx inhibitors of wide clinical use. They are partially charged at neutral pH and exhibit amphiphilic properties. The noncharged species can easily cross the lipid membrane. We have measured with solid-state NMR the structural changes induced by verapamil upon incorporation into phospholipid bilayers and have compared them with earlier data on amlodipine and nimodipine. Verapamil and amlodipine produce a rotation of the phosphocholine headgroup away from the membrane surface and a disordering of the fatty acid chains. We have determined the thermodynamics of verapamil partitioning into neutral and negatively charged membranes with isothermal titration calorimetry. Verapamil undergoes a pK-shift of ΔpKa = 1.2 units in neutral lipid membranes and the percentage of the noncharged species increases from 5% to 45%. Verapamil partitioning is increased for negatively charged membranes and the binding isotherms are strongly affected by the salt concentration. The electrostatic screening can be explained with the Gouy-Chapman theory. Using a functional phosphate assay we have measured the affinity of verapamil, amlodipine, and nimodipine for P-glycoprotein, and have calculated the free energy of drug binding from the aqueous phase to the active center of P-glycoprotein in the lipid phase. By combining the latter results with the lipid partitioning data it was possible, for the first time, to determine the true affinity of the three drugs for the P-glycoprotein active center if the reaction takes place exclusively in the lipid matrix.
INTRODUCTION
Broad-spectrum resistance to chemotherapeutic agents has been termed multidrug resistance (MDR). Although several mechanisms may contribute to MDR in mammalian cells, the best characterized is the efflux or flippase activity of the 170 kDa plasma membrane protein P-glycoprotein (Pgp, MDR1, or ABCB1). Pgp binds its substrates in the cytosolic leaflet of the lipid membrane and flips them to the extracellular leaflet or exports them to the extracellular environment (for review see (2)). Substrate binding to Pgp is best described by a two-step mechanism consisting of 1), a lipid-water partitioning step followed by 2), a binding to the transporter in the lipid phase (l) (3,4). The overall binding constant Ktw for the binding from the aqueous phase (w) to the transporter (t) can thus be expressed as product of the lipid-water partition coefficient, Klw, and the transporter binding constant in the lipid phase, Ktl (5). We measured the transporter-water binding constant, Ktw, and the lipid-water partition coefficient, Klw, for several structurally different drugs and derived the corresponding free energies of binding 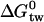 and
and 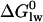 . The free energy of substrate binding to Pgp in the lipid membrane,
. The free energy of substrate binding to Pgp in the lipid membrane, 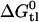 , cannot be measured directly but was determined as the difference
, cannot be measured directly but was determined as the difference 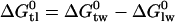 (6). The value
(6). The value 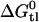 can be rationalized with a modular binding concept based on hydrogen-bond formation (6–8).
can be rationalized with a modular binding concept based on hydrogen-bond formation (6–8).
The quantitative understanding of the two-step Pgp binding mechanism is of importance for efficient pharmacotherapy as well as for drug design. We therefore have selected three calcium channel blockers (verapamil, amlodipine, and nimodipine) of chemically different structure but similar numbers of hydrogen-bond modules (Fig. 1) for a detailed thermodynamic and functional study. Verapamil (pKa 8.9 (9)) and amlodipine (pKa 8.6 (10)) are positively charged at pH 7.4 whereas nimodipine is electrically neutral. Using thermodynamic and spectroscopic techniques we examine the partitioning of verapamil into phospholipid membranes and compare it to previous studies on amlodipine and nimodipine (11). The structure of the lipid membrane at different concentrations of verapamil was elucidated with solid-state NMR methods using selectively deuterated lipids. The influence of verapamil on the order of the lipid membrane is of special interest since it was claimed that a decrease in membrane order would reduce the activity of Pgp (for review, see (12,13)). The thermodynamic results are correlated with a functional assay for the binding of the three calcium-channel antagonists to Pgp in inside-out vesicles of MDR1-transfected mouse embryo fibroblasts (NIH-MDR1-G185) (14,15) and compared to extracellular acidification rate measurements performed with living cells (6,16,17).
FIGURE 1.

Chemical structures and conformational models of three calcium channel antagonists: verapamil, nimodipine, and amlodipine. The three-dimensional structures were obtained by searching the most amphiphilic, energy-minimized conformation with the minimal cross-sectional area, AD. Oxygen and nitrogen molecules are shown in red and blue, respectively. Hydrogen-bond acceptors, constituting the binding modules for P-glycoprotein, are connected with dotted yellow lines. Pgp does not accept secondary amino groups (−NHR) or −NO2 groups (for details, see (7)).
MATERIALS AND METHODS
Materials
Verapamil hydrochloride was purchased from Fluka Biochemika (Buchs, Switzerland), amlodipine maleate from Sequoia Research Products (Pangbourne, United Kingdom), and nimodipine from Sigma-Aldrich (Sternheim, Germany). 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC), 1,2-dioleoyl-trimethylammonium-propane (DOTAP), and 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphoglycerol (POPG) were from Avanti Polar Lipids (Alabaster, AL). All other chemicals were purchased at highest purity from commercial sources.
Preparation of lipid vesicles
Small unilamellar vesicles (SUVs) of ∼30 nm diameter were prepared as follows. Defined amounts of lipid were dissolved in chloroform and were dried first with a stream of N2 and then overnight under high vacuum. For binary lipid mixtures the second lipid was added in chloroform solution to the dried film of the first lipid and treated as before. Subsequently, buffer solution (typically 50 mM HEPES, pH 7.4, plus various NaCl concentrations) was added to the lipid film and the mixture was vortexed extensively. Next, the lipid dispersion was sonicated under a nitrogen atmosphere for 10–25 min (at 10°C) until a clear solution was obtained. Metal debris from the titanium tip was removed by centrifugation at 14,000 g for 10 min.
Cell lines and cell culture
The mouse embryo fibroblast cell lines NIH3T3 and NIH3T3 transfected with the human MDR1 gene, NIH-MDR-G185, were generously provided by Dr. M. M. Gottesman, National Institutes of Health, Bethesda, MD. Cells were maintained as described earlier (16,17). From these cells crude membranes were prepared as described elsewhere (17,18).
NMR measurements
POPC was deuterated either at the α- or β-position of the choline headgroup or at the cis-double bond of the oleic acyl chain (carbon atoms C-9′, C-10′) (19,20),
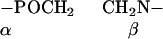 |
A defined amount of deuterated lipid was transferred into a NMR sample tube (typically 10–20 mg lipid) and drug/buffer solution was added to achieve a predefined drug/lipid ratio. For all NMR samples we used 25 mM MES, pH 5.5, and 100 mM NaCl as buffer. The concentration of verapamil was determined before mixing by UV spectroscopy at λ = 277 nm (ɛ = 5818.8 M−1 cm−1). To achieve a homogeneous suspension, the sample was extensively vortexed at room temperature with several freeze-thaw cycles in-between. Centrifugation at 30,000 g for 60 min at room temperature led to a clear supernatant. To calculate the molar amount of verapamil bound per mol of POPC, Xb (mol/mol), the verapamil concentration in the supernatant was determined again. A flat baseline above 380 nm was used as a criterion for complete lipid removal. All 2H-NMR experiments were performed on a Bruker Avance 400 MHz spectrometer (Bruker AXS, Berlin, Germany). 2H-NMR spectra were recorded at 64 MHz with the quadrupole echo technique. The lipid pellets were used without further manipulations. 2H-NMR spectra simulation was done with the NMR WebLab V.4.0 program (21). 31P-NMR spectra were recorded at 161 MHz using a Hahn echo sequence with broadband proton decoupling (WALTZ-16) and a recycle delay of 6 s. The chemical shielding anisotropy, Δσ, was measured as full width at 10% maximum intensity.
Isothermal titration calorimetry
Isothermal titration calorimetry was performed with a VP ITC instrument (Microcal, Northampton, MA). Unless noted otherwise, measurements were made at 37°C. Appropriate buffer solutions were freshly prepared and the pH was properly adjusted when the temperature was changed. The sample cell contained the verapamil solution at a concentration of typically 100 μM. Lipid vesicles suspended in the same buffer as verapamil (lipid concentration of ∼25–30 mM) were placed in a 300 μL syringe. Five microliter injections were made every 5 min. As a control, lipid vesicles were injected into the calorimeter cell containing buffer without verapamil.
Analysis of the ITC data
The classical way to describe drug partitioning into the lipid phase is to use the bulk concentration, CD,eq. Here CD,eq is the equilibrium drug concentration far from the membrane surface. The amount of bound drug,  , is then given by
, is then given by
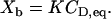 |
(1) |
Here nD is the molar amount of bound drug and  is the total lipid available for binding (for charged drugs that cannot permeate the lipid membrane,
is the total lipid available for binding (for charged drugs that cannot permeate the lipid membrane, 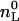 is the lipid on the outer vesicle surface). This simple partitioning law is valid for neutral drug molecules such as nimodipine. A more complex situation is encountered for charged drugs as the adsorption of the cationic amphiphiles leads to a positive surface potential ψ, repelling ions of like charge. Alternatively, the membrane may contain negatively charged lipids, producing a negative surface potential. Under these conditions, drug binding is increased because of electrostatic attraction. In both cases, the drug concentration at the plane of binding, CD,M, is not identical to the bulk concentration, CD,eq, but is given by
is the lipid on the outer vesicle surface). This simple partitioning law is valid for neutral drug molecules such as nimodipine. A more complex situation is encountered for charged drugs as the adsorption of the cationic amphiphiles leads to a positive surface potential ψ, repelling ions of like charge. Alternatively, the membrane may contain negatively charged lipids, producing a negative surface potential. Under these conditions, drug binding is increased because of electrostatic attraction. In both cases, the drug concentration at the plane of binding, CD,M, is not identical to the bulk concentration, CD,eq, but is given by
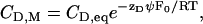 |
(2) |
where zDF0 is the molar electric charge, and RT is the thermal energy. The partition equilibrium Eq. 1 can then be modified as
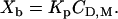 |
(3) |
Electrostatic attraction/repulsion effects are thus explicitly taken into account. Equation 3 predicts a linear relationship between the extent of membrane-bound drug and the drug surface concentration. Comparison with Eq. 1 demonstrates that K is not constant for charged drugs but depends on the surface potential and the effective charge according to  . Consequently, K varies with the drug and salt concentration. For a monovalent drug such as verapamil and anionic POPC/POPG (75:25 mol/mol) membranes in 0.1 M NaCl, K is typically 5–10 times larger than Kp (verapamil concentration is in the mM-range). The surface potential, ψ, can be evaluated with the Gouy-Chapman theory (22,23) and the details of this analysis have been described previously (11,24). The analysis includes the binding of Na+ ions to phosphatidylglycerol using the Langmuir adsorption isotherm with a Na+ binding constant of 0.6 M−1. In this evaluation the HEPES buffer was counted as a 1:1 salt. The sulfonic acid has a pKa ∼ 3.6 (25) and is fully charged; the counterion is Na+. The piperazin ring is 50% charged. This latter effect was not included.
. Consequently, K varies with the drug and salt concentration. For a monovalent drug such as verapamil and anionic POPC/POPG (75:25 mol/mol) membranes in 0.1 M NaCl, K is typically 5–10 times larger than Kp (verapamil concentration is in the mM-range). The surface potential, ψ, can be evaluated with the Gouy-Chapman theory (22,23) and the details of this analysis have been described previously (11,24). The analysis includes the binding of Na+ ions to phosphatidylglycerol using the Langmuir adsorption isotherm with a Na+ binding constant of 0.6 M−1. In this evaluation the HEPES buffer was counted as a 1:1 salt. The sulfonic acid has a pKa ∼ 3.6 (25) and is fully charged; the counterion is Na+. The piperazin ring is 50% charged. This latter effect was not included.
Surface activity measurements
Surface activity measurements were performed at ambient temperature and pH 7.4 (50 mM Tris buffer, 114 mM NaCl) (26,27). Due to the low solubility of amlodipine and nimodipine in water, stock solutions were prepared in methanol. The total concentration of methanol in the Langmuir trough was, however, <5% v/v. The surface activity was corrected for the effect of methanol. Despite this correction, the surface activity of charged drugs dissolved in methanol tends to be slightly higher than that of drugs dissolved in water due to a small pKa shift. To obtain comparable experimental conditions, verapamil was also injected as methanolic stock solution, despite its better water solubility.
Pgp-ATPase activation assay
The P-glycoprotein associated ATPase activity was measured according to Litman et al. (15) in a 96-well microtiter plate. The ATPase assay buffer contained 25 mM Tris-HCl, 50 mM KCl, 3 mM ATP, 2.5 mM MgSO4, 3 mM DTT, 0.5 mM EGTA, 2 mM ouabain, and 3 mM sodium azide, where the latter three compounds were used to inhibit the Ca-, the Na/K-, and the mitochondrial ATPase, respectively. The assay buffer was adjusted to pH 7.4 at 37°C. Membrane vesicles were diluted to a protein concentration of 0.1 mg/ml in ice-cold ATPase assay buffer. Each series of experiments contained 5 μg protein in a total assay volume of 60 μl. Incubation with the various drugs was started by transferring the plate from ice to a water bath, where it was kept 1 h at 37°C. The reaction was terminated by rapidly cooling the plate on ice. The released inorganic phosphate was determined by adding to each well an ice-cold stopping medium (200 μl) containing the color reagent (sulfuric acid 1.43% v/v; and SDS, 0.9% w/v), ammonium molybdate (0.2% (w/v)), and freshly prepared ascorbic acid (1% (w/v))). After incubation at room temperature for 30 min, the released phosphate was quantified calorimetrically at 820 nm using a microplate reader Spectramax M2 (Molecular Devices, Sunnyvale, CA). To determine the vanadate-sensitive Pgp ATPase activity, control samples were incubated in parallel with 500 μM vanadate and the values were subtracted from the values of the Pgp ATPase activation measurements. For stock solutions, drugs were dissolved in DMSO. The DMSO content of the sample was 1.67% (v/v). The data were analyzed according to the model proposed by Litman et al. (15),
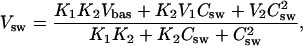 |
(4) |
where Vsw is the rate of Pi release as a function of Csw, the substrate concentration in aqueous solution; Vbas is the basal activity of Pgp in the absence of drug; V1 is the maximum transporter activity (if only activation occurred); and V2 is the minimum activity at infinite substrate concentration. At a substrate concentration, Csw = K1, half-maximum binding of the activating binding region is reached and at a substrate concentration, Csw = K2, half-maximum binding of the inhibitory binding region is reached.
RESULTS
Verapamil-induced structural changes of the lipid bilayer
Fig. 2 shows 2H-NMR spectra obtained with coarse liposomes composed of POPC, deuterated at the α-segment of the choline moiety (−POCD2CH2N−) and measured in buffer at pH 5.5. Under these conditions, verapamil carries a charge of z = +1 (pKa 8.9, at 25°C (9)).
FIGURE 2.

Deuterium and phosphorus-31 NMR spectra of POPC liposomes deuterated at the α-position of the choline headgroup (−POCD2CH2N+). Approximately twenty-five milligrams of POPC was suspended in 50 μL buffer (25 mM, MES 0.1 M NaCl, pH 5.5, deuterium-depleted water) containing different concentrations of verapamil. The two top spectra correspond to pure POPC membranes without verapamil. The spectra below are characterized by increasing drug concentrations. The verapamil/POPC molar ratio from top to bottom is: 0, 0.02, 0.04, 0.07, 0.11, and 0.14. Virtually all verapamil is incorporated into the membrane. (Number of FIDs: 2H NMR spectra 8 K, 31P NMR 2 K.)
All spectra are characteristic of liquid crystalline bilayers with a single quadrupole splitting seen at all drug concentrations. A single, time-averaged quadrupole splitting is also found for the β-segment of the choline headgroup and the cis-double bond of the oleic acyl chain (carbon atoms C-9′, C-10′). Apparently, the mobility of verapamil in the lipid membrane is fast enough so that the presence of the drug is sensed by all lipids in the bilayer within 10−5 s even at low drug concentration. The quadrupole splitting, ΔνQ, which is defined as the separation of the two most intense peaks in the deuterium NMR spectrum, is gradually changed as the molar verapamil/POPC ratio increases. The quadrupole splittings of the labeled carbon atoms are plotted in Fig. 3 as a function of bound drug, Xb (mol drug bound per mol lipid), revealing a linear relationship between the two parameters. For the two headgroup segments, linear regression analysis yields
 |
(5) |
and
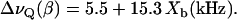 |
(6) |
Fig. 2 also demonstrates that the quadrupole splitting of the α-segment collapses to a single line at a mole fraction of ∼Xb ∼ 0.14. The bilayer structure remains, however, unchanged as evidenced by the phosphorus-31 NMR spectra shown next to the corresponding deuterium NMR spectra. The phosphorus-31 NMR spectra with and without verapamil are very similar and exhibit the typical signature of the bilayer phase (28). The chemical shielding anisotropy is Δσ = −47.9 ppm and remains approximately constant in the concentration range investigated. From the shape of the phosphorus-31 NMR spectra it can be concluded that the long-range structure of the bilayer remains unaltered. The deuterium NMR spectra, on the other hand, provide evidence for a change in the orientation of the choline dipole.
FIGURE 3.

Variation of the deuterium NMR quadrupole splittings of POPC membranes with the verapamil/lipid molar ratio. (A) Phosphocholine headgroup segments: (▪) α-CD2 POPC (−POCD2CH2N); (•) β-CD2-POPC (−POCH2CD2N+). (B) POPC deuterated at the cis-double bond of the oleic acyl chain: (▪) C-9′ deuteron, (•) C-10′ deuteron. Measurements at 22°C in buffer (MES 25 mM + 0.1 M NaCl, pH 5.5).
The verapamil-induced orientational change of the phosphocholine headgroup can be specified in more detail. Binding of the cationic verapamil to neutral POPC membranes imparts a positive electric charge onto the membrane surface. The orientation of choline headgroup P-N+ vector with respect to the membrane surface is, however, dependent on the membrane surface charge density (29). In particular, a positive surface charge moves the N+ end of the P-N+ dipole toward the water phase. This change in orientation entails a counterdirectional response of the quadrupole splittings of the α- and β-segment such that ΔνQ(α) decreases and ΔνQ(β) increases. Indeed, this counterdirectional change of the quadrupole splitting of the two choline segments is also observed for verapamil (Fig. 3). The extent of the out-of-plane rotation cannot yet be quantitated but exceeds that induced by amlodipine (11) or other monovalent hydrophobic drugs when applied at a similar membrane concentration (30).
The influence of verapamil on the hydrophobic part of the bilayer membrane was also examined with deuterium NMR. The deuterium labels were attached to the cis-double bond (C-9′ and C-10′ segment) of the sn-2 oleic acyl chain of POPC. The two deuterons give rise to two separate quadrupole splittings with separations of 13.5 (C-9′) and 2.3 kHz (C-10′), even though they are connected to the same rigid structure (see Fig. 4). The molecular origin of this effect is a tilting of the cis-double bond with respect to the bilayer normal (20).
FIGURE 4.

2H-NMR spectra of POPC membranes deuterated at the cis-double bond of the sn-2-oleic acyl chain and suspended in buffer with various verapamil concentrations. Approximately twenty-five milligrams of lipid suspended in 50 μL buffer (MES 25 mM + 0.1 M NaCl, pH 5.5) were used. The verapamil/lipid molar ratios from bottom-to-top are 0.003, 0.021, 0.037, and 0.048. The smooth lines are the simulated deuterium NMR spectra. The lower panel shows the loss in signal intensity of the C-9′ and C-10′ deuteron as a function of the verapamil/lipid molar ratio referenced to the pure POPC spectrum. The C-9′ deuteron with a 13 kHz splitting shows a much steeper intensity loss than the C-10′ deuteron with a 2 kHz splitting. (4 K free induction decays for all spectra.)
The variation of these quadrupole splittings with the mole fraction of bound verapamil is included in Fig. 3. The quadrupole splittings of both deuterons are moderately decreased upon increasing the verapamil concentration, according to
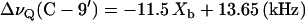 |
(7) |
and
 |
(8) |
The fact that both quadrupole splittings decrease simultaneously suggests a disordering of the hydrophobic core upon verapamil intercalation, that is, a more random motion of the cis-double bond. This is quite different from amlodipine and nimodipine, which induce a small increase in the C-9′ and C-10′ quadrupole splittings (11).
Fig. 4 illustrates a second effect induced by the incorporation of verapamil into the lipid bilayer but limited to the hydrophobic region. All spectra in Fig. 4 were measured under identical conditions, in particular, the same number of free induction decays. Inspection of Fig. 4 nevertheless reveals a loss in signal intensity with increasing amount of verapamil in the membrane, which can be quantified by spectral simulation (smooth lines in Fig. 4). The spectrum with the lowest verapamil concentration serves as a reference spectrum and the areas under the two quadrupole splittings of this spectrum are identical (1:1 intensity ratio, Fig. 4, lower panel). At the highest verapamil concentration (Xb = 0.43) the intensity of the C-10′ deuteron is still 83% of the initial intensity; that of the C-9′ deuteron, however, is reduced to 33%. The most likely explanation of this effect is a reduction in the rate of motion of the cis-double bond caused by a weak complex formation with the aromatic ring system of verapamil. The reduced rate of motion makes the refocusing of the quadrupole echo more difficult, particularly for large quadrupole splittings. Similar effects have been observed for amlodipine in POPC membranes (11) and for reconstituted lipid-protein systems such as sarcoplasmic reticulum membranes, cytochrome c-oxidase, and rhodopsin (31–33). No intensity losses are observed for the α- and β-segments of the choline headgroup, indicating that the dynamics of the headgroup is not affected by the presence of verapamil.
Verapamil binding to lipid bilayers: variation of salt concentration and membrane charge
The adsorption of charged, amphipathic molecules to membranes involves electrostatic and hydrophobic interactions, protonation reactions, and dehydration effects. They contribute, to differing extent, to the heat measured in an isothermal titration calorimetry (ITC) experiment. As an example, Fig. 5 shows the titration of a 100 μM verapamil solution in buffer (50 mM HEPES, 50 mM NaCl, pH 7.4) with sonicated lipid vesicles composed of POPC/POPG (75/25 mol/mol). Verapamil is contained in the calorimeter cell (Vcell = 1.4037 mL) while the lipid suspension is injected at 5 μL aliquots. The total lipid concentration is 25 mM. Each lipid injection causes an exothermic binding reaction as revealed by the heat flow in Fig. 5 A. The size of the titration peak becomes smaller with increasing number of injections as less and less verapamil is available for binding. The integration of the heat flow peaks yield the heats of reaction, hi, and Fig. 5 B displays the cumulative heat of reaction, Σhi, as a function of the injection number i.
FIGURE 5.

Titration of a 100 μM verapamil solution in 50 mM HEPES, pH 7.4 with 30 nm unilamellar lipid vesicles in the same buffer. Lipid composition is POPC/POPG (75:25 mol/mol). The injection of the lipid vesicles was in 5 μL steps. Measuring temperature 37°C. (A) Heat flow and (B) cumulative heat of reaction as a function of injection number.
Fig. 5 further demonstrates that the reaction comes to completion after n ∼ 30 injections. The molar binding enthalpy, 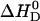 , can then be calculated according to
, can then be calculated according to
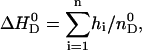 |
(9) |
where  is the total molar amount of verapamil in the calorimeter cell. Fig. 6 and Table 1 summarize the binding enthalpies,
is the total molar amount of verapamil in the calorimeter cell. Fig. 6 and Table 1 summarize the binding enthalpies, 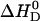 , of verapamil binding to POPC/POPG, POPC, and POPC/DOTAP membranes, as a function of total salt concentration (at 37°C). HEPES buffer was treated as a monovalent salt.
, of verapamil binding to POPC/POPG, POPC, and POPC/DOTAP membranes, as a function of total salt concentration (at 37°C). HEPES buffer was treated as a monovalent salt.
FIGURE 6.

Reaction enthalpies of verapamil binding to phospholipid vesicles (30 nm) of different lipid composition. Variation of the binding enthalpy, 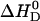 , with the salt concentration. (▪) POPC/POPG 75:25 mol/mol, (•) pure POPC, and (▵) POPC/DOTAP 95:5 mol/mol.
, with the salt concentration. (▪) POPC/POPG 75:25 mol/mol, (•) pure POPC, and (▵) POPC/DOTAP 95:5 mol/mol.
TABLE 1.
Thermodynamics of verapamil binding to phospholipids bilayer vesicles of different charge and size
| SUV 30 nm | POPC mol % | POPG mol % | HEPES mM | NaCl | Drug electric charge near membrane surface 〈z〉 | 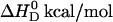 |
Kp M−1 | 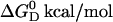 |
|---|---|---|---|---|---|---|---|---|
| 75 | 25 | 50 | 100 | 0.84–0.89 | −3.2 | 520 | −3.84 | |
| 75 | 25 | 50 | 50 | 0.87–0.93 | −2.1 | 400 | −3.75 | |
| 75 | 25 | 50 | 25 | 0.88–0.94 | −2.8 | 450 | −3.75 | |
| 75 | 25 | 50 | 0 | 0.91–0.96 | −3.3 | 300 | −3.50 | |
| 80 | 20 | 25 | 0 | 0.91–0.97 | −2.5 | 380 | −3.65 | |
| 80 | 20 | 5 | 0 | 0.96–0.99 | −2.9 | 330 | −3.56 | |
| 100 | 50 | 100 | 0.57–0.63 | −1.1 | 900 | −4.18 | ||
| 100 | 50 | 50 | 0.57–0.62 | −0.9 | 800 | −4.1 | ||
| 100 | 50 | 25 | 0.53–0.61 | −0.4 | 1200 | −4.35 | ||
| TRIS | ||||||||
| 100 | 50 | 100 | 0.59–0.64 | −3.1 | 900 | −4.18 | ||
 |
||||||||
| 100 | 50 | 100 | 0.59–0.64 | +1.4 | 750 | −4.06 | ||
| DOTAP | HEPES | |||||||
| 94.2 | 5.8 | 50 | 100 | 0.49–0.51 | −1.1 | 500 | −3.81 | |
| LUV | ||||||||
| 100 nm | 100 | 50 | 100 | 0.61–0.64 | −1.0 | 470 | −3.78 | |
| 100 | 50 | 50 | 0.61–0.63 | −1.1 | 400 | −3.68 | ||
| 100 | 50 | 25 | 0.59–0.63 | −0.6 | 500 | −3.81 |
Note that all measurements are at 37°C, pH 7.4.
For negatively charged POPC/POPG vesicles the binding enthalpy is almost independent of the salt concentration and averages to 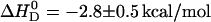 . Neutral POPC and cationic POPC/DOTAP vesicles have
. Neutral POPC and cationic POPC/DOTAP vesicles have 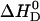 values in the range of −0.4 to −1.1 kcal/mol, which decrease with increasing salt concentration. At salt concentrations <50 mM for POPC and <75 mM for POPC/DOTAP (95:5 mol/mol), no heat of reaction could be observed. Also, no drug binding could be measured when the DOTAP molar percentage was raised above 10 mol %.
values in the range of −0.4 to −1.1 kcal/mol, which decrease with increasing salt concentration. At salt concentrations <50 mM for POPC and <75 mM for POPC/DOTAP (95:5 mol/mol), no heat of reaction could be observed. Also, no drug binding could be measured when the DOTAP molar percentage was raised above 10 mol %.
It is possible to deduce the amount of bound drug directly from the calorimetric titration according to
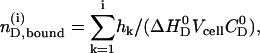 |
(10) |
where 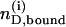 is the molar amount of bound drug after i injections,
is the molar amount of bound drug after i injections, 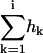 is the cumulative heat of the first i injections, Vcell is the volume of drug solution in the calorimeter cell, and
is the cumulative heat of the first i injections, Vcell is the volume of drug solution in the calorimeter cell, and 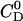 is the total drug concentration. The amount of free drug is then given from mass conservation as
is the total drug concentration. The amount of free drug is then given from mass conservation as
 |
(11) |
The degree of binding was defined above as
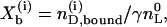 |
(12) |
where 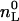 is the total molar amount of lipid and
is the total molar amount of lipid and 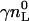 is the fraction of lipid accessible to the drug. It is hence possible to deduce the complete binding isotherm Xb = f (ceq) from the calorimetric titration without invoking a specific binding model.
is the fraction of lipid accessible to the drug. It is hence possible to deduce the complete binding isotherm Xb = f (ceq) from the calorimetric titration without invoking a specific binding model.
For POPC/POPG vesicles at pH 5.5, 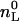 is only 60% of the total lipid (γ = 0.6), since the drug is fully charged and cannot translocate across the membrane. For all other measurements described in this study, a rapid translocation of verapamil across the membrane was assumed (γ = 1). For example, POPC/POPG vesicles at 150 mM salt and pH 7.4 exhibit a surface potential of ∼−30 mV, which reduces the pH near the membrane surface to pH 6.8. At this pH, the neutral form of verapamil accounts for only 1% of the total drug concentration. However, upon entering the membrane, verapamil experiences a pKa shift by 1.2 units (see below). Both effects together are sufficient to establish a rapid trans-membrane equilibrium.
is only 60% of the total lipid (γ = 0.6), since the drug is fully charged and cannot translocate across the membrane. For all other measurements described in this study, a rapid translocation of verapamil across the membrane was assumed (γ = 1). For example, POPC/POPG vesicles at 150 mM salt and pH 7.4 exhibit a surface potential of ∼−30 mV, which reduces the pH near the membrane surface to pH 6.8. At this pH, the neutral form of verapamil accounts for only 1% of the total drug concentration. However, upon entering the membrane, verapamil experiences a pKa shift by 1.2 units (see below). Both effects together are sufficient to establish a rapid trans-membrane equilibrium.
Fig. 7 then displays ITC-derived binding isotherms for POPC/POPG bilayers at various salt concentrations. The strongest binding is observed for the lowest salt concentration where the electrostatic attraction between the anionic membrane and the cationic drug is maximal. All binding isotherms have a curved appearance and the binding constant defined according to Eq. 1 varies with the verapamil and salt concentration. At a verapamil concentration of CD,eq = 10 μM, apparent binding constants are K ∼ 4 × 103 M−1. In contrast, the solid lines in Fig. 7 were calculated with the Gouy-Chapman theory and a common binding constant Kp = 410 ± 30 M−1 describes all three binding isotherms over the whole concentration range. The value Kp refers to the binding of the charged form of verapamil. Table 1 summarizes the Kp values for the different systems investigated. The value Kp is distinctly smaller than the apparent binding constant K, since the electrostatic attraction is not included.
FIGURE 7.

Verapamil binding isotherms for POPC/POPG membranes (75:25 mol/mol) at three different salt concentrations. All measurements made at pH 7.4 and 37°C. 50 mM HEPES + 50 mM NaCl; 50 mM HEPES; and 25 mM HEPES. The solid lines were calculated with the partition constants, Kp, given in Table 1 and the Gouy-Chapman theory. A rapid translocation of the neutral form of verapamil across the membrane was assumed.
For neutral POPC and cationic POPC/DOTAP vesicles at pH 7.4, the pH increases near the membrane surface and the fraction of the neutral form also increases. Membrane translocation of the drug is thus easily possible. Fig. 8 shows binding isotherms for POPC SUVs (three different salt concentrations) and for POPC/DOTAP SUVs. The extent of drug binding to POPC and POPC/DOTAP SUVs is clearly smaller than that observed for POPC/POPG SUVs at the same salt concentration. Electrostatic attraction/repulsion is the dominant factor for verapamil binding to charged membranes. For anionic POPC/POPG membranes the electrostatic interaction is favorable, for cationic POPC/DOTAP membranes it is repulsive. However, even for neutral membranes, electrostatics is important since the membrane surface becomes positively charged as soon as the first cationic drug molecule is incorporated into the membrane. Further binding of drug molecules thus becomes increasingly more difficult. The solid lines in Fig. 8 were again calculated with the Gouy-Chapman theory.
FIGURE 8.

Binding of verapamil to POPC SUVs and mixed POPC/DOTAP (94.2:15.8 mol/mol) SUVs at various salt concentrations. (○) Pure POPC SUVs; (□) POPC/DOTAP SUVs. All measurements in 50 mM Tris or HEPES buffer + various concentrations of NaCl at 37°C: The solid lines are theoretical binding isotherms calculated with the partition coefficients listed in Table 1.
The results described above were obtained with vesicles prepared by sonication, having an average diameter of ∼30 nm (SUV). Verapamil binding to POPC membranes was also studied with unilamellar vesicles of 100 nm diameter (LUVs) prepared by extrusion through polycarbonate filters. LUVs exhibit a tighter lipid packing than SUVs and resemble more closely planar lipid bilayers. The ITC data were analyzed with the same model as described for SUVs. Again fast translocation of the drug (in its uncharged form) across the membrane was assumed. The experimental results are included in Table 1. The reaction enthalpy at 37°C is 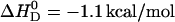 for SUVs and
for SUVs and 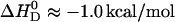 for LUVs. The binding constants, again deduced with the Gouy-Chapman theory, are somewhat smaller than those of SUVs (see Table 1).
for LUVs. The binding constants, again deduced with the Gouy-Chapman theory, are somewhat smaller than those of SUVs (see Table 1).
Membrane-induced pKa shift of verapamil
The ionization constant of verapamil in aqueous solution at 25°C is pKa 8.9 (9). It is temperature-dependent and decreases with increasing temperature (see below). A decrease in pKa can also be expected for verapamil entering the lipid membrane, because the neutral form is favored in the nonpolar environment. The pKa shift was quantified by measuring the binding reaction in buffers of different dissociation enthalpies, since the reaction enthalpy is the sum of the verapamil deprotonation and the buffer protonation. Verapamil binding to POPC vesicles yields  in 50 mM Tris buffer,
in 50 mM Tris buffer, 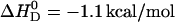 in 50 mM HEPES buffer, and
in 50 mM HEPES buffer, and 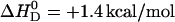 in phosphate buffer (all measurements at 100 mM NaCl, pH 7.4). This provides evidence for a deprotonation reaction (34–36) of verapamil as it enters the membrane. Fig. 9 shows a plot of the binding enthalpy,
in phosphate buffer (all measurements at 100 mM NaCl, pH 7.4). This provides evidence for a deprotonation reaction (34–36) of verapamil as it enters the membrane. Fig. 9 shows a plot of the binding enthalpy, 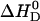 , versus the buffer dissociation enthalpy, ΔHbuffer, yielding
, versus the buffer dissociation enthalpy, ΔHbuffer, yielding
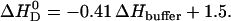 |
(13) |
From the slope it can be deduced that 0.41 H+ dissociate upon verapamil insertion into the neutral POPC membrane. While the average charge of verapamil in solution at pH 7.4 is 〈zD〉 = 0.97 e.u., the membrane-bound drug carries an average charge of 〈zD〉 ≈ 0.56 e.u. The reduction by δz = 0.41 e.u. can be traced back to two sources. Upon binding to the membrane, verapamil repels H+ ions from the membrane surface. At 150 mM salt and 0.1 mM verapamil, the pH at the membrane surface increases to ∼pH 7.6 and the electric charge of verapamil decreases concomitantly to z = 0.95 e.u. (δz = 0.02 e.u.). However, from the buffer dependence one deduces a much larger change of δz = 0.41 e.u., attesting to a pKa shift as the drug enters the hydrophobic environment. At pH 7.6, a pKa shift from pKa ∼ 8.9 to pKa ∼ 7.7 reduces the charge from 〈z〉 = 0.95 to z = 0.56 (δz = 0.39). The two effects together (pH change plus pKa shift) then explain the experimental data. Based on chemical equilibria considerations it can be calculated that the uncharged drug has a 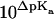 ∼16-times higher partition-coefficient for the lipid membrane than the protonated species. The binding constant of uncharged verapamil is thus Kp = 6.5 × 103 M−1 for POPC/POPG membranes and Kp ∼ 1.6 × 104 M−1 for POPC vesicles (at pH 7.4 and 37°C).
∼16-times higher partition-coefficient for the lipid membrane than the protonated species. The binding constant of uncharged verapamil is thus Kp = 6.5 × 103 M−1 for POPC/POPG membranes and Kp ∼ 1.6 × 104 M−1 for POPC vesicles (at pH 7.4 and 37°C).
FIGURE 9.

Variation of the verapamil binding enthalpies with the buffer dissociation enthalpies. POPC vesicles with 30 nm diameter. Measurements made in Tris (ΔHDiss = 11.51 kcal/mol), HEPES (ΔHDiss = 4.9 kcal/mol), and phosphate (ΔHDiss = 1.22 kcal/mol) at 37°C (36).
For membranes composed of diphytanoyl PC, a dissociation constant for uncharged verapamil was estimated as Kd = 0.061 ± 0.01 mM at pH 10.5 based on electrophoretic and membrane potential measurements (37). This translates into a binding constant of Kp = 1.6 × 104 M−1.
We also measured the molar reaction enthalpy of verapamil binding to POPC vesicles at pH 5.5. At this pH, verapamil is fully charged and no deprotonation takes place upon binding to the membrane. The binding enthalpy was found to be 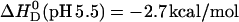 . On the other hand, the (extrapolated) binding enthalpy at pH 7.4 in the absence of a buffer dissociation reaction is ΔHD (pH 7.4) = 1.5 kcal/mol (see Fig. 9 for ΔHDiss = 0). The value ΔHD (pH 7.4) is thus composed of the binding enthalpy of the fully charged drug, ΔHD (pH 5.5), plus the dissociation enthalpy for 0.41 H+,
. On the other hand, the (extrapolated) binding enthalpy at pH 7.4 in the absence of a buffer dissociation reaction is ΔHD (pH 7.4) = 1.5 kcal/mol (see Fig. 9 for ΔHDiss = 0). The value ΔHD (pH 7.4) is thus composed of the binding enthalpy of the fully charged drug, ΔHD (pH 5.5), plus the dissociation enthalpy for 0.41 H+,
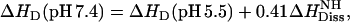 |
(14) |
where 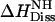 is the dissociation enthalpy of the verapamil amino group. The latter is then calculated as
is the dissociation enthalpy of the verapamil amino group. The latter is then calculated as 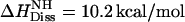 . This result is consistent with data obtained for N-terminal amino group of peptides where the dissociation energy is ∼11 ± 2 kcal/mol (38).
. This result is consistent with data obtained for N-terminal amino group of peptides where the dissociation energy is ∼11 ± 2 kcal/mol (38).
Heat capacity change ΔCP0
We have measured the temperature-dependence of the verapamil-membrane partition equilibrium. For PC/PG (75:25 mol/mol) membranes in 50 mM HEPES at pH 7.4, 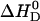 shows only a small temperature-dependence with a molar heat capacity of
shows only a small temperature-dependence with a molar heat capacity of 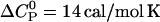 . For pure PC SUVs in buffer (100 mM NaCl, 50 mM HEPES, pH 7.4), the heat capacity change is
. For pure PC SUVs in buffer (100 mM NaCl, 50 mM HEPES, pH 7.4), the heat capacity change is 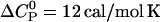 .
.
Molecular cross-sectional area and air-water partition coefficient
Amphiphilic molecules such as verapamil, amlodipine, and nimodipine partition into the air-water and lipid-water interface such that the polar groups remain in either the aqueous phase (ɛ ≈ 80) or the polar headgroup region of the lipid membrane (ɛ ≈ 30). The hydrophobic group is then exposed to air (dielectric constant, ɛ ≈ 1) or inserts into the lipid core region (ɛ ≈ 2). The approximate conformation of the three Ca2+ blockers, calculated by optimizing polar and nonpolar interactions, are shown in Fig. 1 (39). The cross-sectional areas of the three molecules, relevant for membrane insertion, were calculated as AD = 82 Å2 for verapamil, 66.2 Å2 for amlodipine, and 69.4 Å2 for nimodipine.
The surface activities of the three Ca2+ blockers were measured with the Wilhelmy plate method (data not shown) and were described by the Szyszkowski isotherm. The measured cross-sectional area of verapamil in aqueous solution is AD = 82 ± 2 Å2 and in good agreement with the calculated value. The cross-sectional areas of amlodipine (AD = 122 Å2) and nimodipine (AD = 87 Å2) are, however, larger than the calculated data, which is caused by association effects at higher concentrations. For the following calculations, we use the calculated cross-sectional areas.
The air-water partition coefficients are KA/W = 1.7 × 105 M−1 for verapamil, KA/W =1.9 × 106 M−1 for amlodipine, and KA/W = 7.1 × 105 M−1 for nimodipine. Insertion into the lipid bilayer expands the surface area and requires additional expansion work πAD (40) where π is the monolayer-bilayer equivalence pressure (41). The lipid-water partition coefficient can then be calculated from the air-water partition coefficient as KLip/W = KA/W exp (−π AD/kT) (26). For POPC LUVs (100 nm) and planar membranes with an equivalence pressure of π ∼ 30 mN/m, the lipid-water partition coefficient of charged verapamil is predicted to be Klip/W ≈ 540 M−1 in excellent agreement with the ITC measurements. For amlodipine Klip/W = 1.8 × 104 M−1, again in good agreement with previous ITC measurements (Kp = 1.6 × 104 M−1 at pH 7.2, (11)). For nimodipine, the lipid water partition coefficient was calculated as Klip/W = 5.4 × 103 M−1. This value must be considered as a lower limit as nimodipine has a strong tendency to adsorb to the vessel walls. The data are summarized in Table 2.
TABLE 2.
Comparison of verapamil, amlodipine, and nimodipine; bilayer structure, binding thermodynamics, and P-glycoprotein activity
| Nimodipine | Amlodipine | Verapamil | |
|---|---|---|---|
| Deuterium NMR | |||
| Headgroup segments | |||
| mα (kHz/mol) | no effect* | −30.50* | −46.14 |
| mβ (kHz/mol) | no effect* | 15.25* | 15.30 |
| cis-double bond | |||
| mC-9′ (kHz/mol) | −11.50 | ||
| mC-10′ (kHz/mol) | −21.90 | ||
| Surface activity measurements | |||
| Cross-sectional area AD (Å2) | 69.4 | 66.2 | 82 |
| KA/W (M−1)† | 7.1 × 105‡ | 1.9 × 106 | 1.7 × 105 |
| Klip/w (M−1) at 30 mN/m | 5.4 × 103‡ | 1.8 × 104 | 540 |
| ITC measurements | |||
| Kp (M−1) LUVS | nd | 7.6 × 103§ | 470¶ |
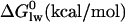 |
−5.275 | −5.484 | −3.776 |
| ΔH0 (kcal/mol) | nd | −8.90§ | −1.0¶ |
| Max. electric charge | 0 | 1 | 1 |
| pK | — | 8.6 | 8.9 |
| Pgp activity | |||
| K1 (M) | 7.9 × 10−7 | 1.1 × 10−7 | 9.5 × 10−7 |
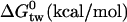 |
−8.624 | −9.834 | −8.511 |
| K2 (M) | — | 3.9 × 10−5 | 3.7 × 10−5 |
| V1 (%) | 2.06 | 2.13 | 2.6 |
| Drug affinity in lipid bilayer | |||
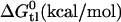 |
−3.35 | −4.350 | −4.735 |
| Ktl (M−1) | 234 | 1200 | 2240 |
| No. of H-bond acceptors | 4 | 4 | 5 |
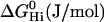 |
−0.84 | −1.088 | −0.947 |
Data taken from Bäuerle et al. (11).
Data obtained with methanolic stock solutions (see Materials and Methods).
Lower limit due to adsorption to the Teflon trough.
Multilamellar POPC liposomes, 0.1 M NaCl, 10 mM Tris, pH 7.25. Bäuerle et al. (11) gives Kp = 1.55 × 104 M−1 and ΔH0 = −8.9 kcal/mol at 23°C. Using van' t Hoff's law and assuming a temperature-independent ΔH0, Kp was recalculated for 37°C.
100 nm POPC LUVs, 0.1 M NaCl, 50 mM HEPES, pH 7.4, 37°C.
P-glycoprotein transporter activity measurements with verapamil, amlodipine, and nimodipine
The P-glycoprotein transporter (Pgp) accepts its substrates from the cytosolic membrane leaflet. The Pgp transport efficiency is determined by 1), the lipid solubility of the substrate; and 2), the affinity of the substrate for the transporter in the lipid phase. The latter can be estimated on the basis of characteristic hydrogen-bonding patterns (6,7). Pgp shows basal activity in the absence of substrates. In the presence of substrates it follows a bell-shaped curve with an initial increase in activation (characterized by the concentration of half-maximum activation, K1) followed by a decrease in activation at higher drug concentrations (characterized by the concentration of half-maximum inhibition, K2). The model is detailed in references (6,15). Experimental results obtained with inside-out vesicles using a phosphate assay are shown in Fig. 10 for both verapamil and amlodipine. The quantitative analysis in terms of the model described by Litman et al. (15) yields the parameters K1, K2 and also the corresponding reaction velocities V1 and V2. The data are summarized in Table 2. As outlined previously, the parameters K1 and K2 can be interpreted as dissociation constants. Table 2 reveals that amlodipine has a higher affinity to Pgp than verapamil during the activation phase. Whereas the inhibition phase can be well measured for verapamil it cannot be measured to high concentrations for amlodipine and nimodipine due to drug association in solution. Partitioning into the lipid membrane requires the monomeric form of drugs and is not possible at concentrations which are higher than the critical micelles concentration.
FIGURE 10.

Pgp activation profiles obtained by a phosphate release assay with inside-out vesicles prepared from NIH-MDR-G185 cells. (▪) Verapamil; (○) amlodipine.
DISCUSSION
Structural aspects of verapamil binding
Verapamil HCl is a calcium-ion influx inhibitor of wide clinical use. It is administered as a racemic mixture of R- and S-enantiomers. At low drug concentration and pH 7.4, a neutral (3%) and a charged species (97%) are in equilibrium in aqueous solution. The molecule exhibits amphiphilic properties and is well soluble in water, organic solvents, and lipid membranes. The pKa-value of verapamil decreases distinctly with increasing temperature as the present ITC data show that the dissociation enthalpy is endothermic with 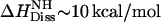 .
.
Fig. 1 displays the conformations of verapamil, amlodipine, and nimodipine in the lipid membrane obtained by an energy minimization calculation (39). Verapamil adopts a folded conformation such that both aromatic ring systems can be inserted into the hydrophobic core of the membrane whereas the charged amino group remains at the lipid-water interface. Partitioning of verapamil into the lipid membrane modulates the lipid bilayer structure, as evidenced by deuterium NMR. The predominant effect in the hydrocarbon region is a disordering of the hydrocarbon chains and a weak complex formation of the cis-double bond with the aromatic rings of verapamil. Hydrocarbon chain disordering is a common phenomenon when proteins or other nonlipid components are inserted into the lipid bilayer (11,32). Particularly effective are detergent molecules (42,43), whereas cholesterol has the opposite effect; that is, the rodlike molecule induces a stiffening of the hydrocarbon chain (44–47). Membrane disordering has been implied in Pgp inactivation but cannot play an important role for verapamil as this molecule is considered as the substrate per-excellence for specific drug-Pgp interactions. At the headgroup level, verapamil moves the +N end of the choline dipole toward the water phase. Verapamil is more efficient than amlodipine in turning the −P-N+ dipole since the change of the ΔνQ(α) quadrupole-splitting-per-mole-incorporated drug is mα = −46.1 kHz/mol for verapamil and only mα = −30.5 for amlodipine. Nimodipine as a noncharged molecule has no effect (11). The consequence of this change in dipole orientation is a change in the electric field across the bilayer membrane. The −P-N+ dipole has a large dipole moment of ∼25 Debye (48). If the −P-N+ dipole is approximately parallel to the membrane surface, as in a pure POPC bilayer (49), the electric dipole field cannot penetrate deeply into the membrane. An orientation 20° away from the membrane surface can, however, creates a field of ∼100 mV in the adjacent hydrophobic part of the membrane with its low dielectric constant of ɛ = 2. Dipole fields are not efficiently screened by salt and are thus long-range. The electric effect of a single verapamil molecule will therefore extend over several layers of surrounding phospholipids. Electric fields of 100 mV across a distance of 2 nm thickness correspond to a field strength of 5 × 107 V/m and can induce conformational changes in proteins.
Thermodynamic binding parameters and Pgp activation
The extent of partitioning of verapamil into a lipid bilayer membrane is influenced by the electric charge of the membrane surface and the screening of Coulombic interactions through inert electrolytes. This is illustrated with the binding isotherms obtained for negatively charged (Fig. 7) and neutral (Fig. 8) membranes. In both cases, increasing NaCl concentrations decrease the amount of bound verapamil. Negatively charged POPC/POPG (75/25 mol/mol) membranes exhibit a surface potential of −40 mV in 150 mM NaCl, decreasing to −115 mV in 5 mM NaCl. The verapamil binding-affinity increases in parallel with the electrostatic attraction (Fig. 7). By using surface concentrations, CD,M, instead of bulk concentrations, the variation of electrostatic attraction can be accounted for, leading to constant intrinsic binding constant. The solid lines in Fig. 7 were simulated with similar binding constants (Kp = 410 ± 30 M−1; see Table 1). The average electric charge of verapamil is 〈z〉 ∼ 0.85–0.99.
A different situation is encountered for pure POPC bilayers. They are noncharged in the absence of verapamil and become positively charged upon drug binding. Under the present experimental conditions the surface potentials are small (ψ ∼ 5–15 mV) and the binding constants determined with and without electrostatic correction differ by ∼0–20% only. The insertion into the hydrophobic membrane entails a distinct pKa-shift and the electric charge of verapamil in the membrane is only 〈z〉 ∼ 0.5. The binding constant of the charged species is Kp ∼ 400–500 M−1 for LUVs but Kp∼ 900–1200 for SUVs. Verapamil binding to phosphatidylcholine bilayers was determined previously by a centrifugation assay at 22°C (1). For multilamellar liposomes composed of egg lecithin (which typically contains 40% POPC), a verapamil partition coefficient of PLipid ∼ 267 was determined (see also Fig. 4).
Verapamil binding to POPC LUVs can be compared to related data obtained for amlodipine and nimodipine leading to the following order of partition coefficients Kp, for POPC LUVs (at 37°C): verapamil 470 M−1 < nimodipine ∼ 5.4 × 103 M−1 < amlodipine 7.6 × 103 M−1 (see Table 2).
Lipid solubility is a prerequisite for a drug to be recognized by Pgp, since the active center of this enzyme is located in the inner part of the lipid membrane. The Pgp activity can be measured with a phosphate release assay or, alternatively, a Cytosensor assay (6). Both assays refer to the overall process, that is, the binding of the drug from the aqueous phase to the active center of the transporter in the lipid phase. The corresponding free energy,  , is given by
, is given by  , where K1 has been defined above as the dissociation constant derived from the activation part of the bell-shaped curve given by Eq. 4. The value
, where K1 has been defined above as the dissociation constant derived from the activation part of the bell-shaped curve given by Eq. 4. The value 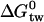 can be divided into two physically distinct processes, namely 1), the partitioning of the drug from water into the lipid membrane,
can be divided into two physically distinct processes, namely 1), the partitioning of the drug from water into the lipid membrane, 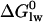 , followed by 2), the binding of the drug to the transporter in the lipid matrix,
, followed by 2), the binding of the drug to the transporter in the lipid matrix, 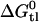 (6):
(6):
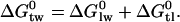 |
(15) |
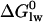 is related to the lipid-water partition coefficient according to
is related to the lipid-water partition coefficient according to 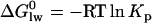 . In this and previous studies we have measured Kp by ITC and by surface activity measurements. We thus know
. In this and previous studies we have measured Kp by ITC and by surface activity measurements. We thus know 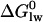 for all three drugs from physical-chemical experiments. On the other hand, Pgp activation has been measured in the present study for verapamil, amlodipine, and nimodipine with the phosphate release assay leading to the overall free energy
for all three drugs from physical-chemical experiments. On the other hand, Pgp activation has been measured in the present study for verapamil, amlodipine, and nimodipine with the phosphate release assay leading to the overall free energy  (Table 2). Knowledge of
(Table 2). Knowledge of 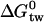 and
and  then allows the evaluation of
then allows the evaluation of 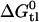 , i.e., the affinity of the drug for the transporter in the lipid phase. The corresponding results for verapamil, amlodipine, and nimodipine are
, i.e., the affinity of the drug for the transporter in the lipid phase. The corresponding results for verapamil, amlodipine, and nimodipine are 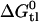 = −4.7, −4.4, and −3.4 kcal/mol, respectively.
= −4.7, −4.4, and −3.4 kcal/mol, respectively.
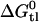 can be further converted to lipid binding constants and it is thus possible, for the first time, to derive the binding constants of verapamil and amlodipine to Pgp in the lipid phase. The corresponding numbers are Ktl = 2.24 × 103 for verapamil, 1.2 × 103 for amlodipine, and 230 for nimodipine.
can be further converted to lipid binding constants and it is thus possible, for the first time, to derive the binding constants of verapamil and amlodipine to Pgp in the lipid phase. The corresponding numbers are Ktl = 2.24 × 103 for verapamil, 1.2 × 103 for amlodipine, and 230 for nimodipine.
We have previously proposed a model which explains the substrate versatility of Pgp on the basis of a modular binding concept, that is, Pgp recognizes well-defined hydrogen-bond acceptor groups (7). Not all hydrogen-bond acceptor groups form hydrogen bonds with same free energy. We distinguish among strong (oxygen atoms), intermediate (nitrogen and sulfur atoms, phenyl groups), and weak (fluorine atoms) hydrogen-bond acceptors, weighted with hydrogen-bond energy units of EUH = 1, 0.5, and 0.25, respectively. This model leads to EUH = 5 for verapamil and EUH = 4 for amlodipine and nimodipine, respectively. Based on the above free energy of binding in the lipid phase, 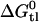 , the average bonding free energy per hydrogen bond is thus
, the average bonding free energy per hydrogen bond is thus 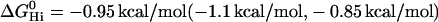 for verapamil (amlodipine, nimodipine). This is in good agreement with a larger set of 15 drug molecules where the free energy of binding from water to the transporter was derived from Cytosensor measurements monitoring the extracellular acidification rate (see (6), Table 1). The average free energy per hydrogen bond was found to be
for verapamil (amlodipine, nimodipine). This is in good agreement with a larger set of 15 drug molecules where the free energy of binding from water to the transporter was derived from Cytosensor measurements monitoring the extracellular acidification rate (see (6), Table 1). The average free energy per hydrogen bond was found to be 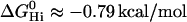 .
.
Transport of calcium channel antagonists by Pgp
As Pgp accepts its substrates from the cytosolic leaflet, lipid solubility is an important prerequisite for a substrate to be recognized by Pgp. The second factor is the binding affinity of the drug to Pgp determined by the respective hydrogen-bonding patterns. The above analysis shows that verapamil exhibits the lowest lipid solubility but has the highest binding affinity to the transporter. If dissolved at equal concentrations in the lipid phase, verapamil is binding more efficiently than amlodipine or nimodipine. However, if dissolved at equal concentrations in the aqueous phase, amlodipine and nimodipine are more efficient in saturating Pgp because of their better lipid solubility. On the other hand, amlodipine and nimodipine diffuse more rapidly across the lipid membrane than verapamil due to the smaller cross-sectional area of the former two. Thus they can escape transport by Pgp more easily. Nimodipine, moreover, lacks the cationic charge and is thus not retained at the inner negatively charged membrane leaflet from which Pgp takes its substrates. Nimodipine can therefore be expected to cross the blood-brain barrier more easily. Indeed, in animal experiments, nimodipine had a strong effect on dilating the cerebral arteries, whereas the other two agonists act mainly on peripheral and cardiac vessels.
In conclusion, we have provided a complete binding analysis of verapamil and amlodipine to P-glycoprotein. We have dissected drug binding into a partitioning step into the lipid membrane, followed by the actual binding to the Pgp active site in the hydrophobic membrane. We could thus deduce the intrinsic binding constants of verapamil and amlodipine to Pgp in the lipid phase. All three drugs produce some disordering of the hydrocarbon chains. However, Pgp is not deactivated by membrane disordering as proposed previously.
Acknowledgments
G. Gerebtzoff was helpful in providing the computer figures and his scientific support is gratefully acknowledged.
This work was supported by the Swiss National Science Foundation grant No. 3100-107793.
References
- 1.Romsicki, Y., and F. J. Sharom. 1999. The membrane lipid environment modulates drug interactions with the P-glycoprotein multidrug transporter. Biochemistry. 38:6887–6896. [DOI] [PubMed] [Google Scholar]
- 2.Higgins, C. F., and M. M. Gottesman. 1992. Is the multidrug transporter a flippase? Trends Biochem. Sci. 17:18–21. [DOI] [PubMed] [Google Scholar]
- 3.Lu, P., R. Liu, and F. J. Sharom. 2001. Drug transport by reconstituted P-glycoprotein in proteoliposomes. Effect of substrates and modulators, and dependence on bilayer phase state. Eur. J. Biochem. 268:1687–1697. [PubMed] [Google Scholar]
- 4.Omote, H., and M. K. Al-Shawi. 2006. Interaction of transported drugs with the lipid bilayer and P-glycoprotein through a solvation exchange mechanism. Biophys. J. 90:4046–4059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Seelig, A., and E. Gatlik-Landwojtowicz. 2005. Inhibitors of multidrug efflux transporters: their membrane and protein interactions. Mini Rev. Med. Chem. 5:135–151. [DOI] [PubMed] [Google Scholar]
- 6.Gatlik-Landwojtowicz, E., P. Aanismaa, and A. Seelig. 2006. Quantification and characterization of P-glycoprotein-substrate interactions. Biochemistry. 45:3020–3032. [DOI] [PubMed] [Google Scholar]
- 7.Seelig, A. 1998. A general pattern for substrate recognition by P-glycoprotein. Eur. J. Biochem. 251:252–261. [DOI] [PubMed] [Google Scholar]
- 8.Sauna, Z. E., M. B. Andrus, T. M. Turner, and S. V. Ambudkar. 2004. Biochemical basis of polyvalency as a strategy for enhancing the efficacy of P-glycoprotein (ABCB1) modulators: stipiamide homodimers separated with defined-length spacers reverse drug efflux with greater efficacy. Biochemistry. 43:2262–2271. [DOI] [PubMed] [Google Scholar]
- 9.Hasegawa, J., T. Fujita, Y. Hayashi, K. Iwamoto, and J. Watanabe. 1984. pKa determination of verapamil by liquid-liquid partition. J. Pharm. Sci. 73:442–445. [DOI] [PubMed] [Google Scholar]
- 10.Kass, R. S., J. P. Arena, and S. Chin. 1989. Cellular electrophysiology of amlodipine: probing the cardiac L-type calcium channel. Am. J. Cardiol. 64:35I–41I (discussion 41I–42I). [DOI] [PubMed] [Google Scholar]
- 11.Bäuerle, H. D., and J. Seelig. 1991. Interaction of charged and uncharged calcium channel antagonists with phospholipid membranes. Binding equilibrium, binding enthalpy, and membrane location. Biochemistry. 30:7203–7211. [DOI] [PubMed] [Google Scholar]
- 12.Eytan, G. D., R. Regev, G. Oren, and Y. G. Assaraf. 1996. The role of passive transbilayer drug movement in multidrug resistance and its modulation. J. Biol. Chem. 271:12897–12902. [DOI] [PubMed] [Google Scholar]
- 13.Ferte, J. 2000. Analysis of the tangled relationships between P-glycoprotein-mediated multidrug resistance and the lipid phase of the cell membrane. Eur. J. Biochem. 267:277–294. [DOI] [PubMed] [Google Scholar]
- 14.Al-Shawi, M. K., M. K. Polar, H. Omote, and R. A. Figler. 2003. Transition state analysis of the coupling of drug transport to ATP hydrolysis by P-glycoprotein. J. Biol. Chem. 278:52629–52640. [DOI] [PubMed] [Google Scholar]
- 15.Litman, T., T. Zeuthen, T. Skovsgaard, and W. D. Stein. 1997. Structure-activity relationships of P-glycoprotein interacting drugs: kinetic characterization of their effects on ATPase activity. Biochim. Biophys. Acta. 1361:159–168. [DOI] [PubMed] [Google Scholar]
- 16.Landwojtowicz, E., P. Nervi, and A. Seelig. 2002. Real-time monitoring of P-glycoprotein activation in living cells. Biochemistry. 41:8050–8057. [DOI] [PubMed] [Google Scholar]
- 17.Gatlik-Landwojtowicz, E., P. Aanismaa, and A. Seelig. 2004. The rate of P-glycoprotein activation depends on the metabolic state of the cell. Biochemistry. 43:14840–14851. [DOI] [PubMed] [Google Scholar]
- 18.Ambudkar, S. V. 1998. Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells. Methods Enzymol. 292:504–514. [DOI] [PubMed] [Google Scholar]
- 19.Gally, H. U., W. Niederberger, and J. Seelig. 1975. Conformation and motion of the choline headgroup in bilayers of dipalmitoyl-3-sn-phosphatidylcholine. Biochemistry. 14:3647–3652. [DOI] [PubMed] [Google Scholar]
- 20.Seelig, J., and N. Waespe-Sarcevic. 1978. Molecular order in cis and trans unsaturated phospholipid bilayers. Biochemistry. 17:3310–3315. [DOI] [PubMed] [Google Scholar]
- 21.Macho, V., L. Brombacher, and H. W. Spiess. 2001. The NMR-WEBLAB: an internet approach to NMR lineshape analysis. Appl. Magn. Reson. 20:405–432. [Google Scholar]
- 22.Aveyard, R., and D. A. Haydon. 1973. Cambridge Chemistry Tests: An Introduction to the Principles of Surface Chemistry. Cambridge University Press, New York.
- 23.McLaughlin, S. 1977. Electrostatic potentials at membrane-solution interfaces. Curr. Top. Membr. Transp. 9:71–144. [Google Scholar]
- 24.Seelig, J., S. Nebel, P. Ganz, and C. Bruns. 1993. Electrostatic and nonpolar peptide-membrane interactions. Lipid binding and functional properties of somatostatin analogues of charge z = +1 to z = +3. Biochemistry. 32:9714–9721. [DOI] [PubMed] [Google Scholar]
- 25.Goldberg, R. N., N. Kishore, and R. M. Lennen. 2002. Thermodynamic quantities for the ionization reactions of buffers. J. Phys. Chem. Ref. Data. 31:231–370. [Google Scholar]
- 26.Fischer, H., R. Gottschlich, and A. Seelig. 1998. Blood-brain barrier permeation: molecular parameters governing passive diffusion. J. Membr. Biol. 165:201–211. [DOI] [PubMed] [Google Scholar]
- 27.Gerebtzoff, G., X. Li-Blatter, H. Fischer, A. Frentzel, and A. Seelig. 2004. Halogenation of drugs enhances membrane binding and permeation. ChemBioChem. 5:676–684. [DOI] [PubMed] [Google Scholar]
- 28.Seelig, J. 1978. 31P nuclear magnetic resonance and the headgroup structure of phospholipids in membranes. Biochim. Biophys. Acta. 515:105–140. [DOI] [PubMed] [Google Scholar]
- 29.Seelig, J., P. M. Macdonald, and P. G. Scherer. 1987. Phospholipid headgroups as sensors of electric charge in membranes. Biochemistry. 26:7535–7541. [DOI] [PubMed] [Google Scholar]
- 30.Seelig, A., and J. Seelig. 2002. Membrane structure. In Encyclopedia of Physical Science and Technology, 3rd Ed. Academic Press, New York.
- 31.Seelig, J., L. Tamm, L. Hymel, and S. Fleischer. 1981. Deuterium and phosphorus nuclear magnetic resonance and fluorescence depolarization studies of functional reconstituted sarcoplasmic reticulum membrane vesicles. Biochemistry. 20:3922–3932. [DOI] [PubMed] [Google Scholar]
- 32.Tamm, L. K., and J. Seelig. 1983. Lipid solvation of cytochrome c oxidase. Deuterium, nitrogen-14, and phosphorus-31 nuclear magnetic resonance studies on the phosphocholine headgroup and on cis-unsaturated fatty acyl chains. Biochemistry. 22:1474–1483. [DOI] [PubMed] [Google Scholar]
- 33.Bienvenue, A., M. Bloom, J. H. Davis, and P. F. Devaux. 1982. Evidence for protein-associated lipids from deuterium nuclear magnetic resonance studies of rhodopsin-dimyristoylphosphatidylcholine recombinants. J. Biol. Chem. 257:3032–3038. [PubMed] [Google Scholar]
- 34.Flogel, M., and R. L. Biltonen. 1975. The pH dependence of the thermodynamics of the interaction of 3′-cytidine monophosphate with ribonuclease A. Biochemistry. 14:2610–2615. [DOI] [PubMed] [Google Scholar]
- 35.Biltonen, R. L., and N. Langerman. 1979. Microcalorimetry for biological chemistry: experimental design, data analysis, and interpretation. Methods Enzymol. 61:287–318. [DOI] [PubMed] [Google Scholar]
- 36.Morin, P. E., and E. Freire. 1991. Direct calorimetric analysis of the enzymatic activity of yeast cytochrome c oxidase. Biochemistry. 30:8494–8500. [DOI] [PubMed] [Google Scholar]
- 37.Pohl, E. E., A. V. Krylov, M. Block, and P. Pohl. 1998. Changes of the membrane potential profile induced by verapamil and propranolol. Biochim. Biophys. Acta. 1373:170–178. [DOI] [PubMed] [Google Scholar]
- 38.Martin, B. R. 1964. Introduction to Biophysical Chemistry. McGraw-Hill, New York.
- 39.Gerebtzoff, G., and A. Seelig. 2006. In-silico prediction of blood-brain barrier permeation using the calculated molecular cross-sectional area as main parameter. J. Chem. Inf. Model. In press. [DOI] [PubMed]
- 40.Boguslavsky, V., M. Rebecchi, A. J. Morris, D. Y. Jhon, S. G. Rhee, and S. McLaughlin. 1994. Effect of monolayer surface pressure on the activities of phosphoinositide-specific phospholipase C-β 1, -γ1, and -δ1. Biochemistry. 33:3032–3037. [DOI] [PubMed] [Google Scholar]
- 41.Seelig, A. 1987. Local anesthetics and pressure: a comparison of dibucaine binding to lipid monolayers and bilayers. Biochim. Biophys. Acta. 899:196–204. [DOI] [PubMed] [Google Scholar]
- 42.Heerklotz, H., T. Wieprecht, and J. Seelig. 2004. Membrane perturbation by the lipopeptide surfactin and detergents as studied by deuterium NMR. J. Phys. Chem. B. 108:4909–4915. [Google Scholar]
- 43.Wenk, M. R., and J. Seelig. 1997. Interaction of octyl-β-thioglucopyranoside with lipid membranes. Biophys. J. 73:2565–2574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blume, A., and R. G. Griffin. 1982. Carbon-13 and deuterium nuclear magnetic resonance study of the interaction of cholesterol with phosphatidylethanolamine. Biochemistry. 21:6230–6242. [DOI] [PubMed] [Google Scholar]
- 45.Gally, H. U., A. Seelig, and J. Seelig. 1976. Cholesterol-induced rod-like motion of fatty acyl chains in lipid bilayers a deuterium magnetic resonance study. Hoppe Seylers Z. Physiol. Chem. 357:1447–1450. [PubMed] [Google Scholar]
- 46.Haberkorn, R. A., R. G. Griffin, M. D. Meadows, and E. Oldfield. 1977. Deuterium nuclear magnetic resonance investigation of the dipalmitoyl lecithin-cholesterol-water system. J. Am. Chem. Soc. 99:7353–7355. [DOI] [PubMed] [Google Scholar]
- 47.Oldfield, E., M. Meadows, D. Rice, and R. Jacobs. 1978. Spectroscopic studies of specifically deuterium labeled membrane systems. Nuclear magnetic resonance investigation of the effects of cholesterol in model systems. Biochemistry. 17:2727–2740. [DOI] [PubMed] [Google Scholar]
- 48.Shepherd, J. C., and G. Buldt. 1978. Zwitterionic dipoles as a dielectric probe for investigating headgroup mobility in phospholipid membranes. Biochim. Biophys. Acta. 514:83–94. [DOI] [PubMed] [Google Scholar]
- 49.Buldt, G., H. U. Gally, A. Seelig, J. Seelig, and G. Zaccai. 1978. Neutron diffraction studies on selectively deuterated phospholipid bilayers. Nature. 271:182–184. [DOI] [PubMed] [Google Scholar]