

Abstract
The molecular mechanisms of airway smooth muscle hypertrophy, a feature of severe asthma, are poorly understood. We previously established a conditionally immortalized human bronchial smooth muscle cell line with a temperature-sensitive SV40 large T antigen. Temperature shift and loss of large T cause G1-phase cell cycle arrest that is accompanied by increased airway smooth muscle cell size. In the present study, we hypothesized that phosphorylation of eukaryotic initiation factor-4E (eIF4E)-binding protein (4E-BP), which subsequently releases eIF4E and initiates cap-dependent mRNA translation, was required for airway smooth muscle hypertrophy. Treatment of cells with chemical inhibitors of PI 3-kinase and mammalian target of rapamycin blocked protein synthesis and cell growth while decreasing the phosphorylation of 4E-BP and increasing the binding of 4E-BP to eIF4E, consistent with the notion that 4E-BP1 phosphorylation and eIF4E function are required for hypertrophy. To test this directly, we infected cells with a retrovirus encoding a phosphorylation site mutant of 4E-BP1 (AA-4E-BP-1) that dominantly inhibits eIF4E. Upon temperature shift, cells infected with AA-4E-BP-1, but not empty vector, failed to undergo hypertrophic growth. We conclude that phosphorylation of 4E-BP, eIF4E release, and cap-dependent protein synthesis are required for hypertrophy of human airway smooth muscle cells.
Keywords: translation, protein synthesis, phosphatidylinositol 3-kinase, mammalian target of rapamycin
Increased airway smooth muscle mass has been shown in fatal and nonfatal asthma. Ebina and coworkers (1) examined airway tissues of patients with fatal asthma using stereologic methods. Two asthmatic subtypes were found, one in which increased airway smooth muscle cell volume (i.e., hypertrophy) was noted throughout the airways, and another in which increased cell number (i.e., hyperplasia) was noted in central bronchi. More recently, Benayoun and colleagues (2), examining bronchial biopsies, found that patients with severe persistent asthma had larger airway smooth muscle cell diameter and increased expression of α-smooth muscle actin and myosin light chain kinase (MLCK) compared with patients with mild disease or chronic obstructive pulmonary disease, further evidence that airway smooth muscle hypertrophy contributes to airway remodeling in asthma.
Despite studies indicating the presence of airway smooth muscle hypertrophy in human asthma, little data exist on the cellular and molecular mechanisms regulating hypertrophy or the accumulation of contractile proteins in these cells. We (3–5) and others (6) have found that prolonged serum deprivation (up to 19 d) of canine airway smooth muscle cells induces about one-sixth of the cells to increase in size and reacquire the contractile protein content and shortening capacity characteristic of contractile cells within intact tissue. Long-term serum deprivation paradoxically reduces the transcriptional activity of the smooth muscle (SM)-22α and smooth muscle myosin heavy chain (smMHC) promoters by 80%, and there is no difference in SM22 promoter activity between nonenlarged and enlarged myocytes (4). These data suggest that cellular hypertrophy and contractile protein expression are regulated in a post-transcriptional manner. In a second model of airway smooth muscle hypertrophy, we developed clonal cell lines of human bronchial smooth muscle origin by retroviral transduction with a temperature-sensitive simian virus (SV)-40 large tumor (T) antigen (7). SV40 large T binds and inactivates the tumor suppressor protein p53. Thus, at the permissive temperature of 33°C, cells demonstrate increased proliferative potential. However, shift to the nonpermissive temperature of 39°C, with subsequent degradation of large T and release of p53, induces expression of p21Cip1, p57Kip1, and cyclin D1 but downregulation of cyclin-dependent kinase (cdk)-2, cyclin A, and cyclin E2, leading to cell cycle arrest in mid-G1 (7). Temperature shift also increases cell size and protein expression of α-smooth muscle actin and MLCK and SM22, paralleling changes observed in the airways of patients with severe asthma (2). Because serum-induced cell growth may continue even when the cell cycle is blocked (8), airway smooth muscle hypertrophy in this system likely results from continued cell growth in the absence of cell division and proliferation. While contractile protein abundance is increased in this model, α-smooth muscle actin and MLCK mRNA are not elevated, suggesting that changes in contractile protein expression, as in the first model, are regulated by translational control mechanisms.
There are two highly regulated steps in the translation initiation pathway. The first step involves the binding of initiator methionyl tRNA (Met-tRNAi) to the 40S ribosomal subunit to form the 43S preinitiation complex, which requires formation of the eukaryotic initiation factor (eIF)-2/GTP/Met-tRNAi ternary complex. eIF2 GTP loading is determined by the activity of eIF2B, a guanine nucleotide exchange factor. eIF2Bε phosphorylation by GSK3β inhibits its GDP/GTP exchange activity, thereby limiting binding of methionyl tRNA to the 40S ribosomal subunit (9). However, phosphorylation by Akt inactivates GSK3β, leading to eIF2B dephosphorylation and activation and a general enhancement of translation initiation. The second step involves the binding of mRNA to the 43S preinitiation complex, which is in turn mediated by through a 7-methylguanosine cap structure at the 5′ end of mRNA. The cap is recognized and “clamped” by the 24-kD eIF4E. The scaffolding protein eIF4G binds to and stabilizes eIF4E as well as eIF4A, a RNA helicase which serves to unwind secondary mRNA structure. In addition to interacting with eIF4G, eIF4E associates with inhibitory proteins termed 4E-binding proteins (4E-BPs). 4E-BP1 undergoes phosphorylation at multiple sites, which results in its release from eIF4E, thereby increasing the availability of eIF4E for binding to eIF4G, eIF4F complex formation, and cap-dependent translation (10, 11). Unlike eIF2 activation, which regulates overall translational activity, the eIF4F complex regulate the translation of specific mRNAs. Regions of hairpin loops and other stable intermolecular secondary structures within the 5′ untranslated region (UTR) impede the initiation of protein synthesis. Translation of mRNAs with regions of stable secondary structure within the 5′ UTR are particularly dependent on eIF4F (12, 13). 4E-BP is typically phosphorylated by the 290-kD kinase mammalian target of rapamycin (mTOR) (10, 11), though other 4E-BP kinases exist. For example, phosphatidylinositol (PI) 3-kinase, which serves as an upstream activator of mTOR, may also phosphorylate 4E-BP directly (14).
Finally, the translation of mRNAs with 5′ terminal oligopyrimidine (TOP) tracts, many of which encode elongation factors and ribosomal proteins involved in mRNA translation, is upregulated by phosphorylation of the S6 ribosomal protein. S6 ribosomal protein, in turn, is phosphorylated by the mitogen- and amino acid–sensitive serine/threonine kinase p70 ribosomal S6 kinase (S6K)-1, which is in turn phosphorylated by mTOR. In contrast to the above pathways, which regulate translation efficiency, this pathway regulates translational capacity by increasing the synthesis of ribosomes.
In serum-deprived canine tracheal myocytes, phophatidylinositol (PI) 3-kinase and S6K1 activities were increased in serum-starved cultures, and immunohistochemical studies of serum-deprived cells showed selective phosphorylation of Akt and S6K1 in elongated cells expressing smMHC. Pretreatment with rapamycin blocked S6K1 phosphorylation and phenotypic change in these cells, implying that PI 3-kinase, mTOR, and S6K1 are responsible for airway smooth muscle hypertrophy and contractile protein accumulation (5). However, because mTOR may phosphorylate either 4E-BP or S6K, the precise translational control mechanism responsible for airway smooth muscle hypertrophy in this model remains unknown. Similarly, little is known about the translational control mechanisms regulating airway smooth muscle hypertrophy in human bronchial smooth muscle cell lines. Activation of PI 3-kinase is increased in temperature-shifted cells, and inhibition of PI 3-kinase blocks α-smooth muscle actin protein expression.
In the present study, we tested the hypothesis that 4E-BP phosphorylation and eIF4E release, which initiate cap-dependent protein synthesis, are required for airway smooth muscle hypertrophy in the human bronchial smooth muscle cell lines described above, a cell culture model which recapitulates airway smooth hypertrophy in severe asthma.
MATERIALS AND METHODS
Use of Temperature-Sensitive SV40 Large T Antigen to Establish Human Bronchial Smooth Muscle Cell Lines
We established this cell line as described (7). Briefly, PA317 and ψ2crip helper cells producing replication-deficient retrovirus carrying the G418 resistance gene and either tsA58 or U19tsa temperature-sensitive mutants of SV40 large T antigen were grown in Dulbecco's modified Eagle's medium with 10% fetal bovine serum, penicillin-streptomycin, and G418 (200 μg/ml; Invitrogen, Carlsbad, CA). Viral supernatant was collected, filtered, and supplemented with polybrene (8 μg/ml; Sigma Chemical, St Louis, MO). Primary human bronchial smooth muscle cells (supplied by Dr. Julian Solway, University of Chicago, Chicago, IL) were incubated with viral supernatant for 48 h. Individual clones were selected at the permissive temperature (33°C) by treatment with G418 (200 μg/ml) and isolated using cloning disks (PGC Scientifics, Frederick, MD). For experiments, low passage (< 20) cells from three different clones were passaged at 50% confluence into 6-well plates and incubated at either 33°C or the nonpermissive temperature (39°C).
Retroviral Transduction of Human Bronchial Cell Lines
The plasmid pACTAG2/hemaggluttinin (HA)-AA-4E-BP1 (Thr37/46Ala) (15) was generously provided by Nahum Sonenberg (McGill University, Montreal, PQ, Canada). cDNA encoding AA-4E-BP-1 was subcloned into the pMSCVpuro retroviral vector (BD Biosciences Clontech, Palo Alto, CA). The Phoenix-GP retrovirus packaging cell line, a 293-cell derivative line that expresses only the gag-pol viral components (provided by G. Nolan, Stanford University, Palo Alto, CA) was transiently transfected with either pMSCVpuro-AA-4E-BP-1 or pMSCV alone, along with pHCMV-G, which contains the VSV envelope glycoprotein. Viral supernatant was collected, filtered, and supplemented with polybrene (8 μg/ml). Human bronchial smooth muscle cell lines at the permissive temperature (33°C) were infected with viral supernatant (4 h × 4). Infected cells were selected with puromycin (2 μg/ml). After selection, cells were grown to confluence, split into 6-well plates, and incubated at either the permissive (33°C) or nonpermissive temperature (39°C).
Cell Lysis
Cells were washed twice with PBS, scraped in lysis buffer containing 50 mM Tris (pH 7.5), 100 mM NaCl, 2 mM EDTA, 50 mM NaF, 1% Triton, 40 mM β-glycerophosphate, 0.2 mM Na3VO4, 1 mM PMSF, and 1% proteinase inhibitor cocktail (Sigma Chemical). Lysates were centrifuged (10 min at 4°C) to remove cellular debris. Protein concentration was measured by using Bradford assay (Bio-Rad, Hercules, CA).
Protein Synthesis
Cells were seeded into 12-well plates (4 × 104 per well) and incubated at either 33°C or 39°C. [3H]-leucine (2 μCi/ml; Perkin Elmer, Boston, MA) was added. Twenty-four hours later, the medium was aspirated and proteins were precipitated with ice-cold 10% trichloroacetic acid (4°C for 30 min). After washing once with ice-cold 95% ethanol, precipitates were dissolved with 0.5 M NaOH containing 0.1% Triton X-100. Radioactivity was then measured by liquid scintillation counter. In selected wells, cells were treated with either LY294002 (10 nM-25 μM), wortmannin (10 nM-10 μM), rapamycin (10 nM-10 μM), SB202190 (10 μM), or PD98059 (30 μM), all from Sigma Chemical.
Cell Size
Cell size was determined by fluorescence-activated cell sorting (Coulter Epics Elite ESP flow cytometer, Fullerton, CA), as described (7, 16). Cells were sorted by cell size (high forward versus sidescatter), autofluorescence (excitation at 488 nm; emission at 525 nm), and cell length (time of flight). To more clearly resolve cell borders and to stain filamentous actin, selected cells were grown on glass coverslips, fixed in 3.7% paraformaldehyde, and incubated with Alexa Fluor 488–conjugated phalloidin (Molecular Probes, Eugene, OR).
7-Methylguanosine-GTP Cap-Binding Assays
Cells were plated on 100-mm dishes at 33°C, grown to 70–80% confluence, and shifted to 39°C. After 24 h, cells were incubated in serum-free media. Selected cultures were treated with LY294002 (25 μM), rapamycin (25 nM), or SB202190 (10 μM). Twenty-four hours later, cells were lysed as described above, and equal cell extracts incubated with 20 μl of m7GTP-Sepharose 4B beads (Pharmacia, Ann Arbor, MI) at 4°C for 4 h (8). After washing twice in lysis buffer, sepharose beads were resuspended in 1× loading buffer with 2% β-mercaptoethanol and resolved by SDS-PAGE. The amounts of endogenous eIF4E and 4E-BP1 bound to beads were determined by immunoblotting using anti-eIF4E and –4E-BP1 antibodies (Cell Signaling Technology, Beverly, MA).
Immunoblotting
Cells were treated in the same manner as 7-methylguanosine-GTP cap-binding assays, above. Lysates were subjected to SDS-PAGE and transferred to nitrocellulose. After blocking, membranes were incubated with primary antibody at 4°C overnight. After washing, blots were incubated with horseradish peroxidase–conjugated secondary antibody at room temperature for 1.5 h and then developed by enhanced chemiluminescence (Pierce, Rockford, IL). Phosphorylation of 4E-BP was assessed by immunoblotting with anti–4E-BP antibody, with inspection for gel shift. The α-band is the least phosphorylated form and migrates most slowly, whereas the β- and γ-bands are more highly phosphorylated and exhibit slower electrophoretic mobility. In selected experiments, phosphorylation was also assessed using anti–phospho-Thr37/Thr46 4E-BP. Phosphorylation of eIF4E, Akt, and mTOR was assessed using anti–phospho-Ser209 eIF4E, anti–phospho-Ser473 Akt, and anti–phospho-Ser2448 mTOR, respectively. To check for protein loading in these experiments, lysates were also probed for total eIF4E, Akt, and mTOR using the appropriate antibodies. Finally, p70 ribosomal S6 kinase phosphorylation was assessed by immunoblotting with anti-p70 ribosomal S6 kinase antibody, with inspection for gel shift. All antibodies were from Cell Signaling Technology.
Statistical Analysis
Changes in protein synthesis were assessed by one-way ANOVA. Differences established by ANOVA were pinpointed by the Student-Newman-Keuls multiple comparison test.
RESULTS
Change to the Nonpermissive Temperature Increases Human Bronchial Smooth Muscle Cell Size
To understand the biochemical pathways that control airway smooth muscle cell size, primary human bronchial smooth muscle cells were conditionally immortalized by infection with retrovirus encoding a temperature-sensitive SV40 large T antigen, as described (7). SV40 large T binds and inactivates p53. Thus, at the permissive temperature of 33°C, cells demonstrate increased proliferative potential. However, shift to the nonpermissive temperature of 39°C, with subsequent degradation of large T and release of p53, induces expression of p21Cip1, p57Kip1, and cyclin D1 but downregulation of cdk2, cyclin A, and cyclin E2, leading to cell cycle arrest in mid-G1 (7).
Conditionally immortalized human bronchial smooth muscle cells incubated at the permissive temperature show the typical “hill and valley” form of primary cells (Figures 1A and 1B). Interestingly, temperature shift increases cell size (Figure 1C). To further assess apparent changes in cell size, myocytes were sorted according to length (time of flight) and forward scatter (Figures 1D and 1E). Doublet discrimination was used to discriminate hypertrophy from doublets, triplets, or damaged cells. Cells incubated at 39°C showed rightward shifts (increases) in time of flight and forward scatter, indicative of hypertrophy. Because serum-induced cell growth may continue even when the cell cycle is blocked (8), airway smooth muscle hypertrophy in this system likely results from continued cell growth in the absence of cell division and proliferation. Culture of primary cells at 33° and 39°C showed no change in phenotype (not shown).
Figure 1.

Change to the non-permissive temperature increases human bronchial smooth muscle cell size. Primary human bronchial smooth muscle cells (A) and cell lines incubated at 33°C (B) each showed the typical “hill and valley” appearance. In C, cells were seeded at half-confluence and shifted from 33°C to 39°C 1 d after plating. Three days later, cell size is increased. Cells incubated at 39°C showed rightward shifts (increases) in time of flight (D) and forward scatter (E), indicative of hypertrophy. Results shown are typical of three experiments.
Time Course of Hypertrophy
We examined the time course of cell size change following temperature shift. Protein synthesis, which in the absence of cell proliferation is an index of hypertrophy, reached a plateau 3 d after temperature shift (Figure 2A). Cell size, as assessed by time of flight, an indicator of cell length, substantially increased 1 d after temperature shift and was near maximal by 2 d after shift (Figure 2B). This rapid time course is consistent with regulation by a fast-acting phosphorylation cascade.
Figure 2.
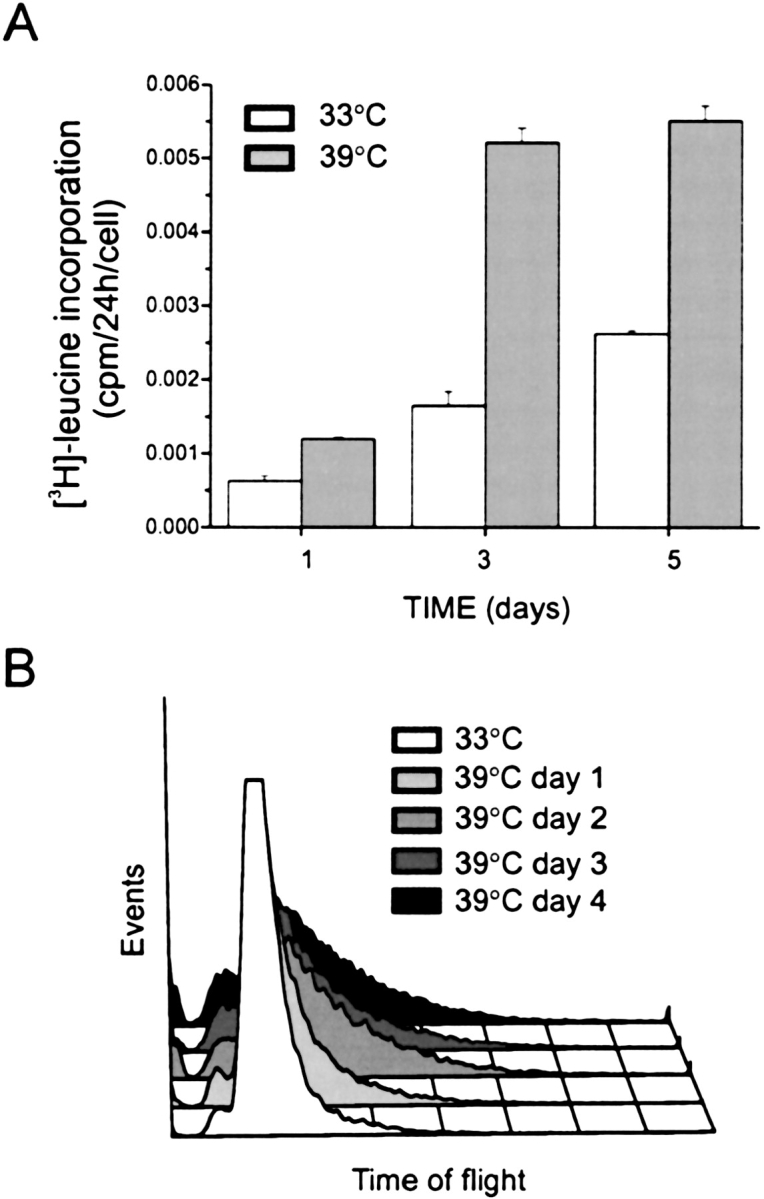
Time course of hypertrophy. Temperature shift-induced changes in protein synthesis (A) and cell size, as assessed by time of flight (B). Human smooth muscle cells were seeded into 6-well plates at 50% confluence and incubated at 33°C overnight. Selected cultures were then shifted to 39°C. For measurement of protein synthesis, cells were incubated with [3H]-leucine (2 μCi/ml) in the last 24 h before harvest, and data normalized by cell number. For time of flight, cells were harvested at designated times for fluorescence activated cell sorting. Protein synthesis reached a plateau 3 d after temperature shift (A). Results shown are mean ± SEM for three experiments. Cell size, as assessed by time of flight, substantially increased 1 d after temperature shift and was near maximal by 2 d after shift (B). Results shown are typical of three experiments.
Attenuation of Cell Size Change by Chemical Inhibitors of PI 3-Kinase, mTOR, and p38 MAP Kinase
We examined the effects of LY294002, a flavonoid-based synthetic PI 3-kinase inhibitor; rapamycin, a macrolide antibiotic inhibitor of mTOR; and SB202190, an imidazole inhibitor of p38 MAP kinase, on the temperature shift-induced change in cell size, as assessed by total protein synthesis and time of flight (Figures 3A and 3B). Rapamycin and LY294002 each significantly reduced protein synthesis and blocked the growth to increased cell size induced by temperature shift, consistent with the notion that activation of PI 3-kinase and mTOR are required for airway smooth muscle hypertrophy. SB202190 had a small but significant effect on both protein synthesis and cell size, suggesting a minor role for p38 MAP kinase. LY294002, and rapamycin also inhibited the protein synthesis of 33°C cells (Figure 3A).
Figure 3.
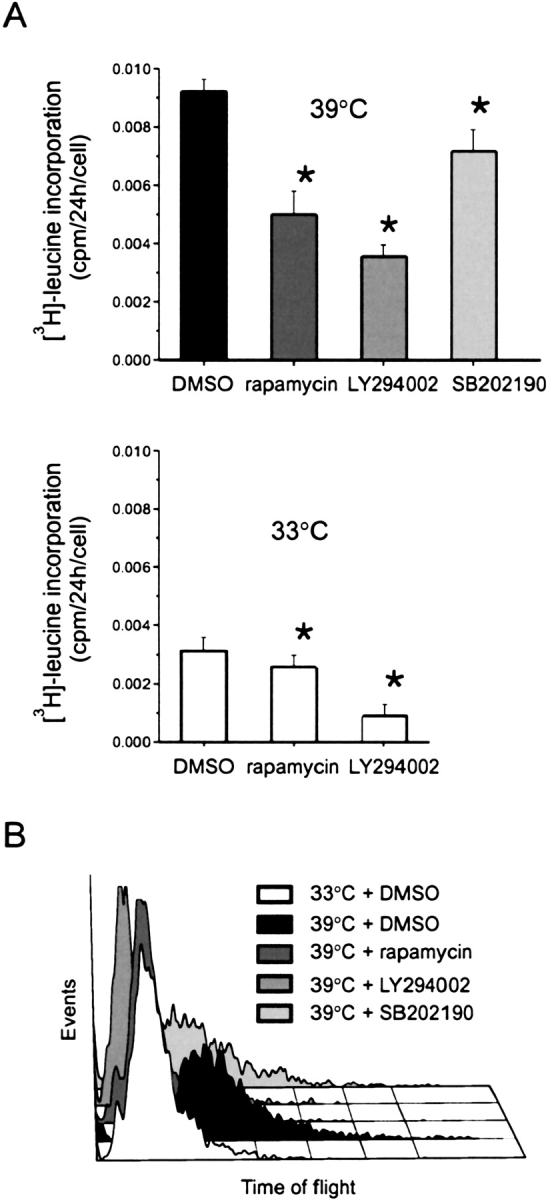
Attenuation of cell size change by chemical inhibitors. The effects of LY294002, rapamycin, and SB202190 on the temperature shift-induced changes in protein synthesis (A) and cell size, as assessed by time of flight (B). Human smooth muscle cells were seeded into 6-well plates at 50% confluence and incubated at 33°C overnight. Selected cultures were then shifted to 39°C. Selected cultures were pretreated with LY294002 (25 μM), rapamycin (25 nM), or SB202190 (10 μM) for 72 h. For measurement of protein synthesis, inhibitors were included with [3H]-leucine (2 uCi/ml) in the last 24 h of the 3-d incubation, and data normalized for cell number. For time of flight, cells were harvested after 3 d for fluorescence activated cell sorting. Protein synthesis results are mean ± SEM for four experiments; * different from DMSO, P < 0.05, ANOVA with repeated measures. Histogram shown is typical of three experiments.
PI 3-Kinase and mTOR, but Not p38 MAP Kinase, Regulate 4E-BP Phosphorylation and Binding to eIF4E
We examined the effect of chemical inhibitors of PI 3-kinase, mTOR, and p38 MAP kinase on the phosphorylation of 4E-BP by immunoblotting with anti–4E-BP antibody and inspecting for gel shift, as well as by immunoblotting with an anti–phospho-Thr37, Thr46 4E-BP antibody. Cells undergoing airway smooth muscle hypertrophy after temperature shift showed substantial 4E-BP phosphorylation, as evidenced by the presence and phosphorylation of the α, β, and γ species (Figure 4A). As expected, pretreatment with the mTOR inhibitor rapamycin decreased phosphorylation of 4E-BP, with near disappearance of the γ species. Pretreatment with LY294002 had even greater effects, with complete loss of the γ species. These data suggest that PI 3-kinase and mTOR are each required for maximal 4E-BP phosphorylation. Finally, inhibition of p38 MAP kinase had no discernable effect on 4E-BP phosphorylation.
Figure 4.

PI 3-kinase and mTOR, but not p38 MAP kinase, regulate 4E-BP phosphorylation and binding to eIF4E. (A) The effect of chemical inhibitors on the phosphorylation of 4E-BP was assessed by immunoblotting with anti–4E-BP antibody and inspecting for gel shift (lower panel), as well as by immunoblotting with an anti–phospho-Thr37, Thr46 4E-BP antibody (upper panel). Three species of phosphorylated 4E-BP, α, β and γ, were detected. (B). Binding of 4E-BP to eIF4E was assessed by m7GTP cap-binding assays. Cell extracts were incubated with 7-methylguanine-GTP-Sepharose 4B beads. Beads were washed, resuspended in sample buffer, and resolved by SDS-PAGE. The amount of eIF4E (upper panel) and 4E-BP (lower panel) bound to beads was determined by immunoblotting using an anti-eIF4E and anti-4E-BP1 antibodies. (C). The effect of chemical inhibitors on the phosphorylation of eIF4E was assessed by immunoblotting with anti–phospho-Ser209 eIF4E antibody (upper panel) and anti-eIF4E (lower panel). Results shown are typical of three experiments.
Binding of 4E-BP to eIF4E was assessed by 7-methylguanine-GTP cap-binding assays. Cell extracts were incubated with 7-methylguanine-GTP-Sepharose 4B beads. Beads were washed, resuspended in sample buffer, and resolved by SDS-PAGE. The amount of eIF4E and 4E-BP bound to beads was determined by immunoblotting using an anti-eIF4E and anti–4E-BP1 antibodies. Cells undergoing airway smooth muscle hypertrophy following temperature shift showed a modest amount of 4E-BP bound to eIF4E (Figure 4B). LY294002 and rapamycin each increased the quantity of 4E-BP bound to eIF4E. SB202190 did not increase the amount of 4E-BP bound to eIF4E, consistent with the absence of an effect on 4E-BP phosphorylation. Because 4E-BP binding reduces the availability of eIF4E for eIF4F complex formation, inhibition of either PI 3-kinase or mTOR would be expected to reduce cap-dependent translation initiation. Taken together with our findings that PI 3-kinase and mTOR are each required for the development of airway smooth muscle hypertrophy in this system, these data suggest that 4E-BP phosphorylation and cap-dependent translation initiation may be required for airway smooth muscle hypertrophy.
eIF4E itself is phosphorylated at Ser209 by the MAP kinase signal integrating kinases (MNK)-1 and -2. MNK1 is activated upon treatment with agents that activate MAP kinases including growth factors and anisomycin, whereas MNK2 has high basal activity (17, 18). Because Ser209 phosphorylation may alter the binding affinity of eIF4E for 7-methylguanosine (19), we examined the effects of the three chemical inhibitors on eIF4E phosphorylation (Figure 4C). LY294002, rapamycin, and SB202190 each reduced eIF4E phosphorylation, with LY294002 again having the maximum effect. Because the SB202190 had only modest inhibitory effects on airway smooth muscle cell size, these data suggest that eIF4E phosphorylation may play a relatively minor role in the development of airway smooth muscle hypertrophy. Pretreatment with a chemical inhibitor of the extracellular signal–regulated kinase pathway, PD98059, had no effect on protein synthesis (data not shown).
To define the relationships between PI 3-kinase, mTOR, and 4E-BP, we examined the effects of various concentrations of the chemical PI 3-kinase inhibitors LY294002 and wortmannin on the phosphorylation of Akt, mTOR, and 4E-BP, as well as on protein synthesis. Submaximal concentrations of LY294002 (1–2.5 μM) reduced Akt phosphorylation, 4E-BP phosphorylation (Figure 5A), and protein synthesis (Figure 5B) while having no discernable effect on mTOR phosphorylation (Figure 5A). Similar results were obtained for wortmannin (see Figure 5B for protein synthesis; for brevity, effects on Akt, 4E-BP, and mTOR phosphorylation are not shown). These data suggest that PI 3-kinase regulates 4E-BP phosphorylation and protein synthesis by a mTOR-independent pathway. On the other hand, the highest concentrations of LY294002 and wortmannin reduced mTOR phosphorylation and further decreased protein synthesis (Figures 5A and 5B), suggesting that PI 3-kinase also regulates protein synthesis by a 4E-BP–independent, mTOR-dependent pathway. Consistent with this, the mTOR inhibitor rapamycin, in addition to decreasing 4E-BP phosphorylation and protein synthesis, attenuated p70 ribosomal S6 kinase phosphorylation at the lowest concentration tested (Figure 5C).
Figure 5.
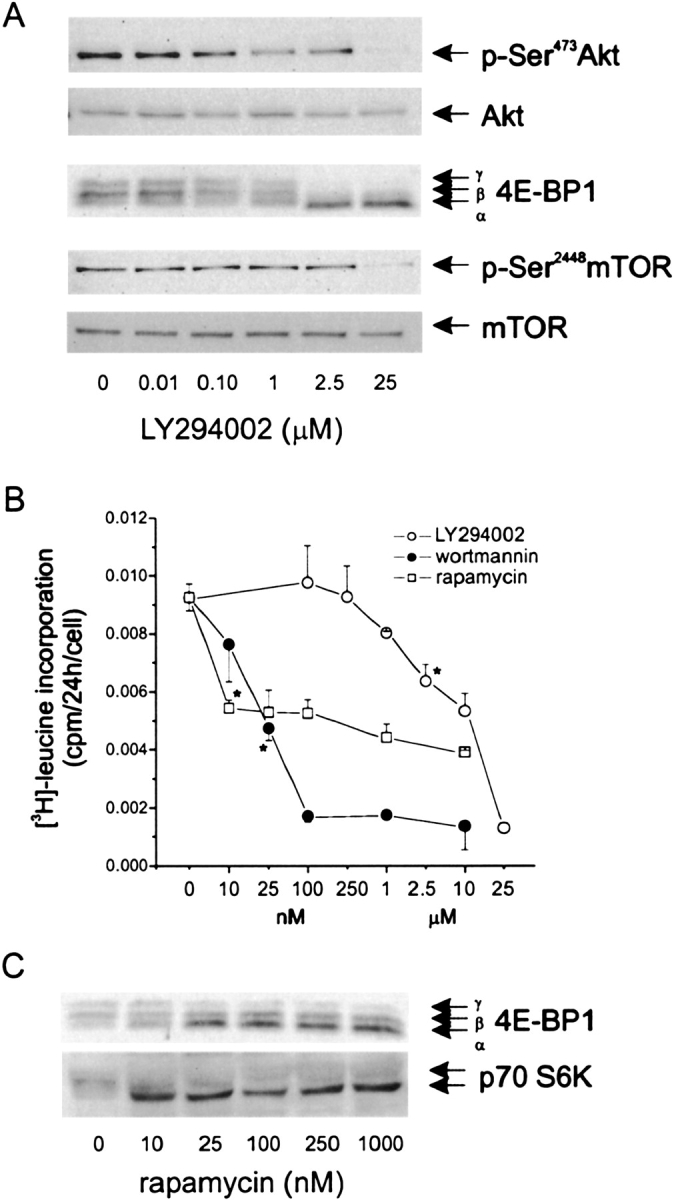
PI 3-kinase regulates protein synthesis via mTOR-dependent and -independent pathways. (A) Effects of LY294002 on phosphorylation of Akt, 4E-BP, and mTOR. (B) Effects of LY294002, wortmannin, and rapamycin on protein synthesis. (C) Effect of rapamycin on phosphorylation of 4E-BP and p70 ribosomal S6 kinase. Blots shown are typical to two experiments. Protein synthesis was assessed by [3H]-leucine incorporation. Protein synthesis results shown are mean ± SEM for three to four experiments; *P < 0.05, one-way ANOVA with repeated measures.
Requirement of 4E-BP Phosphorylation and eIF4E for Airway Smooth Muscle Hypertrophy
As noted above, LY294002 and rapamycin each increased the quantity of 4E-BP bound to eIF4E, suggesting that eIF4E-dependent translation is required for airway smooth muscle hypertrophy. To test this directly, stable cell lines expressing either HA-AA-4E-BP-1 or empty vector were created by retroviral infection of human bronchial smooth muscle cell lines. HA-AA-4E-BP-1 encodes an mTOR-insensitive mutant of 4E-BP1 that dominantly binds to and constitutively inhibits eIF4E and therefore cap-dependent translation. AA-4E-BP1 contains alanine substitution mutations at threonines 37 and 46, which are mTOR-dependent priming phosphorylation sites, and thus cannot be phosphorylated by mTOR (15). At the permissive temperature, all cells assumed a proliferative phenotype (Figures 6A and 6B). Cells expressing the empty retroviral vector, pMSCV, underwent hypertrophy upon temperature shift (Figure 6C). However, human airway smooth muscle cells expressing HA-AA-4E-BP-1 did not change phenotype (Figure 6D). Immunoblots using anti-HA and –4E-BP antibodies confirmed the presence of the HA-tagged mutant in these cells (Figure 6E). Expression of HA-AA-4E-BP-1 was associated with a reduction in protein synthesis (Figure 6F). Taken together, these data suggest that eIF4E is required for the development of airway smooth muscle hypertrophy.
Figure 6.
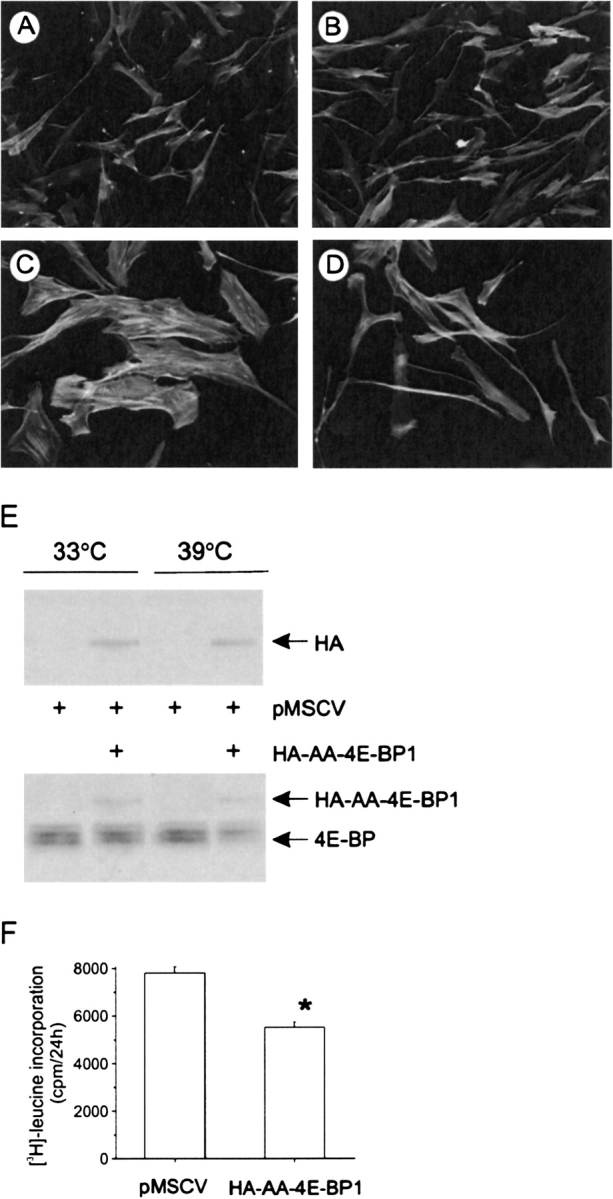
Requirement of 4E-BP phosphorylation and eIF4E for airway smooth muscle hypertrophy. Stable cell lines expressing either HA-AA-4E-BP-1 or empty vector were created by retroviral infection of human bronchial smooth muscle cell lines. At the permissive temperature, all cells assumed a proliferative phenotype (A, B). Cells expressing the empty retroviral vector, pMSCV, underwent hypertrophy upon temperature shift (C). However, human airway smooth muscle cells expressing HA-AA-4E-BP-1 did not change phenotype (D). Results shown are typical of three experiments. The presence of the HA-tagged mutant was confirmed by immunoblots using anti-HA (E, upper panel) and anti–4E-BP antibodies (lower panel). The effect of HA-AA-4E-BP1 expression on protein synthesis was assessed by [3H]-leucine incorporation (F). Protein synthesis results shown are mean ± SEM for three experiments; *P = 0.011, paired t test.
DISCUSSION
Increased airway smooth muscle mass has been shown in nonfatal (2, 20) and fatal asthma (1, 21–25). Ebina and coworkers (1) studied the airway thickness and smooth muscle cell number of patients with fatal asthma with state-of-the-art stereologic methods. Two asthmatic subtypes were found: one in which smooth muscle thickness and cell number were increased only in the central bronchi (Type I), and another in which the quantity of smooth muscle was increased throughout the airway tree (Type II). In Type II, there was no increase in airway smooth muscle cell number, suggesting the presence of cellular hypertrophy. More recently, Benayoun and coworkers (2) examined bronchial biopsies from patients with asthma and chronic obstructive pulmonary disease, as well as from control subjects. They found that larger airway smooth muscle diameter and increased expression of α-smooth muscle actin and MLCK distinguished patients with severe persistent asthma from patients with milder disease or with chronic obstructive pulmonary disease. Finally, Woodruff and colleagues (26) found that patients with mild asthma showed no increase in cell size, though cell number was 2-fold higher. Interestingly, while smooth muscle mass increased by 50–83% (as assessed by α-smooth muscle actin immunoreactivity), the mRNA expression of contractile protein genes, as assessed by real time PCR, was not increased. Taken together, these data highlight the potential importance of hypertrophy and post-transcriptional mechanisms in the pathogenesis of asthma, and the need for a precise understanding of the biochemical events involved in airway smooth muscle hypertrophy as well as mitogenesis.
To this end, we previously established a conditionally immortalized human bronchial smooth muscle cell line with a temperature-sensitive SV40 large T antigen (7). Temperature shift and degradation of large T result in cell cycle arrest and inhibition of cell proliferation. In addition, we found that temperature shift also increases airway smooth muscle cell size and contractile protein expression, similar to changes observed in severe asthma. As mRNA expression is not increased, protein expression is likely under translational control. In the present study, we examined the translational control mechanism regulating airway smooth muscle cell size in this model.
Translation of most eukaryotic mRNAs is initiated through a 7-methylguanosine cap structure at the 5′ end of mRNA. The cap is recognized and “clamped” by the 24-kD eIF4E. Another factor, the scaffolding protein eIF4G, binds to and stabilizes eIF4E and poly-A binding protein, which in turn associates with the poly-A tail at the 3′ end of the mRNA. Because hypophosphorylated forms of 4E-BP prevent the association of eIF4E with eIF4G, phosphorylation of 4E-BP is required for cap-dependent translation initiation. We hypothesized that phosphorylation of 4E-BP is required for airway smooth muscle hypertrophy. To test this, we pretreated cells with chemical inhibitors of PI 3-kinase and mTOR, each of which have been shown to attenuate angiotensin II–induced 4E-BP phosphorylation and protein synthesis in vascular smooth muscle cells (27). We found that, as in vascular smooth muscle, PI 3-kinase and mTOR are required for 4E-BP phosphorylation and hypertrophy in airway smooth muscle. LY294002 appeared to have a greater inhibitory effect on 4E-BP phosphorylation than rapamycin, suggesting the possibility that PI 3-kinase may regulate 4E-BP in an mTOR-independent manner. Consistent with this, moderate concentrations of the chemical PI 3-kinase inhibitors attenuated protein synthesis without decreasing mTOR phosphorylation. Indeed, it has recently been shown that 4E-BP1 may be directly phosphorylated by the p110α and p110γ isoforms of the class I PI 3-kinases (14).
In the present study, LY294002 and rapamycin also increased the binding of 4E-BP with eIF4E, suggesting that eIF4E- and cap-dependent translation is required for airway smooth muscle hypertrophy. To test this directly, cells were infected with retrovirus encoding 4E-BP1 mutant containing alanine substitution mutations at threonines 37 and 46, which are mTOR-dependent priming phosphorylation sites, and thus cannot be phosphorylated by mTOR. This phosphorylation site-defective mutant constitutively binds the eIF4E–cap complex. Expression of HA-AA-4E-BP prevented airway smooth muscle hypertrophy, demonstrating that eIF4E-dependent translation is required for this process. The requirement of 4E-BP phosphorylation and cap-dependent translation initiation in the regulation of cell size has been demonstrated only once previously, in human U2OS osteosarcoma cells expressing the same phosphorylation site-defective mutant of 4EBP1 (8). In the latter study, the 4E-BP mutant blocked the ability of eIF4E overexpression to increase cell growth.
Once activated, mTOR phosphorylates 4E-BP and S6K1, a mitogen- and amino acid–sensitive kinase which increases the translation of mRNAs with 5′ terminal oligopyrimidine (TOP) tracts, many of which are involved in mRNA translation like elongation factors and ribosomal proteins. Our previous studies in serum-deprived canine airway smooth muscle cells have shown that rapamycin blocks both S6 kinase activation and cell hypertrophy. Rapamycin also blocks S6 kinase phosphorylation in the current model system. However, our finding that eIF4E is required for airway smooth muscle hypertrophy is consistent with the notion that S6 kinase phosphorylation and translation of 5′ TOP mRNAs are insufficient to fully drive hypertrophy in the absence of mTOR/4E-BP1 signal.
We also showed that an inhibitor of p38 MAP kinase, SB202190, had small but significant inhibitory effects on airway smooth muscle protein synthesis and cell size. SB202190 had no effect on the phosphorylation of 4E-BP, but instead decreased eIF4E phosphorylation. Together, these results suggest that the phosphorylation state of eIF4E may play a minor role in the development of airway smooth muscle hypertrophy. MNK1 activation and eIF4E phosphorylation have recently been shown to be required for angiotensin II–induced protein synthesis in vascular smooth muscle cells (28). LY294002 and rapamycin also decreased eIF4E phosphorylation and the amount of eIF4E bound to 7-methylguanosine GTP. However, this is likely to be an indirect effect, as binding of 4E-BP to eIF4E has been shown to inhibit the phosphorylation of eIF4E by MNK1 (29).
In conclusion, our data demonstrate that inhibition of 4E-BP phosphorylation by chemical inhibitors of PI3 kinase and mTOR increases the binding of 4E-BP with eIF4E while decreasing cell size and protein synthesis, suggesting that eIF4E- and cap-dependent translation is required for airway smooth muscle hypertrophy. Further, expression of a nonphosphorylatable 4E-BP1 mutant that constitutively binds the eIF4E–cap complex also blocks phenotypic change, confirming that 4E-BP phosphorylation and eIF4E-dependent translation are required for airway smooth muscle hypertrophy. Further studies examining the translational control pathways regulating airway smooth muscle hypertrophy may provide new insight into the pathogenesis of severe asthma, and lead to new therapeutic interventions against this prevalent chronic disease.
These studies were supported by National Institutes of Health grants HL54685 and HL63314 (M.B.H.).
Conflict of Interest Statement: L. Z. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. A.M.G. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. J.K.B. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. Y.J. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.L.R. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.K.A. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. D.C.F. does not have a financial relationship with a commercial entity that has an interest in the subject of this manuscript. M.B.H. is the recipient of a research grant from GlaxoSmithKline.
References
- 1.Ebina M, Takahashi T, Chiba T, Motomiya M. Cellular hypertrophy and hyperplasia of airway smooth muscles underlying bronchial asthma: a 3-D morphometric study. Am Rev Respir Dis 1993;148:720–726. [DOI] [PubMed] [Google Scholar]
- 2.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 2003;167:1360–1368. [DOI] [PubMed] [Google Scholar]
- 3.Halayko AJ, Camoretti-Mercado B, Forsythe SM, Vieira JE, Mitchell RW, Wylam ME, Hershenson MB, Solway J. Divergent differentiation paths in airway smooth muscle culture: induction of functionally contractile myocytes. Am J Physiol Lung Cell Mol Physiol 1999;276:L197–L206. [DOI] [PubMed] [Google Scholar]
- 4.Camoretti-Mercado B, Liu H, Halayko A, Forsythe S, Kyle J, Li B, Fu Y, McConville J, Kogut P, Vieira J, et al. Physiological control of smooth muscle-specific gene expression through regulated nuclear translocation of serum response factor. J Biol Chem 2000;275:30387–30393. [DOI] [PubMed] [Google Scholar]
- 5.Halayko AJ, Kartha S, Stelmack GL, McConville J, Tam J, Camoretti-Mercado B, Forsythe SM, Hershenson MB, Solway J. Phophatidyl-inositol-3 kinase/mammalian target of rapamycin/p70S6K regulates contractile protein accumulation in airway myocyte differentiation. Am J Respir Cell Mol Biol 2004;31:266–275. [DOI] [PubMed] [Google Scholar]
- 6.Ma X, Wang Y, Stephens NL. Serum deprivation induces a unique hypercontractile phenotype of cultured smooth muscle cells. Am J Physiol Cell Physiol 1998;274:C1206–C1214. [DOI] [PubMed] [Google Scholar]
- 7.Zhou L, Li J, Goldsmith AM, Newcomb DC, Giannola DM, Vosk RG, Eves EM, Rosner MR, Solway J, Hershenson MB. Human bronchial smooth muscle cell lines show a hypertrophic phenotype typical of severe asthma. Am J Respir Crit Care Med 2004;169:703–711. [DOI] [PubMed] [Google Scholar]
- 8.Fingar DC, Salama S, Tsou C, Harlow E, Blenis J. Mammalian cell size is controlled by mTOR and its downstream targets S6K1 and 4EBP1/eIF4E. Genes Dev 2002;16:1472–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Welsh GI, Miller CM, Loughlin AJ, Price NT, Proud CG. Regulation of eukaryotic initiation factor eIF2B: glycogen synthase kinase-3 phosphorylates a conserved serine which undergoes dephosphorylation in response to insulin. FEBS Lett 1998;421:125–130. [DOI] [PubMed] [Google Scholar]
- 10.Beretta L, Gingras AC, Svitkin YV, Hall MN, Sonenberg N. Rapamycin blocks the phosphorylation of 4E–BP1 and inhibits cap-dependent initiation of translation. EMBO J 1996;15:658–664. [PMC free article] [PubMed] [Google Scholar]
- 11.Brunn GJ, Hudson CC, Sekuli A, Williams JM, Hosoi H, Houghton PJ, Lawrence JC Jr, Abraham RT. Phosphorylation of the translational repressor PHAS-I by the mammalian target of rapamycin. Science 1997;277:99–101. [DOI] [PubMed] [Google Scholar]
- 12.Sonenberg N, Gingras AC. The mRNA 5′ cap-binding protein eIF4E and control of cell growth. Curr Opin Cell Biol 1998;10:268–275. [DOI] [PubMed] [Google Scholar]
- 13.Svitkin YV, Pause A, Haghighat A, Pyronnet S, Witherell G, Belsham GJ, Sonenberg N. The requirement for eukaryotic initiation factor 4A (elF4A) in translation is in direct proportion to the degree of mRNA 5′ secondary structure. RNA 2001;7:382–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Foukas LC, Shepherd PR. eIF4E binding protein 1 and H-Ras are novel substrates for the protein kinase activity of class-I phosphoinositide 3-kinase. Biochem Biophys Res Commun 2004;319:541–549. [DOI] [PubMed] [Google Scholar]
- 15.Gingras A-C, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R, Sonenberg N. Regulation of 4E–BP1 phosphorylation: a novel two-step mechanism. Genes Dev 1999;13:1422–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Inoki K, Li Y, Zhu T, Wu J, Guan KL. TSC2 is phosphorylated and inhibited by Akt and suppresses mTOR signalling. Nat Cell Biol 2002;4:648–657. [DOI] [PubMed] [Google Scholar]
- 17.Waskiewicz AJ, Flynn A, Proud CG, Cooper JA. Mitogen-activated protein kinases activate the serine/threonine kinases Mnk1 and Mnk2. EMBO J 1997;16:1909–1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Scheper GC, Morrice NA, Kleijn M, Proud CG. The mitogen-activated protein kinase signal-integrating kinase Mnk2 is a eukaryotic initiation factor 4E kinase with high levels of basal activity in mammalian cells. Mol Cell Biol 2001;21:743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Scheper GC, Proud CG. Does phosphorylation of the cap-binding protein eIF4E play a role in translation initiation? Eur J Biochem 2002;269:5350–5359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Carroll N, Elliot J, Morton A, James A. The structure of large and small airways in nonfatal and fatal asthma. Am Rev Respir Dis 1993;147:405–410. [DOI] [PubMed] [Google Scholar]
- 21.Dunhill MS, Massarella GR, Anderson JA. A comparison of the quantitative anatomy of the bronchi in normal subjects, in status asthmaticus, in chronic bronchitis and in emphysema. Thorax 1971;24:176–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Takizawa T, Thurlbeck WM. Muscle and mucous gland size in the major bronchi of patients with chronic bronchitis, asthma and asthmatic bronchitis. Am Rev Respir Dis 1971;104:331–336. [DOI] [PubMed] [Google Scholar]
- 23.James AL, Pare PD, Hogg JC. The mechanics of airway narrowing in asthma. Am Rev Respir Dis 1989;139:242–246. [DOI] [PubMed] [Google Scholar]
- 24.Ebina M, Yaegashi H, Chiba R, Takahashi T, Motomiya M, Tanemura M. Hyperreactive site in the airway tree of asthmatic patients revealed by thickening of bronchial muscles: a morphometric study. Am Rev Respir Dis 1990;141:1327–1332. [DOI] [PubMed] [Google Scholar]
- 25.Saetta M, Di Stefano A, Rosina C, Thiene G, Fabbri LM. Quantitative structural analysis of peripheral airways and arteries in sudden fatal asthma. Am Rev Respir Dis 1991;143:138–143. [DOI] [PubMed] [Google Scholar]
- 26.Woodruff PG, Dolganov GM, Ferrando RE, Donnelly S, Hays SR, Solberg OD, Carter R, Wong HH, Cadbury PS, Fahy JV. Hyperplasia of smooth muscle in mild to moderate asthma without changes in cell size or gene expression. Am J Respir Crit Care Med 2004;169:1001–1006. [DOI] [PubMed] [Google Scholar]
- 27.Yamakawa T, Tanaka S-i, Kamei J, Kadonosono K, Okuda K. Phosphatidylinositol 3-kinase in angiotensin II-induced hypertrophy of vascular smooth muscle cells. Eur J Pharmacol 2003;478:39–46. [DOI] [PubMed] [Google Scholar]
- 28.Ishida M, Ishida T, Nakashima H, Miho N, Miyagawa K, Chayama K, Oshima T, Kambe M, Yoshizumi M. Mnk1 is required for angiotensin II-induced protein synthesis in vascular smooth muscle cells. Circ Res 2003;12:1218–1224. [DOI] [PubMed] [Google Scholar]
- 29.Wang X, Flynn A, Waskiewicz AJ, Webb BLJ, Vries RG, Baines IA, Cooper JA, Proud CG. The Phosphorylation of eukaryotic initiation factor eIF4E in Response to phorbol esters, cell stresses, and cytokines is mediated by distinct MAP kinase pathways. J Biol Chem 1998;273:9373–9377. [DOI] [PubMed] [Google Scholar]