

Abstract
The intraspinal cues that orchestrate T-cell migration and activation after spinal contusion injury were characterized using B10.PL (wild-type) and transgenic (Tg) mice with a T-cell repertoire biased toward recognition of myelin basic protein (MBP). Previously, we showed that these strains exhibit distinct anatomical and behavioral phenotypes. In Tg mice, MBP-reactive T-cells are activated by spinal cord injury (SCI), causing more severe axonal injury, demyelination, and functional impairment than is found in non-Tg wild-type mice (B10.PL). Conversely, despite a robust SCI-induced T-cell response in B10.PL mice, no overt T-cell-mediated pathology was evident. Here, we show that chronic intraspinal T-cell accumulation in B10.PL and Tg mice is associated with a dramatic and sustained increase in CXCL10/IP-10 and CCL5/RANTES mRNA expression. However, in Tg mice, chemokine mRNA were enhanced 2- to 17-fold higher than in B10.PL mice and were associated with accelerated intraspinal T-cell influx and enhanced CNS macrophage activation throughout the spinal cord. These data suggest common molecular pathways for initiating T-cell responses after SCI in mice; however, if T-cell reactions are biased against MBP, molecular and cellular determinants of neuroinflammation are magnified in parallel with exacerbation of neuropathology and functional impairment.
Keywords: macrophage, microglia, neuroinflammation, spinal cord injury, T-lymphocyte, chemokines, myelin basic protein
Introduction
The neuropathology and loss of function associated with spinal cord injury (SCI) is caused initially by mechanical trauma and then by diverse mechanisms of secondary injury that include cells (e.g., neutrophils, macrophages, and T-cells) and mediators (e.g., chemokines, cytokines, and proteases) of the immune system (Popovich, 2000). We showed previously that endogenous myelin-reactive T-cells are activated by SCI and contribute to neurodegeneration (Popovich et al., 1997; Jones et al., 2002). Furthermore, expansion of these T-cells via vaccination induces profound neuroinflammation and exacerbates neuropathology and locomotor dysfunction after SCI (Jones et al., 2004). T-Cells also can inhibit normal repair of injured tissues without directly influencing neuron/glial survival. Specifically, chronic T-cell activation can precipitate pathological fibrosis and scarring (Wynn, 2004).
Still, others have proposed T-cell-mediated neuroprotection in models of CNS injury and neurodegeneration (Schwartz and Kipnis, 2001; Kipnis et al., 2002). Indeed, the functional diversity of lymphocytes within the injury site undoubtedly extends beyond that of cell killing and tissue injury. In all mice that we studied after SCI (four inbred strains), indices of spontaneous functional recovery and wound healing occur concomitant with the intraspinal accumulation of large numbers of T-cells (Sroga et al., 2003). Despite evidence for SCI-induced activation of myelin-reactive T-cells, most T-cells that infiltrate the injury site are likely to be responsive to proteins other than those found in myelin. In experimental autoimmune encephalomyelitis (EAE), a CNS autoimmune demyelinating disease triggered by myelin-reactive T-cells, most (>90%) CNS-infiltrating T-cells are not myelin reactive (Cross et al., 1990; Steinman, 1996). These latter cells are believed to have immune-modulatory functions and could facilitate CNS repair (Hammarberg et al., 2000; Muhallab et al., 2002). In fact, non-CNS-reactive T-cells can be therapeutic in the pathological CNS and are required for healing of peripheral tissues (Barbul et al., 1989a,b; Efron et al., 1990; Schaffer and Barbul, 1998). Thus, how T-cells influence spinal cord integrity and function will be determined in part by the antigen specificity of the responding T-cell population.
T-Cell functions are also controlled by molecular cues that draw them to the injury site and then ultimately by the microenvironment they encounter. Chemokines direct T-cell migration and then modulate their activation and effector potential at sites of inflammation (Sallusto et al., 1998, 2000). The subsequent composition of the cytokine, neurotrophin, and growth factor milieu within inflammatory foci is then influenced by reciprocal interactions between T-cells and CNS macrophages.
Previously, we showed that myelin-reactive T-cells exacerbate the neuropathology and functional outcome caused by SCI (Popovich et al., 1997; Jones et al., 2002, 2004). However, we also showed that spontaneous functional recovery and wound healing occur concomitant with the accumulation of large numbers of intraspinal T-cells after SCI in various inbred mouse strains (Sroga et al., 2003). Whether these divergent T-cell responses are associated with distinct molecular signatures is unknown. Because the functional potential of innate (e.g., macrophages/microglia) and adaptive (e.g., lymphocytes) immune cells are exquisitely controlled by chemokines, cytokines, and intercellular reactions requiring costimulatory molecules, we compared specific subsets of these molecules in wild-type B10.PL [non-transgenic (nTg)] and myelin basic protein (MBP) T-cell receptor (TCR) transgenic (Tg) mice at times coincident with peripheral T-cell activation [7 d postinjury (dpi)] or stabilization of functional recovery (21 dpi) after SCI. A subset of macrophage activation markers and immune-regulatory/growth factors also was compared between strains using real-time PCR and immunohistochemistry. The data reveal similar molecular and cellular neuroinflammatory phenotypes in Tg and nTg mice; however, when the T-cell response is biased toward MBP, these phenotypes are magnified, as is the intraparenchymal distribution of select molecules indicative of enhanced macrophage/T-cell interactions. Importantly, this latter phenotype is associated with marked neurodegeneration and severe functional impairment.
Materials and Methods
Animals and spinal cord injury. MBP TCR Tg mice were generated by breeding Vα4/Vβ8.2 MBP TCR Tg mice with B10.PL mice (Jones et al., 2002). Progeny were screened for expression of the Vβ8.2 transgene (>95% CD4+ T-cells) by flow cytometry of peripheral blood. Wild-type nTg B10.PL littermates served as controls. Anesthetized mice [ketamine (80 mg/kg, i.p.)/xylazine (40 mg/kg, i.p.)] received a spinal contusion injury as described previously (Jones et al., 2002). Briefly, a laminectomy was performed at vertebral level T9/10. The exposed dorsal surface of the spinal cord was displaced 0.8 mm using an electromagnetic SCI device (Jakeman et al., 2000). Manual bladder expression was performed twice daily, and prophylactic antibiotic treatment (Gentocin, 50 mg/d) was maintained throughout the study. Mice were housed in HEPA-filtered Bio-Clean units.
Tissue processing. Spinal cords were removed at 0, 7, or 21 dpi (PCR analyses) or 70 dpi (immunohistochemical analyses). These times coincide with the onset of peripheral T-cell activation (7 dpi) and a time of functional stability (21-70 dpi) in SCI mice (Jones et al., 2002). For quantitative reverse-transcription PCR (Q-RT-PCR) assays, anesthetized animals were perfused intracardially with sterile PBS (0.1 m, pH 7.4). Spinal cords were snap frozen in 2-methylbutane cooled with liquid nitrogen, and RNA was extracted from a 4 mm segment of spinal cord centered on the impact site. For histological and immunohistological analyses, animals were anesthetized and then perfused intracardially with PBS (0.1 m, pH 7.4), followed by 4% paraformaldehyde in 0.1 m PBS. Spinal cords were removed and processed for histological analyses as described previously (Popovich et al., 1997; Jones et al., 2002).
Q-RT-PCR analysis of spinal cord mRNA. Gene-specific primer pairs (Table 1) were used to detect expression of chemokine, costimulatory molecule, and growth factor mRNA in injured (7 and 21 dpi) and uninjured spinal cord (n = 3-4 per group per time point). Briefly, total RNA was purified from spinal cord using Trizol (Invitrogen, Carlsbad, CA), followed by RNeasy (Qiagen, Valencia, CA) binding and quantification by spectrophotometry. cDNA was prepared from RNA by reverse transcription with SuperScript II and random primers (Invitrogen). The PCRs were performed using 10 ng of cDNA, 50 nm each primer, and SYBR Green master mix (Applied Biosystems, Foster City, CA) in 20 μl reactions. Levels of Q-RT-PCR product were measured using SYBR Green fluorescence collected during real-time PCR on an Applied Biosystems 7900HT system (Ririe et al., 1997). Standard curves were generated for each gene using a control cDNA dilution series. Melting point analyses were performed for each reaction to confirm single amplified products. Data are expressed relative to the mean mRNA/glyceraldehyde-3-phosphate dehydrogenase ratio of uninjured spinal cord samples (n = 4 per group).
Table 1.
Primers used for quantitative real-time PCR
|
Mus gene |
Accession number |
Forward primer (5′-3′) |
Reverse primer (5′-3′) |
|---|---|---|---|
| CD80 | AF065894 | CCCCAGAAGACCCTCCTGAT | CTGCCCCAAAGAGCACAAGT |
| CD86 | NM_019388 | CCTCCTTGTGATGCTGCTCA | GGCTGATTCGGCTTCTTGTG |
| CCL5 | NM_013653 | TGCTCCAATCTTGCAGTCGT | ACACACTTGGCGGTTCCTTC |
| CXCL10 | NM_021274 | GGGCCAGTGAGAATGAGGG | GCTCGCAGGGATGATTTCAA |
| CCL2 | NM_010778 | TTCAGTCTGGTAACATGACGGC | GGTGCGTTGAGTCTGGCATAA |
| IGF-1 | NM_010512 | CCTCAGACAGGCATTGTGGA | CCTCAGATCACAGCTCCGGA |
| TGF-β1 | NM_011577 | TGGAGCTGGTGAAACGGAAG | ACAGGATCTGGCCACGGAT |
| IL-1ra |
NM_031167
|
CGCTTTACCTTCATCCGCTC |
TGACTCAAAGCTGGTGGTGG |
Accession numbers are from GenBank.
Immunohistochemistry. Serial sections were cut on a cryostat (14 μm) through the rostrocaudal extent of the lesion as described previously (Sroga et al., 2003) and then were mounted onto Superfrost-Plus slides (Fisher Scientific, Houston, TX). Within the spinal cord, CD4+ T-cells and microglia/macrophages were identified using the following antibodies: anti-CD4 and anti-CD11b (1:200; Serotec, Indianapolis, IN); and anti-I-A/I-E [major histocompatibility complex (MHC) class II; 1:3000; PharMingen, San Diego, CA] and CD86 (B7-2; 1:1000; PharMingen). To minimize nonspecific binding, tissue sections were incubated with 2-4% rabbit serum in PBS before applying primary antisera overnight at 4°C. Sections were rinsed three times in Tris-buffered saline and then incubated with biotinylated secondary antibody for 1 h at room temperature. Endogenous peroxidase was quenched by incubating tissues in 6% H2O2/methanol for 15 min. Bound antibody was visualized using the Elite-ABC reagent for 1 h at room temperature, followed by incubation in DAB substrate (Vector Laboratories, Burlingame, CA).
Image analysis and T-cell counts. Cell counts were obtained using computer-assisted image analysis (MCID Elite; Imaging Research, St. Catherine's, Ontario, Canada). Briefly, digital images were captured at 20×, and labeled cells were detected by electronically establishing the light-intensity threshold at which positively stained profiles were visible. Frequently, T-lymphocytes were found as cell clusters, making it difficult to delineate single cell profiles. When present in clusters, T-cell numbers were estimated from area measurements of labeled clusters using a predetermined mean cell size of 16 μm. This latter value was determined by quantifying the diameter of hundreds of clearly labeled T-cell profiles at high magnification from several spinal injured samples. After SCI, T-cells were not evenly distributed across the rostrocaudal extent of the contusion lesion. Consequently, T-cells were counted in sections spanning 140 μm intervals and then were averaged in 700 μm epochs spanning ∼6 mm of injured spinal cord. Data are plotted as a function of distance from the impact site.
To differentiate between and quantify T-cells within “lesioned” and “nonlesioned” spinal cord, tissue integrity was assessed at high power using luxol fast blue (LFB) or fibronectin-stained (data not shown) sections adjacent to those labeled with anti-CD4 antibodies. Differential interference contrast (DIC/Nomarski) optics also were used to confirm tissue integrity. Lesioned tissue was clearly visible as areas with diffuse myelin pallor (light/patchy LFB staining in regions in which strong labeling typically exists (i.e., white matter) and abnormal cytoarchitecture under DIC (e.g., dense connective tissue formation, cell clustering). Only T-cells with continuous membrane labeling were counted. Because none of the tissue at the site of injury could be designated as nonlesioned, cell counts expressed in Figure 2 A (inset) represent data collapsed across 700 μm epochs spanning ∼6 mm of injured spinal cord excluding the epicenter bin.
Figure 2.
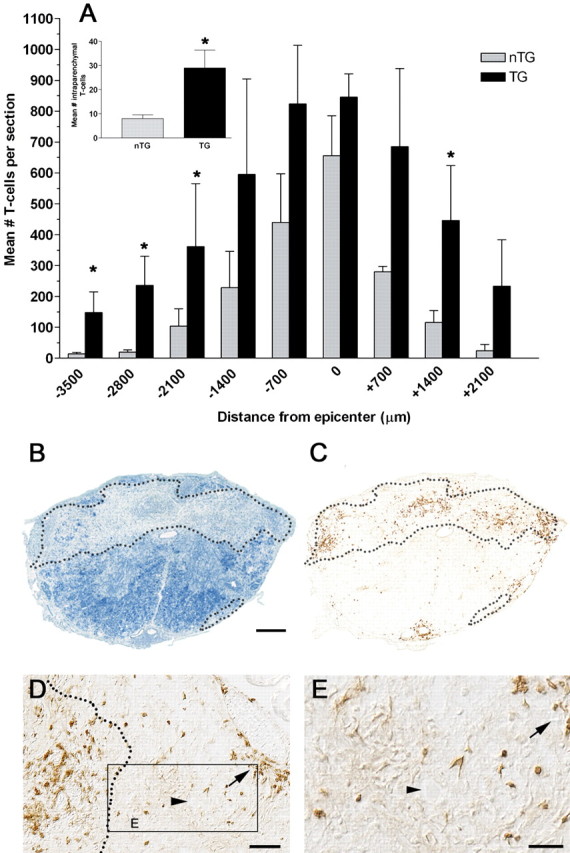
SCI-induced priming of MBP-reactive T-cells increases T-cell infiltration throughout the injured spinal cord. CD4+ T-cells are increased throughout the rostrocaudal spinal cord in Tg and nTg mice at 70 dpi (A). However, more T-cells penetrate both intact (inset in A; p < 0.05) and lesioned spinal parenchyma in Tg mice (A; p < 0.05 vs nTg, two-way ANOVA; n = 4 per group). An example of how lesioned tissue can be distinguished from nonlesioned tissue using luxol fast blue histochemistry is shown in B (∼3 mm rostral to the injury site; dotted line delineates boundary between lesioned and nonlesioned tissue). An adjacent section stained with anti-CD4 antibodies show that CD4+ T-cells are prominent within lesioned tissue (within boundaries of dotted line; nTg mouse; C). In Tg mice, T-cells also penetrate into uninjured parenchyma (boxed region in D shown in high power in E). Arrows highlight a meningeal lymphocyte cluster that appears to migrate into nearby gray matter (arrowheads in D and E point to neuron profile in adjacent ventrolateral gray matter). Scale bars: B, C, 100 μm; D, 50 μm; E, 25 μm.
A series of spinal cord sections containing lesioned and intact spinal cord from all mice (n = 4 per group) was analyzed by two-color immunofluorescence and confocal microscopy (LSM510; Zeiss, Oberkochen, Germany). Images were imported into Adobe Photoshop CS (Adobe Systems, San Jose, CA) in which background illumination was subtracted from all images, and contrast enhancement was selectively applied to recreate the tissue as viewed through the microscope. For confocal analyses, up to 10 sequential optical sections were collected from each sample over an optical scanning plane of 14 μm and then were stacked to create the final image. Stacked images were imported into Adobe Photoshop CS and contrast enhanced, or the red-green-blue channel was split to reveal the staining patterns of the individual fluorophores.
Statistics. Two-way ANOVA with Bonferroni's post hoc analyses were used to analyze T-cell counts (distance from epicenter and group as main effects) and expression of chemokine, costimulatory molecule, and growth factor mRNA (time after injury and group as main effects). Student's t test was used to analyze T-cell numbers in lesioned vs nonlesioned CNS parenchyma. All analyses were considered significant at p < 0.05.
Results
T-cell recruitment and chemokine expression in the injured mouse spinal cord
Previously, we showed that intraspinal T-cell infiltration is delayed up to 2 weeks after SCI in C57BL/6, C57BL/10, BALB/c, and B10.PL mice (Sroga et al., 2003). However, as shown in Figure 1, intraspinal T-cell infiltrates were evident by 7 dpi in Tg mice (Fig. 1B). This accelerated recruitment was paralleled by an increase in expression of the T-cell chemoattractants CXCL10/IP-10 [interferon (IFN)-γ-inducible protein of 10 kDa] (Fig. 1C) and CCL5/RANTES (regulated on activation, normal T-cell expressed and secreted) (Fig. 1D). Significant CXCL10 mRNA induction was present at 7 dpi only in Tg mice (Fig. 1C). Although CCL5 mRNA was increased by 7 dpi and persisted until 21 dpi in both nTg and Tg mice (Fig. 1D), these changes were always larger in Tg spinal cord. Specifically, a 10-fold increase in CXCL10 mRNA expression by 7 dpi was maintained at 21 dpi in Tg mice compared with a maximum fourfold increase in nTg mice. CCL5 mRNA expression increased 180-fold at 7 dpi in Tg mice compared with an 11-fold increase in nTg mice.
Figure 1.

Migration of T-cells to the injured spinal cord is accelerated in Tg mice and is associated with earlier expression of T-cell chemoattractant mRNAs. At 7 dpi, CD4+ T-cells are absent from the injury site of nTg mice (A). In contrast, small T-cell clusters form within the injury site of Tg mice (B). Accelerated T-cell recruitment in Tg mice is associated with increased expression of IP-10/CXCL10 mRNA at 7 dpi in Tg but not nTg mice (C; *p < 0.05 vs uninjured and nTg; n = 4 per group). By 21 dpi, IP-10/CXCL10 mRNA is elevated in Tg and nTg mice; however, the magnitude of change is greater in Tg mice (C; *p < 0.05 vs uninjured and nTg; n = 4 per group). Expression of RANTES/CCL5 mRNA increases as a function of time after injury in both groups but to a greater extent in Tg mice (D; *p < 0.05 vs uninjured and nTg; n = 4 per group). Scale bar, 25 μm.
Sustained T-cell chemokine mRNA was associated with large numbers of infiltrating T-cells in the chronically injured (70 dpi) spinal cord of Tg and nTg mice (Fig. 2). However, the magnitude and tissue distribution of the T-cell infiltrates were distinct. In nTg mice, T-cells were limited to the leptomeninges, perivascular space, or lesioned parenchyma nearby the site of injury. Conversely, in Tg mice, more T-cells infiltrated the spinal cord over greater rostrocaudal extents in both lesioned and intact tissue (Fig. 2).
Cellular and molecular indicators of CNS macrophage activation and antigen presentation within injured spinal cord
SCI activates T-cells and facilitates their migration into the spinal cord (Fig. 2). When the bulk of these cells are myelin reactive, more T-cells penetrate into nonlesioned parenchyma (Fig. 2). Consequently, the ability of these T-cells to influence CNS macrophages and other cells in the parenchyma should be enhanced in Tg mice. Consequently, we evaluated the expression of molecules associated with monocyte recruitment [CCL2/macrophage chemotactic protein-1 (MCP-1)] and antigen presentation (MHC class II, CD80, CD86), i.e., biological processes that are influenced by (or require) T-cell/macrophage interactions.
Previous studies have shown that CCL2/MCP-1 mRNA is increased within 1 dpi in mice with a return to baseline before 7 d (Bartholdi and Schwab, 1997; Ma et al., 2002, 2004). Consistent with this time course, we were unable to detect changes in CCL2 mRNA at 7 dpi for either Tg or nTg mice (Fig. 3A). However, by 21 dpi, CCL2 mRNA expression was increased more than fourfold in Tg mice compared with nTg and uninjured control tissue (p < 0.05) (Fig. 3A).
Figure 3.
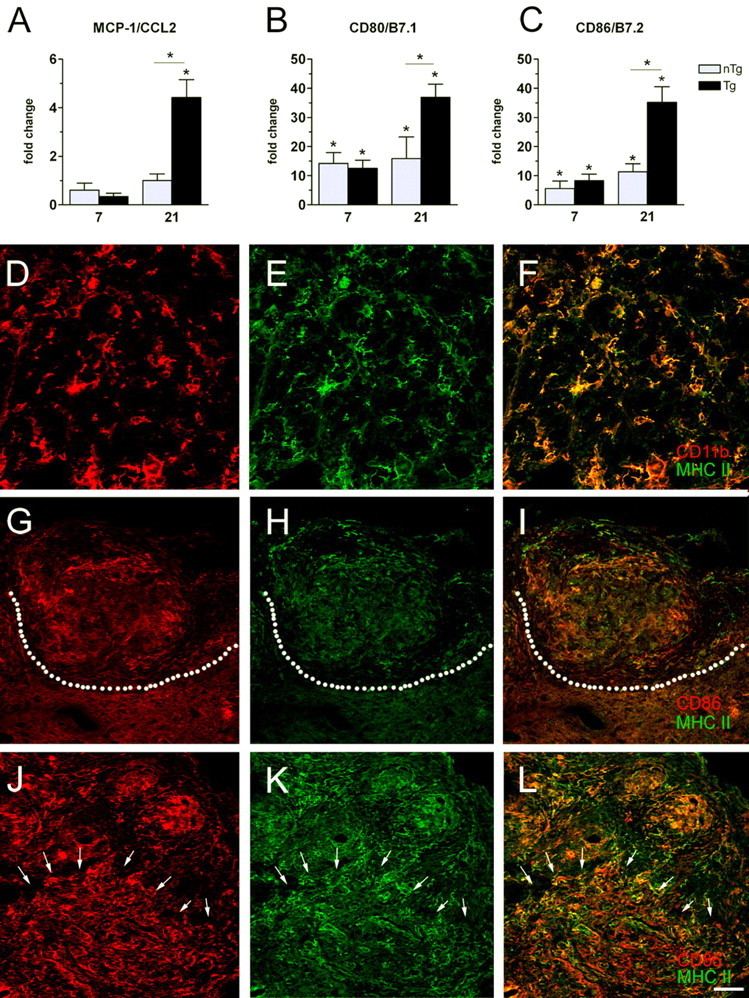
Cellular and molecular indices of CNS macrophage activation are increased in the injured spinal cord of Tg mice. At 7 dpi, MCP-1/CCL2 mRNA is reduced relative to uninjured controls in both groups (not significant); however, by 21 dpi, CCL2 mRNA is increased in Tg mice (A). Expression of CD80/B7.1 and CD86/B7.2 mRNA (B, C) is increased in nTg and Tg mice at 7 and 21 dpi but with 10- to 40-fold greater changes noted in Tg mice at 21 dpi. Confocal analysis of immunofluorescent double-labeled sections reveal MHC class II expressing microglia/macrophages (CD11b+) in and nearby lesioned tissue in Tg and nTg mice (only nTg sample shown; D-F). MHC class II/CD86 double-positive cells were limited in nTg tissues and, when present, were restricted to zones of fibrosis (area above dotted line; G-I). Conversely, MHC class II/CD86 double labeling was seen throughout lesioned and intact spinal cord (boundary demarcated by arrows/lesioned tissue above arrows) of Tg mice (J-L). For D-L, the first two micrographs in a row are single channel (red or green) images with the merged image shown in F, I, or L.(*p < 0.05 vs uninjured or nTg in A-C; two-way ANOVA) Scale bar: D-F, 25 μm; G-L, 50 μm.
Within the CNS, activated CD4+ T-cells need to be restimulated to survive. This requires interaction with antigen-presenting cells, i.e., CNS macrophages expressing costimulatory (CD80/B7.1; CD86/B7.2) and MHC class II molecules. In Tg spinal cord, CD80 and CD86 mRNA expression was increased 10- to 40-fold above nTg and uninjured spinal cord (Fig. 3B,C). Costimulatory mRNAs also were increased in nTg mice, albeit to a lesser extent than in Tg spinal cord (Fig. 3B,C).
Confocal analyses of immunofluorescent double-labeled sections reveal induction of MHC class II on the majority of microglia/macrophages (CD11b+) within lesioned tissue of Tg and nTg mice (Fig. 3D-F). However, in nTg mice, the majority of MHC class II+ and CD86+ CNS macrophages were restricted to zones of fibrosis or within perivascular or submeningeal infiltrates (Fig. 3G-I). Conversely, MHC class II and CD86 were increased throughout injured and intact spinal cord in Tg mice (Fig. 3J-L).
Expression of immunoregulatory/growth factor molecules in the chronically injured mouse spinal cord
Delayed increases in transforming growth factor (TGF)-β1 and endogenous interleukin-1 receptor antagonist (IL-1ra) are typical in inflammatory foci, presumably to limit the destructive potential of inflammation. Given the above data and our previous work showing that MBP-reactive T-cells enhance neuroinflammation, we predicted distinct patterns of growth factor and immunoregulatory molecule expression in Tg and nTg mice. Figure 4 shows that TGF-β1 and insulin-like growth factor (IGF)-1 mRNA are increased in nTg and Tg mice at 7 and 21 dpi. However, in Tg mice, TGF-β1 and IGF-1 mRNA expression continue to increase between 7 and 21 dpi (Fig. 4A,B). Also, IL-1ra mRNA expression is increased at 7 and 21 dpi in both strains; however, the magnitude of this response is dramatically reduced in Tg mice (Fig. 4C).
Figure 4.

mRNAs encoding mediators of tissue repair and immunoregulation are upregulated chronically in the injured mouse spinal cord. Induction of TGF-β1 (A) and IGF-1 (B) mRNA are increased at 7 and 21 dpi in nTg and Tg mice (p < 0.05 vs uninjured controls) but continue to rise at 21 dpi only in Tg mice (p < 0.05 vs nTg). Expression of endogenous IL-1 receptor antagonist (C) is upregulated after SCI in nTg and Tg mice (p < 0.05) but is declining by 21 dpi in Tg mice (p < 0.05 vs nTg). n = 3 per group.
Discussion
Previously, we described marked neuropathological changes and functional impairment after SCI in mice containing large numbers of CNS antigen-specific T-cells (i.e., MBP TCR Tg mice) as opposed to mice with a normal (diverse antigen-specific) T-cell repertoire (nTg/B10.PL mice). In both Tg and wild-type mice, large numbers of intraspinal T-cells were evident. However, only in Tg mice were aberrant neurological and pathological changes associated with a biased production of proinflammatory cytokines in the spinal cord (Jones et al., 2002). Because T-cell migration, activation, and function are controlled by chemokines and cytokines, we compared expression patterns of select chemokines at times before and after the onset of intraspinal T-cell migration. The current data show that marked induction of CXCL10 and CCL5 occurs in both Tg and nTg mice and precedes or parallels the onset of intraspinal T-cell migration. However, accelerated and amplified chemokine signaling in Tg mice is associated with earlier T-cell entry and greater intraparenchymal migration of T-cells than described previously for other mouse strains (Sroga et al., 2003). The robust induction of T-cell chemokines in Tg mice was accompanied by later induction of CCL2 mRNA and widespread CNS macrophage activation. Specifically, only in Tg mice were CD80, CD86, and MHC class II increased throughout the injured and intact spinal cord. Collectively, these data reveal molecular targets that are temporally regulated after SCI and are associated with T-cell recruitment to the site of injury. Whether these molecules predispose the spinal cord to T-cell-mediated injury or repair will depend on the antigen specificity of the responding T-cells and the timing, magnitude, and composition of the chemokines produced.
Chemokines influence T-cell recruitment and function
Inflammatory chemokines coordinate the recruitment of activated leukocytes to sites of injury or infection (Mennicken et al., 1999; Huang et al., 2000; Moser et al., 2004). De novo synthesis of intraspinal chemokines results from acute trauma and subsequent glial activation (Glabinski et al., 1996). To date, chemokine induction has been described in all acute SCI models within hours of injury, with a decline to baseline by 1 week (Bartholdi and Schwab, 1997; McTigue et al., 1998; Ma et al., 2002, 2004). However, there is little information about chemokine signaling beyond the first week after injury. Resolution of acute chemokine signaling is likely attributable to a reduction in inflammatory stimuli and active regulation of chemokine promoter regions (Huang et al., 2000). In the present study, we extended our analyses beyond the first week after injury to coincide with a time of robust T-cell infiltration (Sroga et al., 2003). The data reveal a secondary increase in the expression of chemokines specific for T-cells and monocytes. In the case of Tg mice, these chemokine changes were associated with sustained T-cell infiltration and greater penetration into intact spinal cord tissue along with widespread activation of CNS macrophages. These data suggest that, after injury, silencing of chemokine expression is temporary and/or that new inflammatory signals are present after 7 d. This delayed “secondary” inflammatory signaling cascade could be targeted for therapeutic purposes. Additional studies are needed to address the chronicity of inflammatory signaling mechanisms after SCI and their impact on recovery of function.
In the present study, increased CXCL10 and CCL5 expression preceded or paralleled T-cell influx after SCI in nTg and Tg mice. The larger numbers and more rapid appearance of T-cells in Tg mouse spinal cord is undoubtedly related to the magnitude of chemokine induction and the unique cytokine milieu that develops at the injury site of MBP TCR Tg mice. Specifically, we described previously marked induction of IFN-γ and tumor necrosis factor-α in Tg mice (Jones et al., 2002). Although these cytokines exert little effect on CXCL10 alone, they synergize to promote marked induction of CXCL10 (Huang et al., 2000).
Besides their role as chemoattractants, CXCL10 and CCL5 induce T-cell proliferation and cytokine production (Bacon et al., 1995; Appay et al., 2000; Dufour et al., 2002). Also, CXCL10 is pivotal in generating antigen-specific T-cells (Dufour et al., 2002). A similar role has been reported for CCL2 (Izikson et al., 2000; Tylaska et al., 2002). Thus, the array of chemokines expressed in both nTg and Tg mice would serve diverse roles, including enhancement of T-cell recruitment, activation, and effector potential. Once they are activated, whether responding T-cells exert physiological or pathological effects will depend on a number of variables, including their antigen specificity, whether they become reactivated in the target tissue, and the efficacy of endogenous CNS regulatory cascades.
Antigen specificity and T-cell functions in the CNS
Despite remarkable differences in anatomical and functional outcome in SCI Tg and nTg mice (Jones et al., 2002), large numbers of T-cells infiltrate the injury site of both strains (Fig. 2). A similar situation occurs in rat models of EAE. For example, vaccination of rats with MBP- and myelin oligodendrocyte glycoprotein (MOG)-specific T-cells causes robust neuroinflammation but has divergent effects on clinical outcome. Specifically, MOG-reactive T-cells cause little or no disease, whereas MBP-reactive T-cells exert a lethal pathology (Kawakami et al., 2004). These data indicate that intraspinal accumulation of T-cells is not sufficient to induce CNS pathology. Similarly, delayed and chronic chemokine upregulation need not have pathological implications. Indeed, chemokines can be increased in the CNS without neuropathological consequences. For example, transgenic or viral-mediated overexpression of CXCL10 in the CNS produces robust inflammation without obvious pathology (Boztug et al., 2002; Trifilo and Lane, 2003). Instead, the secretory potential of T-cells and their ability to influence other cells in the CNS is influenced by T-cell antigen specificity and the activation status of the responding T-cells (Kawakami et al., 2004). Moreover, the status of the CNS microenvironment will determine the range and efficacy of T-cell effector functions.
For example, the overwhelming proinflammatory milieu (e.g., IFN-γ, IL-12, CXCL10, and CCL2) that forms in Tg mice as a result of MBP-reactive T-cell activation appears to augment microglia and macrophage recruitment and/or activation (Jones et al., 2002) (Figs. 1, 3). This view is supported by the widespread upregulation of CD80, CD86, MHC class II, and CCL2 only in Tg mice (Fig. 3). Interestingly, MBP-reactive T-cells have been shown to induce CD86 expression on microglia, and this intercellular exchange has been deemed essential for T-cell-mediated neuroprotection (Bechmann et al., 2001; Butovsky et al., 2001). Our data would suggest that expression of CD86 is induced by SCI, regardless of the presence of autoreactive T-cells. However, in the presence of MBP-reactive T-cells (e.g., in Tg mice), de novo expression of CD86 on microglia/macrophages is increased over much greater areas of the spinal cord than would be expected after SCI alone. Importantly, in the context of the MBP TCR Tg model, enhanced CD86 expression is not associated with neuroprotection. These data support the notion that activated macrophages exacerbate acute CNS pathology and loss of function after SCI and EAE (Huitinga et al., 1990; Blight, 1994; Tran et al., 1998; Popovich et al., 1999; Izikson et al., 2000; Mabon et al., 2000; Huang et al., 2001).
Pronounced T-cell pathogenicity in Tg mice may also occur because of a reduction in intrinsic immune regulatory cascades. For example, in SCI Tg mice in which neuropathology is enhanced, one would expect a corresponding reduction in neuronal inhibition of MHC class II expression and endogenous cytokine antagonists (e.g., IL-1ra) (Neumann et al., 1996). Data in Figures 3 and 4 support this hypothesis.
It is clear that models of spinal trauma and autoimmune demyelination (e.g., EAE) can elicit pathology via similar molecular and cellular mechanisms. The present data also suggest that titration of chemokine signaling or selective manipulation of chemokine receptors that recruit/activate T-cells could be therapeutic after SCI. Recent studies indicate that inhibition of CXCL10 limits T-cell accumulation and promotes functional recovery after SCI (Gonzalez et al., 2003). Because pathogenic Th1 cells preferentially express receptors for CXCL10 and CCL5 (i.e., CXCR3 and CCR5, respectively), CXCR3- and CCR5-expressing T-cells are expected to predominate after SCI, just as they do in EAE and multiple sclerosis (Balashov et al., 1997, 1999; Sorensen et al., 1999). Future studies using selective antagonists or mice deficient in CXCL10/CXCR3 or CCL5/CCR5 should reveal what role these signaling cascades play in inflammatory-mediated repair and/or injury.
T-cells, macrophages, and fibrosis in the injured spinal cord
Despite their pathogenic potential, the accumulation of intraspinal T-cells need not imply impending tissue injury. T-cells infiltrate the injured mouse spinal cord in parallel with a plateau in functional recovery in all mice (Sroga et al., 2003). Moreover, T-cell clusters form within zones of dense fibrosis rather than in regions of necrosis. Data from EAE and nerve lesion studies would suggest that most T-cells responding to SCI are not myelin reactive (Steinman, 1996; Cross et al., 1990; Hammarberg et al., 2000; Muhallab et al., 2002). Still, even if neuron and glial survival are not adversely affected by T-cell recruitment/activation, T-cells could inhibit normal CNS repair.
Although matrix deposition is critical for tissue repair, if not balanced by endogenous regulatory signals, pathological fibrosis and scarring can occur. Ultimately, this results in functional impairment. In the injured CNS, fibrosis would be expected to limit cell migration, a necessary step in reparative processes such as angiogenesis. Similarly, fibrosis would restrict the diffusion of metabolites, cytokines, and growth factors necessary for efficient intercellular communication. Recent data suggest that the fibrotic substrate that forms after mouse SCI does not support regeneration of descending axons (Inman and Steward, 2003; Ma et al., 2004). Because reciprocal interactions between T-cells and macrophages influence fibroblast proliferation/migration, collagen and fibronectin synthesis/deposition, and scarring, it is not surprising that blocking T-cell chemotaxis abrogates fibrosis in non-CNS tissues (Huaux et al., 2003). Similar studies are needed in SCI models to determine whether T-cells contribute to pathological neurofibrosis.
In the present study, TGF-β1 and IGF-1 mRNA increased in Tg and nTg mice between 7 and 21 dpi. Both growth factors are increased as a result of inflammation and are known to be involved in normal wound healing and pathological fibrosis (Wynn, 2004). Previously, we and others documented SCI-associated increases in TGF-β1 mRNA colocalized to regions of intraspinal inflammation (McTigue et al., 2000; Lagord et al., 2002). Like IGF-1, TGF-β1 can be neuroprotective and immunosuppressive (Finch et al., 1993; Pu et al., 1999; Pulford et al., 1999). However, the fibrotic pathology that defines the injured mouse spinal cord may suggest a less advantageous role for both growth factors, and studies have shown a direct causal role for TGF-β1 in glial scarring and extracellular matrix deposition after CNS injury (Logan et al., 1994). It is also possible that IGF-1 and TGF-β mediate their effects indirectly by promoting the survival of infiltrating T-cells, which in turn could promote chronic fibrosis (Genestier et al., 1999; Walsh and O'Connor, 2000).
Conclusions
Select chemokine signals are required to initiate T-cell recruitment and then shape their functional repertoire at sites of inflammation. In the present study, the delayed but protracted increase in CXCL10 and CCL5 in SCI mice is undoubtedly responsible for the chronic T-cell recruitment that we observed in both strains. However, the magnitude and kinetics of the T-cell response is accelerated when the predominant antigen specificity of responding T-cells is to CNS myelin. Because the primary insult in Tg and nTg mice was identical (i.e., standardized spinal contusion injury), the distinct cellular and molecular sequelae that we describe results from effector mechanisms unique to MBP-specific T-cells. Specifically, greater axial migration of T-cells occurs in SCI Tg mice and is associated with enhanced macrophage/microglial activation and divergent patterns of growth factor/immune regulatory molecule expression. Because we described previously a pathological phenotype in SCI Tg mice, the present data reveal several potential targets for therapeutic modulation of pathogenic T-cell function in the face of enhanced autoimmune function. Indeed, despite the inherent detrimental effects of myelin-reactive T-cells after SCI, any reparative potential that these cells possess (Schwartz and Kipnis, 2001) might be improved by simultaneously antagonizing chemokines or T-cell-mediated macrophage activation.
Footnotes
This work was supported by the National Institute for Neurological Disorders and Stroke, The New Jersey Commission on Spinal Cord Research, and the National Institutes of Health/National Institute of Neurological Disorders and Stroke/National Institutes on Aging. We thank Dr. Ming Wang, Qin Yin, Pat Walters, Ingrid Gienapp, and Dr. Dana McTigue for their contributions to this work.
Correspondence should be addressed to Dr. Phillip Popovich, Department Molecular Virology, Immunology, and Medical Genetics, 2078 Graves Hall, 333 West 10th Avenue, Columbus, OH 43210. E-mail: popovich.2@osu.edu.
Copyright © 2005 Society for Neuroscience 0270-6474/05/256576-08$15.00/0
References
- Appay V, Dunbar PR, Cerundolo V, McMichael A, Czaplewski L, Rowland-Jones S (2000) RANTES activates antigen-specific cytotoxic T lymphocytes in a mitogen-like manner through cell surface aggregation. Int Immunol 12: 1173-1182. [DOI] [PubMed] [Google Scholar]
- Bacon KB, Premack BA, Gardner P, Schall TJ (1995) Activation of dual T cell signaling pathways by the chemokine RANTES. Science 269: 1727-1730. [DOI] [PubMed] [Google Scholar]
- Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL (1997) Increased interleukin 12 production in progressive multiple sclerosis: induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci USA 94: 599-603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balashov KE, Rottman JB, Weiner HL, Hancock WW (1999) CCR5+ and CXCR3+ T cells are increased in multiple sclerosis and their ligands MIP-1alpha and IP-10 are expressed in demyelinating brain lesions. Proc Natl Acad Sci USA 96: 6873-6878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbul A, Breslin RJ, Woodyard JP, Wasserkrug HL, Efron G (1989a) The effect of in vivo T helper and T suppressor lymphocyte depletion on wound healing. Ann Surg 209: 479-483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbul A, Shawe T, Rotter SM, Efron JE, Wasserkrug HL, Badawy SB (1989b) Wound healing in nude mice: a study on the regulatory role of lymphocytes in fibroplasia. Surgery 105: 764-769. [PubMed] [Google Scholar]
- Bartholdi D, Schwab ME (1997) Expression of pro-inflammatory cytokine and chemokine mRNA upon experimental spinal cord injury in mouse: an in situ hybridization study. Eur J Neurosci 9: 1422-1438. [DOI] [PubMed] [Google Scholar]
- Bechmann I, Peter S, Beyer M, Gimsa U, Nitsch R (2001) Presence of B7-2 (CD86) and lack of B7-1 (CD80) on myelin phagocytosing MHC-II positive rat microglia is associated with nondestructive immunity in vivo. FASEB J 15: 1086-1088. [DOI] [PubMed] [Google Scholar]
- Blight AR (1994) Effects of silica on the outcome from experimental spinal cord injury: implication of macrophages in secondary tissue damage. Neuroscience 60: 263-273. [DOI] [PubMed] [Google Scholar]
- Boztug K, Carson MJ, Pham-Mitchell N, Asensio VC, DeMartino J, Campbell IL (2002) Leukocyte infiltration, but not neurodegeneration, in the CNS of transgenic mice with astrocyte production of the CXC chemokine ligand 10. J Immunol 169: 1505-1515. [DOI] [PubMed] [Google Scholar]
- Butovsky O, Hauben E, Schwartz M (2001) Morphological aspects of spinal cord autoimmune neuroprotection: colocalization of T cells with B7-2 (CD86) and prevention of cyst formation. FASEB J 15: 1065-1067. [DOI] [PubMed] [Google Scholar]
- Cross AH, Cannella B, Brosnan CF, Raine CS (1990) Homing to central nervous system vasculature by antigen specific lymphocytes. I. Localization of C14-labeled cells during acute, chronic, and relapsing experimental allergic encephalomyelitis. Lab Invest 63: 162-170. [PubMed] [Google Scholar]
- Dufour JH, Dziejman M, Liu MT, Leung JH, Lane TE, Luster AD (2002) IFN-gamma-inducible protein 10 (IP-10; CXCL10)-deficient mice reveal a role for IP-10 in effector T cell generation and trafficking. J Immunol 168: 3195-3204. [DOI] [PubMed] [Google Scholar]
- Efron JE, Frankel HL, Lazarou SA, Wasserkrug HL, Barbul A (1990) Wound healing and T-lymphocytes. J Surg Res 48: 460-463. [DOI] [PubMed] [Google Scholar]
- Finch CE, Laping NJ, Morgan TE, Nichols NR, Pasinetti GM (1993) TGF-β1 is an organizer of responses to neurodegeneration. J Cell Biochem 53: 314-322. [DOI] [PubMed] [Google Scholar]
- Genestier L, Kasibhatla S, Brunner T, Green DR (1999) Transforming growth factor beta1 inhibits Fas ligand expression and subsequent activation-induced cell death in T cells via downregulation of c-Myc. J Exp Med 189: 231-239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glabinski AR, Balasingam V, Tani M, Kunkel SL, Strieter RM, Yong VW, Ransohoff RM (1996) Chemokine monocyte chemoattractant protein-1 is expressed by astrocytes after mechanical injury to the brain. J Immunol 156: 4363-4368. [PubMed] [Google Scholar]
- Gonzalez R, Glaser J, Liu MT, Lane TE, Keirstead HS (2003) Reducing inflammation decreases secondary degeneration and functional deficit after spinal cord injury. Exp Neurol 184: 456-463. [DOI] [PubMed] [Google Scholar]
- Hammarberg H, Lidman O, Lundberg C, Eltayeb SY, Gielen AW, Muhallab S, Svenningsson A, Linda H, Der Meide PH, Cullheim S, Olsson T, Piehl F (2000) Neuroprotection by encephalomyelitis: rescue of mechanically injured neurons and neurotrophin production by CNS-infiltrating T and natural killer cells. J Neurosci 20: 5283-5291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Han Y, Rani MR, Glabinski A, Trebst C, Sorensen T, Tani M, Wang J, Chien P, O'Bryan S, Bielecki B, Zhou ZL, Majumder S, Ransohoff RM (2000) Chemokines and chemokine receptors in inflammation of the nervous system: manifold roles and exquisite regulation. Immunol Rev 177: 52-67. [DOI] [PubMed] [Google Scholar]
- Huang DR, Wang J, Kivisakk P, Rollins BJ, Ransohoff RM (2001) Absence of monocyte chemoattractant protein 1 in mice leads to decreased local macrophage recruitment and antigen-specific T helper cell type 1 immune response in experimental autoimmune encephalomyelitis. J Exp Med 193: 713-726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huaux F, Liu T, McGarry B, Ullenbruch M, Xing Z, Phan SH (2003) Eosinophils and T lymphocytes possess distinct roles in bleomycin-induced lung injury and fibrosis. J Immunol 171: 5470-5481. [DOI] [PubMed] [Google Scholar]
- Huitinga I, van Rooijen N, de Groot CJ, Uitdehaag BM, Dijkstra CD (1990) Suppression of experimental allergic encephalomyelitis in Lewis rats after elimination of macrophages. J Exp Med 172: 1025-1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inman DM, Steward O (2003) Ascending sensory, but not other long-tract axons, regenerate into the connective tissue matrix that forms at the site of a spinal cord injury in mice. J Comp Neurol 462: 431-449. [DOI] [PubMed] [Google Scholar]
- Izikson L, Klein RS, Charo IF, Weiner HL, Luster AD (2000) Resistance to experimental autoimmune encephalomyelitis in mice lacking the CC chemokine receptor (CCR)2. J Exp Med 192: 1075-1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakeman LB, Guan Z, Wei P, Ponnappan R, Dzwonczyk R, Popovich PG, Stokes BT (2000) Traumatic spinal cord injury produced by controlled contusion in mouse. J Neurotrauma 17: 299-319. [DOI] [PubMed] [Google Scholar]
- Jones TB, Basso DM, Sodhi A, Pan JZ, Hart RP, MacCallum RC, Lee S, Whitacre CC, Popovich PG (2002) Pathological CNS autoimmune disease triggered by traumatic spinal cord injury: implications for autoimmune vaccine therapy. J Neurosci 22: 2690-2700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TB, Ankeny DP, Guan Z, McGaughy V, Fisher LC, Basso DM, Popovich PG (2004) Passive or active immunization with myelin basic protein impairs neurological function and exacerbates neuropathology after spinal cord injury in rats. J Neurosci 24: 3752-3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami N, Lassmann S, Li Z, Odoardi F, Ritter T, Ziemssen T, Klinkert WE, Ellwart JW, Bradl M, Krivacic K, Lassmann H, Ransohoff RM, Volk HD, Wekerle H, Linington C, Flugel A (2004) The activation status of neuroantigen-specific T cells in the target organ determines the clinical outcome of autoimmune encephalomyelitis. J Exp Med 199: 185-197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipnis J, Mizrahi T, Yoles E, Ben-Nun A, Schwartz M (2002) Myelin specific Th1 cells are necessary for post-traumatic protective autoimmunity. J Neuroimmunol 130: 78-85. [DOI] [PubMed] [Google Scholar]
- Lagord C, Berry M, Logan A (2002) Expression of TGFbeta2 but not TGF-beta1 correlates with the deposition of scar tissue in the lesioned spinal cord. Mol Cell Neurosci 20: 69-92. [DOI] [PubMed] [Google Scholar]
- Logan A, Berry M, Gonzalez AM, Frautschy SA, Sporn MB, Baird A (1994) Effects of transforming growth factor beta 1 on scar production in the injured central nervous system of the rat. Eur J Neurosci 6: 355-363. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei T, Boring L, Charo IF, Ransohoff RM, Jakeman LB (2002) Monocyte recruitment and myelin removal are delayed following spinal cord injury in mice with CCR2 chemokine receptor deletion. J Neurosci Res 68: 691-702. [DOI] [PubMed] [Google Scholar]
- Ma M, Wei P, Wei T, Ransohoff RM, Jakeman LB (2004) Enhanced axonal growth into a spinal cord contusion injury site in a strain of mouse (129X1/SvJ) with a diminished inflammatory response. J Comp Neurol 474: 469-486. [DOI] [PubMed] [Google Scholar]
- Mabon PJ, Weaver LC, Dekaban GA (2000) Inhibition of monocyte/macrophage migration to a spinal cord injury site by an antibody to the integrin alphaD: a potential new anti-inflammatory treatment. Exp Neurol 166: 52-64. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Tani M, Krivacic K, Chernosky A, Kelner GS, Maciejewski D, Maki R, Ransohoff RM, Stokes BT (1998) Selective chemokine mRNA accumulation in the rat spinal cord after contusion injury. J Neurosci Res 53: 368-376. [DOI] [PubMed] [Google Scholar]
- McTigue DM, Popovich PG, Morgan TE, Stokes BT (2000) Localization of transforming growth factor-beta1 and receptor mRNA after experimental spinal cord injury. Exp Neurol 163: 220-230. [DOI] [PubMed] [Google Scholar]
- Mennicken F, Maki R, de Souza EB, Quirion R (1999) Chemokines and chemokine receptors in the CNS: a possible role in neuroinflammation and patterning. Trends Pharmacol Sci 20: 73-78. [DOI] [PubMed] [Google Scholar]
- Moser B, Wolf M, Walz A, Loetscher P (2004) Chemokines: multiple levels of leukocyte migration control. Trends Immunol 25: 75-84. [DOI] [PubMed] [Google Scholar]
- Muhallab S, Lundberg C, Gielen AW, Lidman O, Svenningsson A, Piehl F, Olsson T (2002) Differential expression of neurotrophic factors and inflammatory cytokines by myelin basic protein-specific and other recruited T cells infiltrating the central nervous system during experimental autoimmune encephalomyelitis. Scand J Immunol 55: 264-273. [DOI] [PubMed] [Google Scholar]
- Neumann H, Boucraut J, Hahnel C, Misgeld T, Wekerle H (1996) Neuronal control of MHC class II inducibility in rat astrocytes and microglia. Eur J Neurosci 8: 2582-2590. [DOI] [PubMed] [Google Scholar]
- Popovich PG (2000) Immunological regulation of neuronal degeneration and regeneration in the injured spinal cord. Prog Brain Res 128: 43-58. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Wei P, Stokes BT (1997) The cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol 377: 443-464. [DOI] [PubMed] [Google Scholar]
- Popovich PG, Guan Z, Wei P, Huitinga I, van Rooijen N, Stokes BT (1999) Depletion of hematogenous macrophages promotes partial hindlimb recovery and neuroanatomical repair after experimental spinal cord injury. Exp Neurol 158: 351-365. [DOI] [PubMed] [Google Scholar]
- Pu SF, Zhuang HX, Marsh DJ, Ishii DN (1999) Insulin-like growth factor-II increases and IGF is required for postnatal rat spinal motoneuron survival following sciatic nerve axotomy. J Neurosci Res 55: 9-16. [DOI] [PubMed] [Google Scholar]
- Pulford BE, Whalen LR, Ishii DN (1999) Peripherally administered insulin-like growth factor-I preserves hindlimb reflex and spinal cord noradrenergic circuitry following a central nervous system lesion in rats. Exp Neurol 159: 114-123. [DOI] [PubMed] [Google Scholar]
- Ririe KM, Rasmussen RP, Wittwer CT (1997) Product differentiation by analysis of DNA melting curves during the polymerase chain reaction. Anal Biochem 245: 154-160. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A, Mackay CR (1998) Chemokines and chemokine receptors in T-cell priming and Th1/Th2-mediated responses. Immunol Today 19: 568-574. [DOI] [PubMed] [Google Scholar]
- Sallusto F, Mackay CR, Lanzavecchia A (2000) The role of chemokine receptors in primary, effector, and memory immune responses. Annu Rev Immunol 18: 593-620. [DOI] [PubMed] [Google Scholar]
- Schaffer M, Barbul A (1998) Lymphocyte function in wound healing and following injury. Br J Surg 85: 444-460. [DOI] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J (2001) Protective autoimmunity: regulation and prospects for vaccination after brain and spinal cord injuries. Trends Mol Med 7: 252-258. [DOI] [PubMed] [Google Scholar]
- Sorensen TL, Tani M, Jensen J, Pierce V, Lucchinetti C, Folcik VA, Qin S, Rottman J, Sellebjerg F, Strieter RM, Frederiksen JL, Ransohoff RM (1999) Expression of specific chemokines and chemokine receptors in the central nervous system of multiple sclerosis patients. J Clin Invest 103: 807-815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sroga JM, Jones TB, Kigerl KA, McGaughy VM, Popovich PG (2003) Rats and mice exhibit distinct inflammatory reactions after spinal cord injury. J Comp Neurol 462: 223-240. [DOI] [PubMed] [Google Scholar]
- Steinman L (1996) A few autoreactive cells in an autoimmune infiltrate control a vast population of nonspecific cells: a tale of smart bombs and the infantry. Proc Natl Acad Sci USA 93: 2253-2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran EH, Hoekstra K, van Rooijen N, Dijkstra CD, Owens T (1998) Immune invasion of the central nervous system parenchyma and experimental allergic encephalomyelitis, but not leukocyte extravasation from blood, are prevented in macrophage-depleted mice. J Immunol 161: 3767-3775. [PubMed] [Google Scholar]
- Trifilo MJ, Lane TE (2003) Adenovirus-mediated expression of CXCL10 in the central nervous system results in T-cell recruitment and limited neuropathology. J Neurovirol 9: 315-324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tylaska LA, Boring L, Weng W, Aiello R, Charo IF, Rollins BJ, Gladue RP (2002) Ccr2 regulates the level of MCP-1/CCL2 in vitro and at inflammatory sites and controls T cell activation in response to alloantigen. Cytokine 18: 184-190. [DOI] [PubMed] [Google Scholar]
- Walsh PT, O'Connor R (2000) The insulin-like growth factor-I receptor is regulated by CD28 and protects activated T cells from apoptosis. Eur J Immunol 30: 1010-1018. [DOI] [PubMed] [Google Scholar]
- Wynn TA (2004) Fibrotic disease and the T(H)1/T(H)2 paradigm. Nat Rev Immunol 4: 583-594. [DOI] [PMC free article] [PubMed] [Google Scholar]