

Abstract
Studies on hypoxia-sensitive pathways have revealed a series of Fe(II)-dependent dioxygenases that regulate hypoxia-inducible factor (HIF) by prolyl and asparaginyl hydroxylation. The recognition of these unprecedented signaling processes has led to a search for other substrates of the HIF hydroxylases. Here we show that the human HIF asparaginyl hydroxylase, factor inhibiting HIF (FIH), also efficiently hydroxylates specific asparaginyl (Asn)-residues within proteins of the IκB family. After the identification of a series of ankyrin repeat domain (ARD)-containing proteins in a screen for proteins interacting with FIH, the ARDs of p105 (NFKB1) and IκBα were shown to be efficiently hydroxylated by FIH at specific Asn residues in the hairpin loops linking particular ankyrin repeats. The target Asn residue is highly conserved as part of the ankyrin consensus, and peptides derived from a diverse range of ARD-containing proteins supported FIH enzyme activity. These findings demonstrate that this type of protein hydroxylation is not restricted to HIF and strongly suggest that FIH-dependent ARD hydroxylation is a common occurrence, potentially providing an oxygen-sensitive signal to a diverse range of processes.
Keywords: NF-κB, 2-oxoglutarate-dependent dioxygenase, protein hydroxylation
Cells react to variation in oxygen availability with adaptive responses that involve changes in most basic cellular functions. Analysis of the transcriptional component of this response has defined pathways that regulate hypoxia-inducible factor (HIF) by posttranslational hydroxylation of specific residues. HIF is an α/β heterodimer that binds hypoxia response elements in a range of hypoxia-inducible genes (for review, see ref. 1). Regulation is mediated by the α-subunits and involves dual mechanisms controlling both the abundance and activity of the protein. Thus, hydroxylation of specific prolyl residues promotes interaction with the von Hippel–Lindau E3 ligase and hence proteolysis, whereas hydroxylation of a C-terminal Asn residue blocks recruitment of the coactivators p300/CBP. The prolyl and asparaginyl hydroxylase enzymes that catalyze these reactions are 2-oxoglutarate (2-OG) and Fe(II)-dependent dioxygenases that couple the oxidative decarboxylation of 2-OG with oxidation of peptidyl substrates. Dioxygen is an obligate cosubstrate, and reductions in the rate of hydroxylation during hypoxia allow HIF-α to escape VHL-mediated destruction and to activate transcription (for reviews, see refs. 2 and 3).
HIF prolyl hydroxylation is catalyzed by three enzymes, PHD1, -2, and -3 (equivalent to EGLN2, -1, and -3 and HPH-3, -2, and -1). HIF Asn hydroxylation is catalyzed by a more distantly related 2-OG-dependent dioxygenase, factor inhibiting HIF (FIH) (for reviews, see refs. 2 and 3). A key question raised by these findings is whether the roles of all four dioxygenases are specific to HIF regulation, or whether one or more have alternative substrates. Several studies have identified proteins that interact to modulate HIF hydroxylase activity (4) or to direct other functions (5), but none have been demonstrated to be hydroxylase substrates.
We identified a series of ankyrin repeat domain (ARD)-containing proteins including the NF-κB precursor, p105 (NFKB1), in screens for proteins that interact with FIH. Using in vitro assays of FIH activity and mass spectrometric analysis of transfected and native IκB proteins in vivo, we show that both p105 and IκBα are hydroxylated by FIH at specific residues within their ARDs. The target Asn residues are highly conserved within the ankyrin consensus sequence (6), and peptides from a range of non-IκB ARD-containing proteins supported hydroxylation as efficiently as, or more efficiently than, the known site of FIH-dependent hydroxylation in HIF-α. These findings strongly suggest that FIH-dependent ARD hydroxylation is common, and that the prevalence of intracellular hydroxylation has previously been underestimated.
Results
FIH was first identified in a yeast two-hybrid (Y2H) screen for HIF-1α-interacting proteins (7), indicating that FIH enzyme–substrate interactions can be detected with this methodology. To investigate the possible existence of alternative FIH substrates, a human umbilical vein endothelial cell (HUVEC) and a human testis cDNA library were screened by Y2H by using FIH as bait. From the HUVEC library, 115 positive clones representing nine independent proteins (including HIF-1α) were obtained. Two proteins containing ARDs were isolated: p105 and uveal autoantigen with coiled-coil domains and ankyrin repeats (UACA). From the testis library, three proteins were identified, one of which corresponded to the ARD-containing protein FEM1β. To validate the Y2H interactions, the ability of cell lysate-derived FIH to interact with 35S-labeled candidate polypeptides synthesized in reticulocyte lysate was tested. The ARD-containing polypeptides corresponding to the p105, UACA, and FEM1β clones all interacted with FIH, whereas other polypeptides from the screen did not.
p105 is the precursor of the p50 component of NF-κB and through its ARD can function as an NF-κB inhibitor (8). Given the biological importance of NF-κB regulation and its reported links with oxygen-dependent signaling (9, 10), p105 was chosen for further analysis. Full-length p105 was demonstrated to interact with FIH in vitro, although less efficiently than the isolated ARD (542–803). Deletional analysis identified residues 650–684 as critical for the FIH interaction (Fig. 1A). Within this region, a potential substrate motif was identified by comparison with the known FIH-mediated hydroxylation site in HIF-1α and sequences in UACA and FEM1β (Fig. 1B). Further analyses using the full-length proteins expressed in 293T cells confirmed the importance of the core Asn residue (Asn-678) for interaction with FIH (Fig. 1C). Interestingly, addition of dimethyloxalylglycine (DMOG), a cell-penetrating precursor of an inhibitor of FIH, enhanced the interaction between FIH and p105, consistent with p105 being an FIH substrate. Interaction between endogenous p105 and FIH was confirmed by coimmunoprecipitation of the proteins from HeLa cell extract (Fig. 1D).
Fig. 1.

Interactions between p105 and FIH. (A) Interaction of [35S]methionine-labeled p105 polypeptides with extract from control cells (−) or cells induced with doxycycline (DOX) to express FLAG-FIH (+). Anti-FLAG immunoprecipitations; coprecipitating products are visualized by autoradiography. Residues 650–684 are essential for FIH interaction. (B) Alignment of residues 667–690 (fourth ankyrin repeat) of p105 with the ARD of FEM1β and UACA as well as the HIF-1α hydroxylation site (∗). (C) Interaction between FIH and transfected full-length PK-tagged p105, Asn-678 mutant p105 (N678A), in 293T cells exposed to DMOG (+) or vehicle control (−). EV, empty vector control. Anti-PK immunoprecipitates; coprecipitating FIH is detected by anti-FIH mAb. (D) Coimmunoprecipitation of endogenous FIH with p105 from HeLa cell extract. p105 IP, antibody to the C terminus of p105; CON IP, preimmune serum.
Based on these data, we postulated that p105 was an FIH substrate. To investigate this, a GST-p105 ARD fusion protein was tested for ability to promote 2-OG decarboxylation in an assay of FIH activity. GST-p105 ARD promoted 2-OG decarboxylation to a level that was comparable to, or greater than, the known HIF-1α substrate. In keeping with the interaction studies, mutation of p105 Asn-678 reduced 2-OG decarboxylation to background levels (Fig. 2A), implying that Asn-678 was a bona fide, and probably the only, site of FIH-dependent hydroxylation.
Fig. 2.

Recombinant p105 and IκBα promote decarboxylation of 2-OG in assays of FIH activity. 2-OG decarboxylation is measured by 14CO2 formation. (A) Reactions containing the indicated GST-tagged proteins: HIF-1α 775–826 (HIF-1α C-terminal activation domain); p105 ARD 537–809 (p105 ARD); p105 ARD Asn-678 mutant (p105 ARD(N678A)). Wild type (p105 ARD) but not the N678A mutant promoted 2-OG decarboxylation. Higher background was observed with p105 ARD(N678A) because of less pure preparation of protein. (B) Experiments comparing p105 ARD with full-length IκBα and IκBβ proteins. IκBα, but not IκBβ, promotes 2-OG decarboxylation.
We next sought to determine whether the ability to support FIH activity was unique to the p105 ARD or whether it was also a property of other ARD-containing IκB proteins. Sequence analysis indicated potential hydroxylation motifs in some family members (e.g., IκBα) but not others (e.g., IκBβ). Accordingly, these proteins were produced as N-terminal GST fusions, cleaved to generate untagged full-length proteins, and tested in decarboxylation assays. Full-length IκBα, but not IκBβ, supported 2-OG decarboxylation, with the activity being at least 2-fold greater for IκBα than p105 ARD (Fig. 2B), implying that IκBα might contain more than one site of hydroxylation. Indeed, mutation of Asn-210, the candidate hydroxylation site best aligning with that of p105, reduced but did not ablate activity (data not shown).
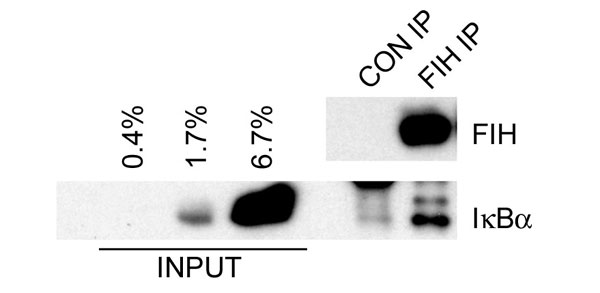
To define the sites of hydroxylation directly, IκBα was reacted with FIH and analyzed by liquid chromatography/mass spectrometry (LC/MS). Incubation with FIH resulted in the appearance of two peaks corresponding to mass shifts of +16 (30–40%) and +32 Da (60–70%) relative to the unreacted IκBα, implying two hydroxylation sites (Fig. 6, which is published as supporting information on the PNAS web site). MS/MS analysis demonstrated hydroxylation of Asn-244 (data not shown). A +16-Da mass shift was also observed on the 41-residue peptide containing the second putative site of hydroxylation, Asn-210, although because of its size assignment of the site by MS/MS, was not possible. To facilitate MS/MS analysis of Asn-210, an additional trypsin cleavage (E200K) site was introduced. The E200K IκBα protein promoted similar levels of 2-OG decarboxylation to wild-type protein, and MS/MS analysis confirmed hydroxylation of Asn-210 (Fig. 7, which is published as supporting information on the PNAS web site). A higher level of hydroxylation was observed at Asn-244 (>95%) than at Asn-210 (≈60%). Interaction between endogenous IκBα and FIH was verified by coimmunoprecipitation (Fig. 8, which is published as supporting information on the PNAS web site). Taken together, these results indicate that interactions with FIH extend from p105 to other members of the IκB family, and that individual proteins may be modified at more than one site.
To determine whether p105 and IκBα are in vivo substrates of endogenous FIH, full-length proteins bearing modified tryptic cleavage sites to facilitate MS/MS analysis of Asn-210 in IκBα and Asn-678 in p105 (see Experimental Procedures) were expressed in 293T cells, purified, and analyzed by MS. LC/MS and MS/MS analysis demonstrated hydroxylation at each of the three Asn residues identified by the in vitro studies (Fig. 3A and B for p105 Asn-678; Fig. 3C and Fig. 9, which is published as supporting information on the PNAS web site, for IκBα Asn-244; Asn-210 data not shown). Under the conditions of these experiments, hydroxylation was incomplete. For IκBα, the level of hydroxylation at Asn-244 was consistently greater (25–45%) than that at Asn-210 (10–20%), correlating with the in vitro data. Hydroxylation was repressed by treatment of cells with hypoxia (Fig. 10, which is published as supporting information on the PNAS web site) and DMOG (data not shown) and clearly dependent on FIH activity, being almost totally ablated by FIH siRNA and enhanced to essentially 100% by FIH overexpression (Fig. 3C).
Fig. 3.

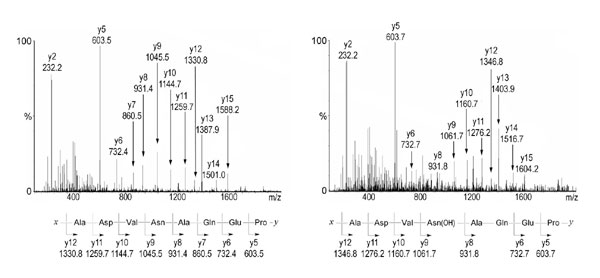
Mass spectrometric analysis of FIH-mediated hydroxylation of p105 and IκBα in vivo. (A) LC/MS spectrum from a tryptic digest of PK-tagged p105 immunopurified from transfected 293T cells. Peaks at m/z = 763.87 and 771.87 correspond to [M + 2H]2+ of unhydroxylated and hydroxylated LLVAAGADVNAQEQK peptides, respectively. Mutations (L668K/R870A) were introduced into p105 to enable LC/MS and MS/MS analysis of tryptic peptides. (B) MS/MS spectra of the m/z 763.87 (Left) and m/z 771.87 (Right) parent ions assign Asn-678 as the site of hydroxylation in p105. A +16-Da shift appears in the y-ion series appearing at y6, corresponding to fragments containing Asn-678. (C) Extracted ion chromatograms illustrating the effect of FIH intervention on IκBα hydroxylation at Asn-244. (Left) Standards for the hydroxylated and unhydroxylated modified tryptic peptides (see Fig. 9 for MS/MS assignments). (Right) Tryptic digest of HA-tagged IκBα immunopurified from transfected 293T cells; FIH overexpression increases, whereas FIH siRNA decreases, the ratio of hydroxylated to unhydroxylated peptide. (∗C indicates derivatization on cysteine; see Experimental Procedures.)
In the case of HIF-α, FIH-dependent hydroxylation of a single Asn residue is proposed to ablate the interaction of the HIF-α C-terminal activation domain with the coactivators p300/CBP (11, 12). One of the best-understood functions of the IκB ARD is to mediate repressive interactions with NF-κB Rel homology domain proteins such as p65 and p50 that are involved in DNA binding and activation of NF-κB transcription (8). This function is most clearly defined in IκBα, which lacks the additional function of p105 as the precursor of p50. We therefore focused analyses on IκBα to determine whether ARD hydroxylation affects inhibition of NF-κB DNA binding. Interestingly, mutation of both target Asn residues to alanine almost completely ablated the ability of IκBα protein to inhibit NF-κB DNA-binding activity (Fig. 11, which is published as supporting information on the PNAS web site). However, further studies using recombinant IκBα exposed to either wild type or the catalytically inactive FIH mutant D201A, and demonstrated to be either fully hydroxylated or unhydroxylated by MS, indicated that hydroxylation at these sites did not ablate the inhibitory activity of IκBα on NF-κB DNA binding. Rather, a small enhancement of inhibitory activity was observed with IκBα that had been exposed to wild type as opposed to the D201A mutant FIH. This effect was seen on NF-κB DNA-binding activity derived from both TNF-α-induced HeLa cells (Fig. 4A) and unprogrammed reticulocyte lysate (data not shown). To pursue this further, we tested for effects of IκBα hydroxylation on interactions with p65 and p50 protein produced by in vitro transcription and translation (IVTT). No effect of FIH treatment on the ability of recombinant IκBα to interact with p65 was observed. However, treatment with wild type but not the FIH D201A mutant apparently enhanced interaction with p50, irrespective of the presence or absence of p65 in the complex (Fig. 4B).
Fig. 4.

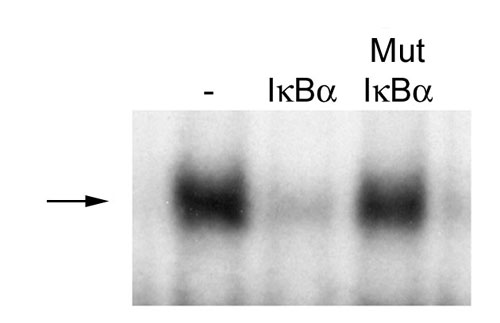
Effects of FIH on IκB/NF-κB complex formation and activity. (A) EMSA of NF-κB DNA-binding activity in nuclear extracts of TNF-α-stimulated HeLa cells. Effect of recombinant IκBα, GST-IκBα, and GST-IκBα incubated in vitro with FIH, FIH plus inhibitor (N-oxalylglycine, NOG), or mutant D201A FIH (mut FIH). Slightly more potent inhibitory effects of hydroxylated IκBα (lane 4) were consistently observed. Arrow indicates the NF-κB DNA-binding activity. IκBα inputs were normalized but not shown. (B) Interaction of recombinant GST-IκBα, pretreated with either wild type or mutant D201A FIH (mut FIH), with 35S-labeled p50 and/or p65 produced by IVTT in wheat germ lysate. Interaction was performed in the presence of BSA, assayed by GST pull-down. Coprecipitating p50 and p65 are detected by autoradiography. GST-IκBα input is normalized but not shown. (C) Association of IκBα with p65 and p50 in cells. U2OS cells were transfected with control or FIH siRNA and stimulated with TNF-α. Anti-IκBα immunoprecipitates were immunoblotted with anti-IκBα, anti-p50, or anti-p65 antibodies. Cells were harvested either at time 0 (untreated), after a 30-min exposure to TNF-α (TNF-α), or at the indicated times (in minutes) after removal of TNF-α. (D) NF-κB DNA-binding activity. EMSA of nuclear extracts from serum-deprived 293T cells transfected with control (−) or FIH-specific (+) siRNA duplexes and treated with TNF-α for 30 min. Arrow indicates the NF-κB DNA-binding activity. (E) NF-κB reporter activity. 293T cells were treated with FIH or control siRNA then transfected with NF-κB (κB LUC) or control (CON LUC) reporter plasmids. Cells were stimulated with TNF-α for 6 h to induce NF-κB activity, and the ability of cotransfected IκBα to suppress reporter gene activity was assessed. Relative luciferase activity is shown as the mean ± 1 SD of triplicate samples from a representative experiment.
To address the potential role of ARD hydroxylation in NF-κB regulation in vivo, we next looked for alterations in IκB/NF-κB complex formation in cells subjected to suppression of FIH by siRNA or up-regulation by transfection, using conditions similar to those that were used to define changes in p105 and IκBα hydroxylation in vivo (Fig. 3C). Because ARD hydroxylation might be irreversible, we considered that the effects of genetic manipulation of FIH on endogenous complexes might be confounded by preexisting hydroxylation and might be more clearly manifest on newly synthesized proteins. Therefore, we studied cells under basal conditions and during activation and recovery from TNF-α stimulation. In immunoprecipitation experiments using antibodies directed against IκBα or the C terminus of p105, we observed no effect of FIH manipulation on capture of p50 or p65 in whole-cell extracts at any stage in the experiments (Fig. 4C for IκBα; p105 data not shown). Furthermore, analysis of nuclear extracts revealed no substantial FIH-dependent changes in levels of nuclear p50 or p65 (data not shown). Based on the in vitro data, it might be predicted that FIH siRNA treatment in the steady state and during TNF-α recovery would manifest as an increase in NF-κB DNA binding. However, no change and, if anything, a slight reduction in NF-κB DNA binding was observed (Fig. 4D). Because in vitro experiments had suggested an effect of FIH on ARD interactions with p50 but not p65, we considered whether the relatively high ratio of p65 to p50 might have obscured any effects. Experiments were therefore repeated using siRNA to suppress p65 levels and using transfection to enhance p50 levels. Manipulations of FIH levels in this setting resulted in no major effects on interactions of p50 with either IκBα or p105, on nuclear abundance of the proteins, or on NF-κB DNA-binding activity (data not shown). Finally, we tested for effects of siRNA-mediated suppression of FIH on NF-κB activity after TNF-α stimulation using an NF-κB transcriptional reporter. A modest reduction in activity was observed after siRNA suppression of FIH. Cotransfection of IκBα reduced the absolute level of TNF-α-induced reporter activity but did not appear to enhance the FIH-dependent effect (Fig. 4E).
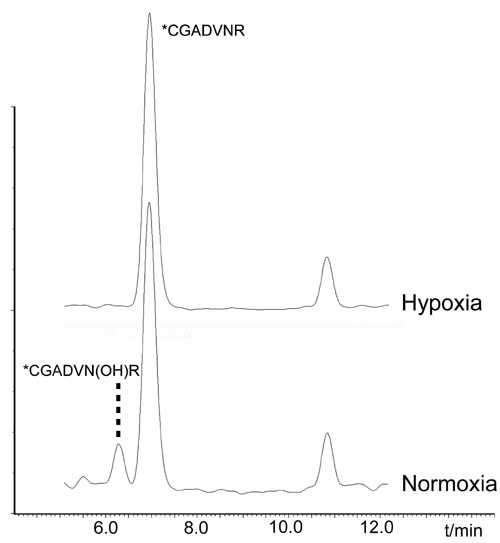
The inability to see major FIH-dependent effects on NF-κB or on IκB-ARD protein interactions in vivo led us to consider whether endogenous IκB proteins might be protected from hydroxylation because of complex formation or other restrictions on FIH activity. We therefore sought to define hydroxylation status of an endogenous target site directly. To this end, Asn-244 in IκBα was best-suited, because the endogenous sequence was directly amenable to MS/MS analysis after tryptic digestion. Accordingly, endogenous IκBα was purified from large-scale cultures of U2OS, 293T (data not shown), and HeLa S3 cells and analyzed by LC/MS. A peptide exhibiting chromatographic properties and a +16-Da mass shift identical to that previously defined for hydroxylated Asn-244 was identified. MS/MS analyses clearly assigned the site of hydroxylation as Asn-244 (Fig. 5). The level of endogenous hydroxylation was suppressed by FIH inhibition with DMOG and varied between the cell lines tested, with protein from HeLa S3 cells having a lower level (between 5% and 20%) of hydroxylation than that observed in U2OS and 293T cells (≈50%).
Fig. 5.

MS demonstrating hydroxylation of endogenous IκBα. MS/MS spectra are depicted showing the N-terminally modified *CGADVNR peptide, derived by 6-exo-trig cyclization from the peptide S-alkylated with iodoacetamide. (Left) Fragments of the unhydroxylated IκBα peptide, [M+H]+ = m/z 774.3. (Right) Fragments of the hydroxylated IκBα peptide, [M+H]+ = m/z 790.3; a +16-Da shift is observed in the y-ion series, appearing at the y2 ion, which corresponds to fragments containing Asn-244.
Because the ARD is a common interaction domain, we sought to determine whether FIH-mediated hydroxylation was likely to affect other ARD proteins. Sequence analysis identified a conserved Asn residue at the hydroxylated position as part of the consensus sequence for ARDs (6). A set of peptides from ARD-containing proteins encompassing predicted sites of modification were compared with HIF-1α, p105, and IκBα ARD sites in assays of FIH-dependent 2-OG decarboxylation. All of the peptides, with the possible exception of ILK-1, were highly active (Table 1), and all were shown to be hydroxylated on the conserved Asn residue by LC/MS (for representative illustration of tankyrase-1 peptide, see Fig. 12, which is published as supporting information on the PNAS web site) and MS/MS analyses, suggesting that hydroxylation is likely a common modification of ARD proteins.
Table 1.
Diverse ankyrin repeat sequences are substrates for FIH
| Peptide sequence | Protein | Activity relative to HIF-1α peptide, % |
|---|---|---|
| DESGLPQLTSYDCEVNAPI | HIF-1α (788–806) | 100 |
| SLPCLLLLVAAGADVNAQEQK | p105 (663–683) | 125 |
| YLGIVELLVSLGADVNAQEPC | IκBα (195–215) | 127 |
| NPDLVSLLLKCGADVNRVTY | IκBα (229–248) | 120 |
| NALVTKLLLDCGAEVNAVDNE | FEM1β (511–531) | 112 |
| FLDTLKVLVEHGADVNVPDG | p19-INK4d (86–105) | 124 |
| HASIVEVLLKHGADVNAKDM | GABP-β (83–102) | 107 |
| NLEVAEYLLEHGADVNAQDK | Tankyrase-1 (849–868) | 107 |
| RDEIVKALLGKGAQVNAVNQ | Gankyrin (85–104) | 116 |
| YTEVLKLLIQAGYDVNIKDV | MYPT1 (211–230) | 122 |
| QLEILEFLLLKGADINAPDK | Myotrophin (46–65) | 124 |
| NTRVASFLLQHDADINAQTK | FGIF (153–172) | 168 |
| HRDIVQKLLQYKADINAVNEH | ILK-1 (79–99) | 45 |
2-OG decarboxylation assays illustrating the ability of peptides derived from different ARD proteins to act as FIH substrates. Activity is normalized to that of the HIF-1α peptide. Target Asn residues are in bold.
Discussion
The identification of a set of 2-OG and Fe(II)-dependent oxygenases that regulate cellular responses to hypoxia by posttranslational hydroxylation of HIF-α has raised a number of important issues. These include whether one or more of the HIF hydroxylases have other substrates, and how HIF signaling interfaces with other pathways. Here we have revealed alternative substrates for FIH as conserved Asn residues present within the ARDs of the IκB proteins, p105 and IκBα. We identified three asparagines, two in IκBα and one in p105, that were readily hydroxylated by FIH both in vitro and in vivo and confirmed hydroxylation of endogenous IκBα at one asparagine that was amenable to MS/MS analysis of the native protein. Based on the data for IκB proteins, sequence alignments with other ARD-containing proteins, and assays on peptides derived from selected examples of these proteins, the results strongly suggest that hydroxylation will occur in many, although not all, ARD-containing proteins.
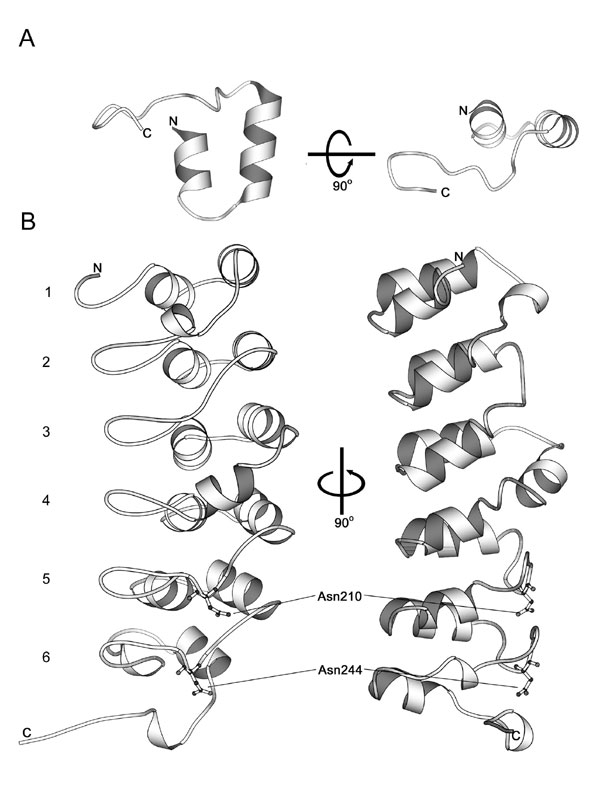
Subsequent to its identification in the yeast cell-cycle regulator Swi6/Cdc10 and the Drosophila signaling protein Notch (13), the canonical ARD has been identified in all kingdoms of life; ≈9,000 sequences have been identified, with ≈450 from the human genome (SMART database; ref. 14). The structure of a single 33-residue ankyrin repeat comprises a helix–loop–helix hairpin-loop fold with the helices arranged in an antiparallel fashion followed by an orthogonally related hairpin loop (6). Sequence alignments revealed that the hydroxylated Asn residues in IκBα and p105 occur in conserved positions in the hairpin loop that links individual repeats (Fig. 13, which is published as supporting information on the PNAS web site).
Interestingly, we observed consistent differences in the extent of hydroxylation at different sites in IκBα, with Asn-244 more efficiently hydroxylated than Asn-210, both in vitro and in vivo. Given that FIH appears to be able to accept a range of ARD sequences and accepts HIF-α variants mutated in the positions surrounding the hydroxylated Asn residue (15), the difference in the level of hydroxylation at the two sites in IκBα likely reflects the position of the individual ARDs in the overall protein rather than the sequences immediately surrounding the hydroxylated residue. Analysis of endogenous IκBα hydroxylation at Asn-244 revealed that, although the extent of hydroxylation was substantial, it varied with cell type and was clearly incomplete even in well oxygenated cultures supplemented with Fe(II) and ascorbate to optimize FIH activity. That hydroxylation of native IκBα is incomplete might suggest a role in signaling. However, despite considerable effort, we have so far been unable to define a major role, at least in regulating interactions with p65/p50 or NF-κB-dependent transcription under standard tissue culture conditions.
Induction of NF-κB signaling by hypoxia has been reported in certain systems (9, 10), although the precise signaling mechanism is unclear. Although it could be argued that our in vitro studies demonstrating a slightly enhanced inhibitory action of hydroxylated IκBα (that might be suppressed by hypoxia) provide a potential explanation, we do not think this is the case, particularly because siRNA-mediated suppression of FIH did not increase, and if anything reduced, NF-κB activity.
At least six ARD proteins (p105, p100, Bcl3, IκBα, IκBβ, and IκBε) are directly involved in the NF-κB transcriptional response (8). A number of other ARD proteins, such as p53BP2/ASPP2 (16) and p16/INK4 (17), are known to complex with members of this family. The abundance of potential interactions with FIH-dependent hydroxylation therefore complicates analysis, and it is possible that analysis in different settings or more accurate assessment of subtle effects on the output would clarify a role in NF-κB signaling.
ARD-containing proteins are also known to function in a range of other cellular processes, including cell adhesion, cell-cycle regulation, cell-fate determination, telomere regulation, transcriptional regulation, and tumor suppression (for review, see ref. 18). It is therefore very possible that ARD hydroxylation has a more direct role in signal transduction within one or more signaling systems than have so far been indicated for NF-κB. Interestingly, in a recent analysis of interactions between HIF and Notch signaling, transfected FIH was reported to down-regulate the activity of a Notch transcriptional reporter gene, although the mechanism was not defined (19).
Alternatively, it is possible that Asn hydroxylation in the ARD proteins has primarily a structural role. Prolyl and lysyl hydroxylation has a well established structural role in extracellular proteins, for instance in the stabilization of collagen (for review, see ref. 20), but has not been described for intracellular proteins. Interestingly, coimmunoprecipitation studies more readily displayed association of endogenous FIH with IκB-ARD proteins than with HIF-α, suggesting an unusually tight association. Given the overall abundance of ARD proteins, their availability for FIH-mediated association and/or hydroxylation might indirectly affect human HIF activity by limiting or regulating the binding of FIH to HIF-α. Such stoichiometric competition could explain the paradox that overexpression of FIH is able to further suppress HIF target gene expression even in well oxygenated cells where endogenous FIH activity ought to be more than sufficient to maximally suppress HIF (21).
Overall, our findings suggest that posttranslational protein hydroxylation of intracellular proteins is more widespread than previously appreciated and open several avenues for investigation. Given the range of proteins containing ankyrin repeats, across species from bacteria to humans, the demonstration of FIH-dependent hydroxylation of specific residues raises major issues concerning the extent of hydroxylation among different proteins, the nature of structural or signaling roles, and the potential for interaction with HIF signaling and for conferring oxygen sensitivity to a variety of biological processes.
Experimental Procedures
Transfection, siRNA, and Reagents.
Plasmid DNA was transfected by using FuGENE 6 (Roche, Indianapolis, IN); siRNA duplexes were transfected using Oligofectamine (Invitrogen, Carlsbad, CA); for details of cells, plasmids, and siRNA sequences, see Supporting Text, which is published as supporting information on the PNAS web site. TNF-α (Sigma, St. Louis, MO) was used at 10–20 ng/ml. DMOG was synthesized in the host laboratory. Synthetic peptides were generated by Peptide Protein Research (Wickham, U.K.).
Recombinant Protein Expression and Purification.
FIH and D201A mutant FIH were prepared by His6 affinity chromatography (11). Full-length human IκBα, IκBβ, and p105 ARD and mutants were prepared to typically >90% purity as GST fusions.
Yeast Two-Hybrid Library Screening and in Vitro Interactions.
A HUVEC cDNA library (a gift from T. Rabbitts, Medical Research Council Laboratory of Molecular Biology, Cambridge, U.K.) was screened by cotransformation with FIH bait by using the Y190 yeast strain. A human testis cDNA library pretransformed in strain Y187 (Clontech, Mountain View, CA) was screened by mating with strain AH109 transformed with FIH. For FIH/IVTT interaction experiments, doxycycline-induced and uninduced whole-cell extracts from U2OS/FLAG-FIH stable transfectants were incubated with 35S-labeled reticulocyte lysate programmed with the relevant cDNA before anti-FLAG IP (21).
2-OG Decarboxylation Assays and FIH Reactions.
Purified recombinant proteins and synthetic peptides were tested for ability to stimulate FIH-dependent decarboxylation of 14C-labeled 2-OG, as described (11). Material reacted with FIH for mass spectrometric analysis was prepared under similar conditions without radioactivity.
In Vitro IκBα/NF-κB Interaction Assays.
GST-IκBα was reacted with either wild type or D201A FIH. Purified material was analyzed for hydroxylation by MS and tested for ability to bind 35S-labeled NF-κB proteins produced by IVTT in wheat germ lysate (Promega, Madison, WI). Binding to IVTTs was performed in IP+ buffer [20 mM Tris·HCl (pH 7.4)/100 mM NaCl/5 mM MgCl2/0.5% (vol/vol) Igepal CA-630/1× Complete protease inhibitor mixture (Roche)] followed by GST pull-down using glutathione Sepharose 4B.
NF-κB DNA-Binding Assays.
EMSAs were performed by using nuclear extracts from HeLa or 293T cells stimulated with TNF-α, or unprogrammed reticulocyte lysate and double-stranded 32P-end-labeled oligonucleotides corresponding to the sequence AGTTGAGGGGACTTTCCCCAGGC. For assays of IκBα activity, nuclear extracts were preincubated (20 min) with IκBα proteins produced by IVTT, or as GST fusions.
Assay of Endogenous Protein Interactions by Immunoprecipitation.
Whole-cell extracts were prepared in IP+ buffer, and 400 μg of cell extract was immunoprecipitated with α-IκBα (mAb 10B), α-p105 (pAb p105c), α-FIH (pAb FIH), or isotype/species-matched control antibody, coupled to Protein A agarose. Immunoblotting of glycine-eluted samples was with α-IκBα (mAb 10B) α-p50 (pAb H119 or N-19), α-p65 (pAb C20), and α-FIH (mAb 162c); sources of antibodies are provided in Supporting Text.
NF-κB Reporter Gene Activity.
293T cells were transfected with control or FIH-specific siRNA duplexes. Twenty-four hours later, cells were cotransfected with a plasmid-expressing IκBα, control plasmid pEF-Bos-β-Gal, and either wild-type NF-κB-Luc or mutant NF-κB-Luc reporter genes. After 24 h, cells were stimulated with TNF-α for 6 h before harvest and assay of luciferase and β-galactosidase activities.
Expression and Purification of p105 and IκBα from 293T Cells.
Cells were transfected with plasmids encoding epitope-tagged and sequence-modified versions of IκBα [IκBα(E200K)-HA] and p105 [p105(L668K/R870A)-PK], in which mutations either introduced tryptic sites to facilitate MS/MS analysis (IκBα, E200K; p105, L668K) or ablated sites generating confounding peptides (p105, R870A). Extracts were prepared in IP+ buffer and immunoprecipitated with anti-HA agarose or anti-V5 (PK) agarose affinity gel, respectively.
Purification of Endogenous IκBα.
Cell pellets derived from 8 liters of HeLa S3 suspension cells [or equivalent, for adherent 293T and U2OS; Cancer Research UK (London, U.K.) cell services] were solubilized in 500 ml of IP+ buffer before large-scale immunopurification by using mAb 10B.
MS.
Proteins were resolved by SDS/PAGE, and excised gel fragments were digested with trypsin. For analysis of the IκBα peptide containing residues 239–245, an additional cyclization step was performed by heating of the alkylated peptide at 50°C for 16 h to generate a δ-lactam form. After separation on an Agilent Technologies (Palo Alto, CA) 1100 capillary LC system, the peptides were analyzed by using positive ion electrospray with a Waters (Milford, MA) QTof micro mass spectrometer. Where required for comparison, synthetic peptides were also run on this system. Synthetic peptides were otherwise analyzed by using a Waters Alliance 2790 analytical HPLC system and a Waters LCT electrospray TOF spectrometer.
Supplementary Material
Acknowledgments
We are grateful to H. Morcrette and S. Webb for technical assistance; B. Kessler for helpful discussions; and Y.-M. Tian (University of Oxford), T. Rabbitts (Medical Research Center Laboratory of Molecular Biology, University of Cambridge, Cambridge, U.K.), M. McCormack (Medical Research Center Laboratory of Molecular Biology, University of Cambridge), I. Udalova (Imperial College, London, U.K.), D. Krappmann (Max-Delbrück Center, Berlin-Buch, Germany), and C. Scheidereit (Max-Delbrück Center, Berlin-Buch, Germany) for reagents. This work was funded by the Wellcome Trust, Cancer Research UK, and Biotechnology and Biosciences Research Council. K.S.H. was supported by a Glasstone Fellowship.
Abbreviations
- ARD
ankyrin repeat domain
- HIF
hypoxia-inducible factor
- FIH
factor inhibiting HIF
- 2-OG
2-oxoglutarate
- DMOG
dimethyloxalylglycine
- LC/MS
liquid chromatography/mass spectrometry
- IVTT
in vitro transcription and translation
References
- 1.Semenza GL. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 2.Kaelin WG., Jr Annu Rev Biochem. 2004;74:115–128. doi: 10.1146/annurev.biochem.74.082803.133142. [DOI] [PubMed] [Google Scholar]
- 3.Schofield CJ, Ratcliffe PJ. Nat Rev Mol Cell Biol. 2004;5:343–354. doi: 10.1038/nrm1366. [DOI] [PubMed] [Google Scholar]
- 4.Baek JH, Mahon PC, Oh J, Kelly B, Krishnamachary B, Pearson M, Chan DA, Giaccia AJ, Semenza GL. Mol Cell. 2005;17:503–512. doi: 10.1016/j.molcel.2005.01.011. [DOI] [PubMed] [Google Scholar]
- 5.Ozer A, Wu LC, Bruick RK. Proc Natl Acad Sci USA. 2005;102:7481–7486. doi: 10.1073/pnas.0502716102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mosavi LK, Minor DL, Jr, Peng ZY. Proc Natl Acad Sci USA. 2002;99:16029–16034. doi: 10.1073/pnas.252537899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mahon PC, Hirota K, Semenza GL. Genes Dev. 2001;15:2675–2686. doi: 10.1101/gad.924501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hayden MS, Ghosh S. Genes Dev. 2004;18:2195–2224. doi: 10.1101/gad.1228704. [DOI] [PubMed] [Google Scholar]
- 9.Schmedtje JF, Jr, Ji YS, Liu WL, DuBois RN, Runge MS. J Biol Chem. 1997;272:601–608. doi: 10.1074/jbc.272.1.601. [DOI] [PubMed] [Google Scholar]
- 10.Zampetaki A, Mitsialis SA, Pfeilschifter J, Kourembanas S. FASEB J. 2004;18:1090–1092. doi: 10.1096/fj.03-0991fje. [DOI] [PubMed] [Google Scholar]
- 11.Hewitson KS, McNeill LA, Riordan MV, Tian YM, Bullock AN, Welford RW, Elkins JM, Oldham NJ, Bhattacharya S, Gleadle JM, et al. J Biol Chem. 2002;277:26351–2635. doi: 10.1074/jbc.C200273200. [DOI] [PubMed] [Google Scholar]
- 12.Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK. Genes Dev. 2002;16:1466–1471. doi: 10.1101/gad.991402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Breeden L, Nasmyth K. Nature. 1987;329:651–654. doi: 10.1038/329651a0. [DOI] [PubMed] [Google Scholar]
- 14.Schultz J, Milpetz F, Bork P, Ponting CP. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Linke S, Stojkoski C, Kewley RJ, Booker GW, Whitelaw ML, Peet DJ. J Biol Chem. 2004;279:14391–14397. doi: 10.1074/jbc.M313614200. [DOI] [PubMed] [Google Scholar]
- 16.Yang JP, Hori M, Takahashi N, Kawabe T, Kato H, Okamoto T. Oncogene. 1999;18:5177–5186. doi: 10.1038/sj.onc.1202904. [DOI] [PubMed] [Google Scholar]
- 17.Wolff B, Naumann M. Oncogene. 1999;18:2663–2666. doi: 10.1038/sj.onc.1202617. [DOI] [PubMed] [Google Scholar]
- 18.Sedgwick SG, Smerdon SJ. Trends Biochem Sci. 1999;24:311–316. doi: 10.1016/s0968-0004(99)01426-7. [DOI] [PubMed] [Google Scholar]
- 19.Gustafsson MV, Zheng X, Pereira T, Gradin K, Jin S, Lundkvist J, Ruas JL, Poellinger L, Lendahl U, Bondesson M. Dev Cell. 2005;9:617–628. doi: 10.1016/j.devcel.2005.09.010. [DOI] [PubMed] [Google Scholar]
- 20.Myllyharju J, Kivirikko KI. Trends Genet. 2004;20:33–43. doi: 10.1016/j.tig.2003.11.004. [DOI] [PubMed] [Google Scholar]
- 21.Stolze IP, Tian YM, Appelhoff RJ, Turley H, Wykoff CC, Gleadle JM, Ratcliffe PJ. J Biol Chem. 2004;279:42719–42725. doi: 10.1074/jbc.M406713200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}