

Abstract
Intestinal barrier dysfunction occurs following hemorrhagic shock and resuscitation (HS/R). High-mobility group B1 (HMGB1) has been shown to increase the permeability of Caco-2 human enterocyte-like epithelial monolayers in vitro. In this study, we found that serum concentrations of HMGB1 were higher in blood samples obtained from 25 trauma victims with hemorrhagic shock than in 9 normal volunteers. We also studied whether treatment with anti-HMGB1 antibody can ameliorate HS/R-induced gut barrier dysfunction in mice. Animals were shocked by withdrawal of blood to maintain mean arterial pressure at 25 to 30 mmHg for 2 h. After resuscitation with shed blood plus Ringer’s lactate solution, the mice were treated with either anti-HMGB1 antibody or nonimmune rabbit IgG. Serum HMGB1 concentrations were significantly higher in trauma victims than control mice. Treatment with anti-HMGB1 antibody improved survival at 24 h and ameliorated the development of ileal mucosal hyperpermeability to FITC-labeled dextran. At 24 h after HS/R, treatment with anti-HMGB1 antibody decreased bacterial translocation to mesenteric lymph nodes and was associated with lower circulating concentrations of IL-6 and IL-10. These data support the notion that HMGB1 is a mediator of HS/R-induced gut barrier dysfunction and suggest that anti-HMGB1 antibodies warrant further evaluation as a therapeutic to ameliorate the morbidity of HS/R in trauma patients.
INTRODUCTION
Trauma ranks fifth as a cause of death among people of all ages living in the United States, and it is the leading cause of death among people less than 45 years of age (1). In the United States, traumatic injuries result in approximately 100,000 deaths per year (1). Early deaths are secondary to exsanguination or overwhelming central nervous system injuries, whereas late deaths are secondary to sepsis and multiple organ system dysfunction syndrome (MODS) (2,3). Massive hemorrhage is a major risk factor for the development of MODS in trauma victims (4,5).
Intestinal barrier dysfunction, manifested by increased mucosal permeability to hydrophilic macromolecules and/or increased bacterial translocation to mesenteric lymph nodes (MLN), occurs following hemorrhagic shock and resuscitation (HS/R) in rodents (6–13). These findings may have clinical implications, because increased intestinal permeability has been shown to be associated with an increased risk of complications, MODS, or even mortality in critically ill patients (14–17). The underlying mechanisms responsible for gut barrier dysfunction after HS/R are not fully understood, but increased production of certain proin-flammatory mediators, such as IL-6 (11) or nitric oxide (18), may be involved.
High-mobility group proteins are small DNA-binding proteins that serve an important role in transcriptional regulation (19). One of these proteins, HMGB1, has been identified as a late-acting mediator of lipopolysaccharide (LPS)-induced (20) or sepsis-induced (21) lethality in mice. Additional studies have documented that HMGB1 is a cytokine-like molecule that can promote TNF release from mononuclear cells (22). HMGB1 is actively secreted by immunostimulated macrophages (20,23–25) and enterocytes (26) and is also released by necrotic but not apoptotic cells (27).
In 1999, Ombrellino and co-workers (28) described a patient with high circulating levels of HMGB1 following an episode of hemorrhagic shock, and Kim et al. (29) recently reported data supporting the view that HMGB1 contributes to the development of acute lung injury in mice subjected to HS/R. These data, along with results from our laboratory showing that exposure to HMGB1 increases the permeability of Caco-2 enterocyte-like monolayers in vitro (30), prompted to us to measure circulating HMGB1 levels in a cohort of adult trauma patients with physiological and/or biochemical evidence of hemorrhagic shock. Because serum HMGB1 concentrations were significantly elevated in patients with trauma-induced hemorrhagic shock, we sought to determine whether HMGB1 contributes to the development of gut barrier dysfunction in a well-characterized murine model of HS/R.
MATERIALS AND METHODS
Materials
All chemicals were purchased from Sigma-Aldrich Chemical Co. (St. Louis, MO, USA) unless otherwise noted. Polyclonal antibodies against HMGB1 were raised in rabbits (Cocalico Biologicals, Reamstown, PA, USA) as previously described (21). Polyclonal antibodies agai nst HMGB1 B box were prepared as described previously (21). Polyclonal antibodies against HMGB1 B box were raised in rabbits, and titers were determined by immunoblotting. Anti–HMGB1 B box antibodies were affinity-purified by using cyanogen bromide–activated Sepharose beads following standard procedures. Neutralizing activity of anti-HMGB1 was confirmed in HMGB1-stimulated macrophage cultures by assay of TNF release. In the presence of anti-HMGB1 antibody, neutralizing antibody was defined as inhibiting > 80% of HMGB1-induced TNF release. Nonimmune rabbit IgG (item I5006) was purchased from Sigma-Aldrich.
Human Subjects Study Design
We carried out a single-center observational study of adult trauma victims. A patient could be enrolled if all of the following criteria were satisfied: 1) admission to the hospital for nonpenetrating trauma other than isolated head injuries; 2) absence of traumatic brain injury, defined as Abbreviated Injury Score < 4 for the head region or Glasgow Coma Scale motor score > 3 within 24 h of injury (31); 3) arrival in the Emergency Department at the University of Pittsburgh Medical Center < 6 h after injury; 4) transfusion of packed red blood cells within 12 h of the time of injury; 5) base deficit ≥6 mEq/L or systolic blood pressure < 90 mmHg within 60 min of arrival in the Emergency Department; and 6) intact cervical spinal cord. Blood samples for determination of base deficit, lactate concentration, and serum HMGB1 concentration were obtained at the time of admission and approximately 24 h later. Control concentrations of HMGB1 also were measured using blood samples obtained from 9 healthy adult volunteers. HMGB1 concentrations were determined by quantitative Western blotting, as previously described (20). The study was approved by the Institutional Review Board for the Protection of Human Subjects in Research at the University of Pittsburgh.
Experimental Protocol for Studies Using Mice
This research protocol complied with the regulations regarding the care and use of experimental animals published by the National Institutes of Health and was approved by the Institutional Animal Use and Care Committee of the University of Pittsburgh School of Medicine. Male C57Bl/6 mice weighing 20 to 25 g (Jackson Laboratories, Bar Harbor, ME, USA) were used in this study. The animals were maintained at the University of Pittsburgh Animal Research Center with a 12-h light-dark cycle and free access to standard laboratory feed and water. Animals were not fasted before the experiments, but were acclimatized for 7 days before being studied.
Cannulae, syringes, and tubing were flushed with heparin sodium (1000 units/mL) before all procedures. For the experiments, the mice were anesthetized with pentobarbital (90 mg/kg intramuscularly). Both femoral arteries were surgically prepared and cannulated. One femoral cannula was used for continuous blood pressure monitoring, and the other was used for blood withdrawal or fluid administration. Animals were shocked by withdrawal of blood over 10 min to achieve a mean arterial pressure (MAP) of 25 mmHg. MAP was maintained at 25 to 30 mmHg for 2 h by withdrawing or returning blood as needed. For resuscitation, MAP was increased to ≥80 mmHg by infusing over a period of 30 min all of the remaining shed blood plus a volume of Ringer’s lactate solution equal to twice the total volume of shed blood. Time-matched sham-operated mice underwent anesthesia and bilateral femoral artery cannulation but were not bled or resuscitated. Immediately after completion of the resuscitation protocol, the mice were injected intraperitoneally with either 600 μg anti-HMGB1 neutralizing antibody (aHMGB1 group) or 600 μg non-immune rabbit IgG (IgG group) in 500 μL PBS. Mice in the sham group were injected with a similar volume of PBS. All animals were returned to their cages and allowed free access to food and water.
In experiment 1, 24 h after resuscitation, all surviving mice were re-anesthetized with sodium pentobarbital (90 mg/kg intramuscularly), and the following procedures were performed: 1) blood was aspirated from the heart for the subsequent measurement of serum concentrations of IL-6, IL-10, nitrite, and alanine aminotransferase (ALT); 2) a segment of ileum was harvested for determination of mucosal permeability; 3) ileal mucosa was scraped for measurements of the expression of TNF, IL-6, and iNOS mRNA and the tight junction proteins, ZO-1 and occludin; and 4) the MLN complex was harvested to measure bacterial translocation.
In experiment 2, the mice were re-anesthetized 4 h after resuscitation, blood was aspirated from the heart to measure the serum concentration of IL-6, IL-10, and ALT, a segment of ileum was harvested for determination of mucosal permeability, and the MLN complex was harvested to measure bacterial translocation.
Determination of Intestinal Mucosal Permeability
Ileal mucosal permeability to the fluorescent tracer, FITC-dextran with a molecular mass of 4000 Da (FD4), was determined using an everted gut sac method, as previously described (7). Briefly, gut sacs were prepared in ice-cold modified Krebs-Henseleit bicarbonate buffer (KHBB), pH 7.4. One end of the gut segment was ligated with suture, and then the segment was everted onto a thin plastic rod. The resulting sac was secured with another suture to the grooved tip of a 3-mL plastic syringe containing KHBB. The sac was gently distended by injecting 1.5 mL KHBB and suspended for 30 min in a 50-mL beaker containing 40 mL KHBB plus FD4 (40 mg/mL). The solution in the beaker was temperature jacketed at 37 °C and was continuously bubbled with a gas mixture containing 95% O2, 5% CO2. The FD4 concentration of the fluid in the beaker and the inside of the sac was determined spectrofluoro-metrically, and permeability was expressed as the mucosal-to-serosal clearance of FD4.
Estimation of Bacterial Translocation
The skin was cleaned with 10% povi-done-iodine. Using sterile technique, the abdominal cavity was opened and the viscera exposed. The MLN complex was removed, weighed, and placed in a grinding tube containing 0.5 mL ice-cold PBS. The MLN were homogenized with glass grinders, and a 250-μL aliquot of the homogenate was plated onto brain-heart infusion and MacConkey’s agar (Becton Dickinson, Sparks, MD, USA). The plates were examined 24 h later after being aerobically incubated at 37 °C. The colonies were counted and results expressed as colony-forming units (CFU) per gram of tissue.
Measurement of IL-6, IL-10, and Nitrite Concentrations in Serum
Blood (200 μL) was obtained by cardiac puncture and placed in a 0.5-mL centrifugation tube on ice. The samples were centrifuged at 5000g for 3 min. The serum was collected and stored frozen at −80 °C until assayed for IL-6 and IL-10 using ELISA kits from R&D Systems (Minneapolis, MN, USA) according to the manufacturer’s instructions. Serum nitrite concentration was measured using the Greiss reaction, as described (32).
Reverse-Transcriptase Polymerase Chain Reaction (RT-PCR)
Total RNA was extracted from thawed ileal mucosal tissue samples with chloroform and TRI Reagent (Molecular Research Center, Cincinnati, OH, USA) exactly as directed by the manufacturer. The total RNA was treated with DNAFree (Ambion, Houston, TX, USA) as instructed by the manufacturer using 10 units of DNase I/10 μg RNA. Total RNA (2 μg) was reverse transcribed in a 40-μL reaction volume containing 0.5 μg oligo(dT)15 (Promega, Madison, WI, USA), 1 mM of each dNTP, 15 units AMV reverse transcriptase (Promega), and 1 unit/μL recombinant RNasin ri-bonuclease inhibitor (Promega) in 5 mM MgCl2, 10 mM Tris-HCl, 50 mM KCl, and 0.1% Triton X-100, pH 8.0. The reaction mixtures were preincubated at 21 °C for 10 min before DNA synthesis. The reverse transcription reactions were carried out for 50 min at 42 °C and were heated to 95 °C for 5 min to terminate the reactions. Reaction mixtures (50 μL) for PCR were assembled using 5 μL cDNA template, 10 units AdvanTaq Plus DNA Polymerase (Clontech, Palo Alto, CA, USA), 200 μM of each dNTP, 1.5 mM MgCl2, and 1.0 μ M of each primer in 1× AdvanTaq Plus PCR buffer. PCR reactions were performed using a Model 480 thermocycler (Perkin Elmer, Norwalk, CT, USA). Amplification of cDNA was initiated with 5 min of denaturation at 94 °C. The PCR conditions for amplifying cDNA for TNF and IL-6 were as follows: denaturation at 94 °C for 45 s, annealing at 61 °C for 45 s, and polymerization at 72 °C for 45 s. To ensure that amplification was in the linear range, we empirically determined that 33 was the optimal number of cycles for TNF and IL-6 cDNA. After the last cycle of amplification, the samples were incubated at 72 °C for 10 min and then held at 4°C. The 5′ and 3′ primers for TNF were GGC AGG TCT ACT TTG GAG TCA TTG C and ACA TTC GAG GCT CCA GTG AAT TCG G, respectively; the expected product length was 307 bp. The 5′ and 3′ primers for IL-6 were TTC CAT CCA GTT GCC TTC TTG G and TTC TCA TTT CCA CGA TTT CCC AG, respectively; the expected product length was 174 bp. 18S ribosomal RNA was amplified to verify equal loading. For this reaction, the 5′ and 3′ primers were CCC GGG GAG GTA GTG ACG AAA AAT and CGC CCG CTC CCA AGA TCC AAC TAC, respectively; the expected product length was 209 bp. Ten micro-liters of each PCR reaction was electrophoresed on a 2% agarose gel and scanned using a NucleoVision imaging workstation (NucleoTech, San Mateo, CA, USA). Densitometry was carried out using GelExpert release 3.5 (NucleoTech).
Western Blotting
For preparation of samples, small intestinal mucosa was gently scraped with a glass microscope slide. The scrapings were homogenized on ice with a Polytron tissue homogenizer in 1 mL RIPA buffer [1× PBS, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 0.1 mg/mL phenylmethylsulfonyl fluoride, 1.0 mM sodium orthovanadate, and 1× mammalian protease inhibitor cocktail (Sigma-Aldrich catalog #P 8340)]. The homogenate was transferred to a 1.5-mL microfuge tube.
The samples were sonicated 3 times for 30 s on ice using a 0.1-W Fisher Scientific Sonic Dismembrator fitted with a microtip on power setting 3. The lysate was transferred to a microcentrifuge tube and incubated for 30 min on ice. The lysate was centrifuged at 10,000g for 20 min at 4 °C, and then the supernatant was transferred to a new tube. Total protein concentration was determined using the Bio-Rad (Hercules, CA, USA) protein reagent.
Equivalent amounts of protein were mixed with Laemmli buffer (20% glycerol; 10% β-mercaptoethanol; 5% SDS; 0.2 M Tris·HCl, pH 6.8; and 0.4% bro-mophenol blue). After boiling for 5 to 10 min, the protein samples were centrifuged for 10 s. Samples of the supernatants containing 100 μg mucosal protein/lane were electrophoresed at 100 mÅ for 40 min on 7.5% precast SDS-polyacrylamide gels (Bio-Rad). The size-fractionated proteins were electroblotted onto a Hybond-P PVDF membrane (Amersham Pharmacia Biotech, Leicester, Denmark) and blocked with Blotto (1× TBS, 5% milk, 0.05% Tween-20, and 0.2% NaN3) for 60 min. The membranes were then incubated at 4 °C overnight with rabbit anti-ZO-1 polyclonal antibody from Zymed Laboratories (South San Francisco, CA, USA) used at a 1:2000 dilution, rabbit anti-occludin polyclonal antibody (Zymed; 1:2000 dilution), rabbit anti-RAGE poly-clonal antibody (product number sc-5563) from Santa Cruz Biotechnology (Santa Cruz, CA, USA) used at a 1:500 dilution, or anti-β-actin monoclonal antibody (Santa Cruz; 1:4000 dilution). Antibodies were diluted using TBST (1× TBS, 0.1% Tween-20, and 5% BSA). After washing 3 times in 1× PBST, immunoblots were exposed at room temperature for 1 h to a 1:10,000 dilution of the appropriate horse-radish peroxidase–conjugated secondary antibody (Jackson ImmunoResearch Laboratories; West Grove, PA, USA). After 3 washes in PBST and 2 washes in PBS, the membranes were impregnated with the enhanced chemiluminescence (ECL) substrate (Amersham Pharmacia Biotech) and used to expose X-ray film according to the manufacturer’s instructions.
Statistical Methods
Results are presented as median values or means ± SEM, as appropriate. Differences in serum HMGB1 concentrations or CFU between groups were analyzed using the Wilcoxon U test. Other continuous data were analyzed using Student t test or analysis of variance followed by Fisher’s LSD test, as appropriate. P values < 0.05 were considered significant. Differences in survival were assessed using Fisher’s exact test. Summary statistics are presented for densitometry results from studies using RT-PCR to estimate TNF and IL-6 mRNA expression, but these results were not subjected to statistical analysis because the method employed was only semi-quantitative and the sample sizes were small (n = 3–4) (7).
RESULTS
Circulating HMGB1 Concentrations in Trauma Victims with Hemorrhagic Shock
Serum concentrations of HMGB1 were determined in blood samples from 25 patients (20 men and 5 women) admitted to the University of Pittsburgh Medical Center from January 2004 through May 2005. The median age of the patients was 32 years (range, 18–54). The median blood lactate concentration measured at the time of presentation in the Emergency Department was 3.5 mM (range, 0.99–6; upper limit of normal is 2 mM). Increased blood lactate concentration is a cardinal biochemical feature of hemorrhagic shock (33). When serum HMGB1 concentration was measured in blood samples from 9 healthy human volunteers, the median was 9.6 ng/mL (range, 0–20.8) (Figure 1). In contrast, the median serum HMGB1 concentration in blood samples obtained within 6 h of injury was 54 ng/mL (range, 25.1–162.9) (P < 0.0001 vs. normal volunteers). The median serum HMGB1 concentration in blood samples obtained about 24 h after injury was 57.2 ng/mL (range, 21.3–112.6) (P < 0.0001 vs. normal volunteers).
Figure 1.

Serum HMGB1 concentrations in trauma victims with hemorrhagic shock and healthy human volunteers (n = 9). Blood samples from the trauma victims were obtained as soon as possible after admission to the Emergency Department and always < 6 h after the time of injury (n = 25) and again the next day (i.e., approximately 24 h after injury; n = 24). Serum HMGB1 concentrations in the trauma victims at either time point were significantly greater than the values measured in the healthy subjects (P < 0.0001).
Survival of Mice Subjected to HS/R
As expected, all 10 mice in the sham group were alive 24 h after HS/R. In contrast, only 6 of 13 IgG group mice (46%) survived after being subjected to HS/R and treated with nonimmune IgG (P = 0.007). When mice subjected to HS/R were treated with anti-HMGB1 antibody, 9 of 10 mice (90%) survived for 24 h (P = 0.037 vs. IgG group).
HS/R-Induced Intestinal Barrier Dysfunction
Relative to values obtained by studying sham-hemorrhaged controls, ileal mucosal permeability to the fluorescent macromolecule, FD4, was significantly increased in surviving mice studied 24 h after being subjected to HS/R and treatment with nonimmune IgG (Figure 2A). Similarly, bacterial translocation to MLN was minimal in the sham group, but was extensive in the IgG group (Figure 2B). Treatment with anti-HMGB1 antibody at the time of resuscitation, however, significantly ameliorated the development of both ileal mucosal hyperpermeability and bacterial translocation.
Figure 2.
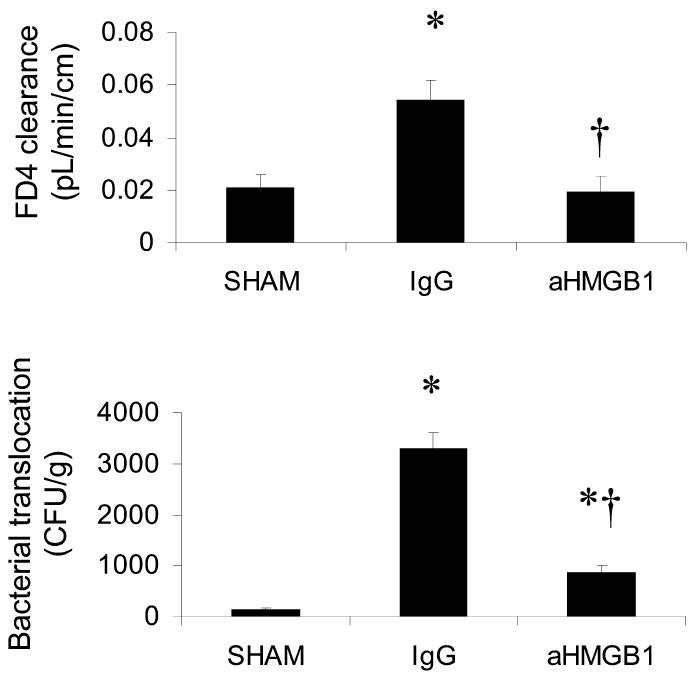
Effect of treatment with anti-HMGB1 antibody on HS/R-induced gut mucosal permeability (A) and bacterial translocation to MLN (B). Ileal mucosal permeability and bacterial translocation were assessed 24 h after HS/R (or the sham procedure). Mice in the sham group (n = 6) were subjected to anesthesia and vascular cannulation but not hemorrhage. Mice in the IgG (n = 6) and aHMGB1 (n = 8) groups were shocked by withdrawal of blood over 10 min until MAP decreased to 25 mmHg. MAP was maintained at 25 to 30 mmHg for 2 h, and then the mice were resuscitated by infusing all of the remaining shed blood plus a volume of Ringer’s lactate solution equal to twice the total volume of shed blood. Mice in the IgG group were treated with a single intraperitoneal dose of rabbit IgG (600 μg in 500 μL) immediately after the resuscitation procedure. Mice in the aHMGB1 group were similarly treated, except these animals received a dose of a polyclonal rabbit neutralizing aHMGB1 antibody. Mice in the sham group were treated with PBS (500 μL intraperitoneally). Results are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. IgG.
Serum IL-6, IL-10, and Nitrite Concentrations
Increased IL-6 synthesis (11) and increased iNOS-derived nitric oxide production (34–37) have been implicated as important factors leading to the development of gut epithelial hyperper-meability or increased bacterial translocation in a variety of inflammatory states. By the same token, increased endogenous production of the anti-inflammatory cytokine, IL-10, has been proposed as a factor that protects gut barrier function under certain conditions (38,39). Accordingly, we measured circulating levels of IL-6, IL-10, and nitrite (NO metabolite) in sham-hemorrhaged mice, mice subjected to HS/R and treated with nonimmune IgG, and mice subjected HS/R and treated with anti-HMGB1 antibody. Mean serum IL-6 (Figure 3A) and IL-10 (Figure 3B) concentrations 24 h after HS/R were significantly greater in the IgG and aHMGB1 groups than in the sham group. However, the mean circulating levels of both of these cytokines were significantly lower in the aHMGB1 group than in the IgG group. Mean serum nitrite concentrations were similar (40–60 μM) in all 3 groups.
Figure 3.
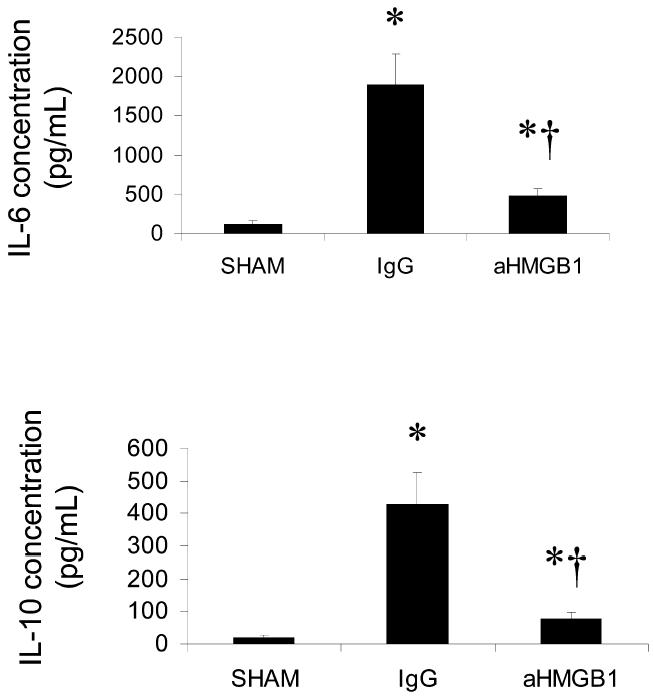
Effect of treatment with anti-HMGB1 antibody on HS/R-induced changes in serum IL-6 (A) and IL-10 (B) concentrations. Cytokine levels were assessed 24 h after HS/R (or the sham procedure). Groups and sample sizes are the same as in Figure 1. Results are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. IgG.
Ileal Mucosal TNF and IL-6 mRNA Expression
A number of studies using in vitro model systems suggest that the pro-inflammatory cytokine, TNF, is capable of disrupting intestinal epithelial barrier function (38–40). Moreover, increased systemic and/or local production of IL-6 has been implicated as being important in the pathogenesis of gut barrier dysfunction associated with HS/R (11). Accordingly, we used semi-quantitative RT-PCR to estimate changes in the expression of TNF (Figure 4A) and IL-6 (Figure 4B) mRNA in ileal mucosal scrapings after HS/R. In mice treated with with nonimmune IgG, steady-state levels of transcripts for both of these cytokines were increased in mucosa 24 h after resuscitation. However, increased expression of TNF or IL-6 mRNA was not detectable in scrapings from mice subjected to HS/R and treated with anti-HMGB1 antibody.
Figure 4.

Effect of treatment with IgG or HMGB1 antibody on the expression of TNF, iNOS, and IL-6 mRNA in samples of intestinal mucosal tissue. RT-PCR was performed using tissue samples obtained 24 h after HS/R. Bands visualized after agarose gel electrophoresis of PCR reaction products were scanned using a NucleoVision imaging workstation and quantified using GelExpert release 3.5. Data are means ± SE (n = 3 per condition). A typical gel is depicted.
ZO-1 and Occludin Expression
The regulation and maintenance of normal intestinal mucosal barrier function depends on the proper assembly and functioning of the tight junctions between adjacent epithelial cells. Formation of tight junctions requires the assembly of several proteins, including the trans-membrane protein, occludin, and the in-tracellular protein, ZO-1 (41,42). Therefore, we used Western blotting of whole-cell extracts prepared from mucosal scrapings to assess changes in the expression of these proteins. Compared with the results obtained by studying ileal mucosal scrapings from sham-hemorrhaged mice, the expression of ZO-1 and occludin was substantially decreased in mice subjected to HS/R and treated with nonimmune IgG (Figure 5). Treatment with anti-HMGB1 antibody, however, preserved the expression of both of these proteins after HS/R.
Figure 5.

Effect of treatment with anti-HMGB1 antibody on HS/R-induced changes in ZO-1, occludin, and RAGE protein expression in gut mucosal tissue lysates. Western blots were performed using mucosal extracts prepared from tissues obtained 24 h after HS/R or the sham procedure. The groups are same as in Figure 4. The figure depicts results from representative assays that were performed 3 or 4 times with comparable findings. Typical gels are depicted.
Ileal Mucosal RAGE Expression
The receptor for advanced glycation end-products (RAGE), a member of the immunoglobulin superfamily of proteins, is activated by a wide variety of ligands, including products of the nonenzymatic oxidation of glucose (AGEs) (43), the amyloid-β peptide cleavage product of β-amyloid precursor protein (44), and the S100/calgranulin family of pro-inflammatory cytokine-like mediators (45). HMGB1 also binds to RAGE with high affinity (46,47), and at least some of the pro-inflammatory and cytopathic effects of HMGB1 appear to be mediated by binding of HMGB1 to RAGE (30,48–50). Therefore, we used Western blotting of whole-cell extracts prepared from mucosal scrapings to assess the expression of RAGE in mice subjected to HS/R or the sham procedure. As shown in Figure 5C, expression of RAGE was barely detectable in the sham group. However, 24 h after HS/R, RAGE expression was upregulated in both the IgG and aHMGB1 groups.
Serum ALT Concentration
Other anti-inflammatory therapeutic agents have been shown to ameliorate hepatocellular injury after HS/R in rodents (7,51). In the present study, however, treatment with anti-HMGB1 antibody was not demonstrably effective in this regard. As expected, the serum concentration of ALT, a marker of hepatocel-lular damage, was significantly increased in mice subjected to HS/R 24 h earlier (Figure 6). Although the mean concentration of ALT was somewhat lower in the aHMGB1 group, the treatment effect was not statistically significant. However, by the same token, the mean serum ALT concentration in the aHMGB1 group was not significantly greater than the mean value measured in the SHAM group.
Figure 6.
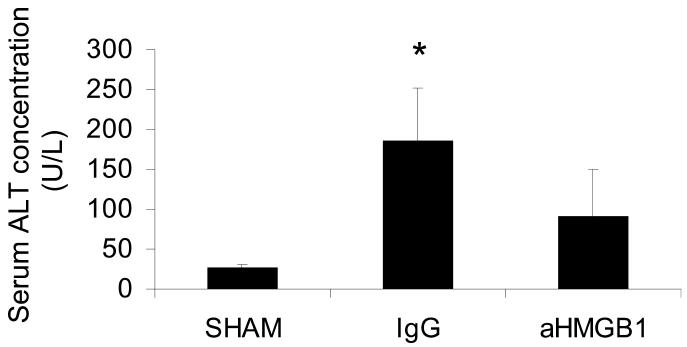
Effect of treatment with anti-HMGB1 antibody on HS/R-induced changes in serum ALT concentration assessed 24 h after HS/R (or the sham procedure). Groups and sample sizes are the same as in Figure 1. Results are means ± SEM. *P < 0.05 vs. sham; †P < 0.05 vs. IgG.
Effect of Anti-HMGB1 4 Hours After Resuscitation
In previous studies, we have shown that HS/R in mice leads to ileal mucosal hyperpermeability and increases bacterial translocation to MLN as early as 4 h after resuscitation (7,8,11). Prompted by the data presented here indicating that treatment with anti-HMGB1 antibody ameliorated gut barrier dysfunction assessed at 24 h after resuscitation, we sought to determine whether this therapy would also ameliorate gut barrier dysfunction at the 4-h time point. Because the sham procedure clearly represents a form of trauma, in this second study we also included a group of completely untreated mice in addition to the sham group. Although the combination of general anesthesia and the trauma associated with vascular cannulation and HS/R clearly caused derangements in gut barrier function 4 h after resuscitation, these changes were not ameliorated by treatment with anti-HMGB1 antibody (Table 1). Similarly, treatment with the antibody also failed to significantly alter circulating concentrations of IL-6, IL-10, or ALT measured 4 h after resuscitation.
Table 1.
Effect of treatment with anti-HMGB1 antibody on several parameters measured at 4 h after HS/R or the sham procedure in mice.
| Parameter | Normal | Sham | IgG | aHMGB1 |
|---|---|---|---|---|
| n | 6 | 6 | 6 | 6 |
| FD4 clearance (nL min−1 cm−1) | 14 ± 1 | 24 ± 2a | 35 ± 6b | 29 ± 6 |
| Bacterial translocation (CFU/g) | 17 ± 17 | 374 ± 251a | 258 ± 216 | 241 ± 198 |
| Serum IL-6 concentration (pg/mL) | 0 ± 0 | 49 ± 8a | 360 ± 161b | 262 ± 54b |
| Serum IL-10 concentration (pg/mL) | 7 ± 2 | 19 ± 5a | 173 ± 35b | 193 ± 40b |
| Serum ALT concentration (pg/mL) | 40 ± 2 | 55 ± 4 | 130 ± 22b | 103 ± 20b |
P < 0.05 sham vs. normal;
P < 0.05 IgG vs. sham. None of the contrasts between the IgG and aHMGB1 groups were statistically significant.
DISCUSSION
HMGB1, which has been shown to be a late mediator of lethality in murine models of endotoxemia and sepsis, does not reach maximal levels in the circulation until about 15 to 18 h after the initiation of these inflammatory processes (20,21). Similarly, in a recent study that used an animal model very similar to the one employed here, Kim and colleagues (29) reported that plasma HMGB1 levels are undetectable 4 h after HS/R but are elevated 24 after HS/R. The clinical data reported here present a somewhat different picture. Our results indicate that circulating HMGB1 levels are increased in human trauma victims with physiological or biochemical evidence of shock (low systolic blood pressure or high base deficit, respectively) within 6 h of injury. We can only speculate about the mechanisms responsible for the observation that circulating HMGB1 levels are increased relatively sooner after hemorrhage in trauma patients compared with mice subjected to HS/R (29). One obvious difference between the murine paradigm and the clinical situation is that hemorrhage in humans is typically associated with extensive soft tissue and/or bony trauma, whereas most hemorrhage models using rodents, including the one employed for the present studies and the one employed by Kim et al. (29), subject the animals to only minimal mechanical tissue damage (from the surgical manipulation needed to gain vascular access). HMGB1 is present in most nucleated cells, and it is known that HMGB1 is released when cells undergo necrosis (27), so it seems probable to us that soft and/or bony tissue trauma leads to HMGB1 release on the basis of direct mechanical damage to cells. Although Ombrellino et al. (28) previously reported a single case of hemorrhagic shock (due to rupture of an abdominal aortic aneurysm) that was associated with increased circulating levels of HMGB1, the data reported by us here are the first to document that serum HMGB1 levels are significantly increased in trauma patients with hemorrhagic shock.
Prompted by our clinical findings and our previous observation that HMGB1 is capable of increasing the permeability of Caco-2 enterocyte-like monolayers in vitro (30), we hypothesized that treatment with a neutralizing polyclonal anti-HMGB1 antibody at the time of resuscitation would ameliorate the deleterious effects of HS/R on gut barrier function. When measurements were made at 24 h after resuscitation, treatment with a single intraperitoneal injection of anti-HMGB1 antibody provided significant protection against the development of ileal mucosal hyperpermeability to the fluorescent macromolecule, FD4, and also significantly decreased the extent of bacterial translocation to MLN relative to that measured in hemorrhaged control mice treated with nonimmune IgG. These beneficial effects of anti-HMGB1 on gut barrier function were associated with a significant improvement in survival. Whereas only 46% of the mice in the IgG group survived to 24 h, 90% of the mice in the aHMGB1 group survived to this time point. Although we did not assess survival past 24 h, our previous experience with this murine model of HS/R indicates that most animals alive at 24 h will be permanent survivors (R.Y. and M.P.F., unpublished observations). Because of the marked difference in survival between the 2 groups, our results probably underestimated the degree of protection against post-HS/R gut barrier dysfunction that was provided by the anti-HMGB1 antibody, because it is likely that the most severely affected animals in the IgG group did not survive long enough to be included among those used to assess ileal mucosal permeability and bacterial translocation at 24 h.
Multiple mechanisms are probably responsible for gut barrier dysfunction after HS/R. One factor that is important in the first few hours after resuscitation is depletion of adenosine triphosphate within mucosal cells (10). By 24 hours after resuscitation, it seems unlikely that derangements in cellular adenosine triphosphate production still play an important role. Deleterious redox-mediated effects related to the formation of toxic reactive oxygen species also probably contribute to the development of post-HS/R intestinal barrier dysfunction (6,7,52,53). Additionally, there is abundant evidence to support the notion that inflammatory processes are very important. This notion is supported by many studies, showing that pro-inflammatory cytokines, such as in-terferon-γ, TNF, and IL-1β, increase the permeability of enterocyte-like monolayers growing on filters in diffusion chambers (35,40,54,55). In the context of the present study, it is worth pointing out that HMGB1 has been added to the list of cytokines that increase intestinal epithelial permeability in vitro, because Sappington et al. (30) reported that both HMGB1 and the B box portion of this molecule increase the clearance of FD4 across monolayers of Caco-2 cells growing in culture. The notion that inflammatory processes contribute to post-HS/R gut barrier dysfunction is also supported by results from a previous study by our laboratory, wherein we showed that IL-6–deficient mice are protected from the development of ileal mucosal hyperpermeability and increased bacterial translocation after HS/R (11). Various anti-inflammatory pharmacological strategies also have been shown to be effective. For example, Luyer et al. (56) recently reported that treatment of rats with a high-fat enteral diet protects against late bacterial translocation after HS/R, probably by downregulating the inflammatory response. Moreover, Goldman et al. (57) reported that treatment with an anti-TNF antibody ameliorates bacterial translocation in rats after HS/R. The data presented herein add strong support for the idea that inflammatory mechanisms contribute to intestinal barrier dysfunction after HS/R, particularly during the later phases of this process (for example, 24 h after resuscitation). Furthermore, our results indicate that increased release of HMGB1 may be a key element in a “final common pathway” that promotes intestinal epithelial barrier dysfunction after hemorrhage.
Although originally described as a nonhistone DNA-binding protein, HMGB1 is now recognized to function in the extracellular milieu as a pro-inflammatory cytokine (58). HMGB1 has been shown to activate DNA binding by the pro-inflammatory transcription factor, NF-κB, in Caco-2 enterocyte-like cells (30), murine neutrophils (59), human microvascular endothelial cells (50), and murine bone marrow–derived macrophages (60). HMGB1 also has been shown to activate pro-inflammatory signaling via p38 mitogen-activated protein kinase in dendritic cells (61), en-dothelial cells (50), and neutrophils (62), and to promote cytokine production by various cell types (22,61). Based on the results from the present study, HMGB1 appears to be an important signal for the release of key pro-inflammatory cy-tokines, namely IL-6 and TNF, and a key anti-inflammatory cytokine, namely IL-10, at 24 h after HS/R in mice. This idea is supported by our data showing that circulating IL-6 and IL-10 levels were decreased in the aHMGB1 group (relative to those measured at the same time point in the IgG group). This idea also is supported by our observation that steady-state IL-6 and TNF mRNA levels were lower in mucosal samples obtained at 24 h from mice in the aHMGB1 group.
It is possible, of course, that HMGB1 is not directly responsible for the release of anti-inflammatory mediators after HS/R, but rather IL-10 (and perhaps other unmeasured counterregulatory factors) is released in response to increased expression of various “classical” pro-inflammatory mediators (exemplified by TNF and IL-6). According to this model, the observed blunting of the IL-10 response after HS/R in the aHMGB1 group could be explained by the effect of the antibody treatment on the release of pro-inflammatory mediators, such as TNF and IL-6.
In the present study, we did not attempt to measure circulating levels of HMGB1 because there was insufficient serum remaining after we ran the assays for IL-6, IL-10, nitrite, and ALT. Furthermore, as noted above, another laboratory using a model very similar to ours recently reported that plasma HMGB1 levels increase dramatically after HS/R in mice, but only at 24 h and not at 4 h (29). These findings in conjunction with our results showing protection at 24 h but not 4 h following treatment with an anti-HMGB1 antibody support the conclusion that HMGB1 is a late-acting mediator of HS/R-induced intestinal barrier dysfunction, but other factors are more important at an earlier time point (4 h).
Even though serum HMGB1 levels increase after hemorrhagic shock in both humans (data presented here) and mice (29), HMGB1 in the circulation may not be the primary or only target that is responsible for the salutary effects of neutralizing anti-HMGB1 antibody treatment. Recently, our laboratory reported that immunostimulated enterocytes (both Caco-2 cells and primary murine gut epithelial cells) secrete HMGB1 (26). Moreover, in this recent report, we showed that the HMGB1 that is secreted by activated enterocytes amplifies the increase in epithelial permeability that is initiated by other pro-inflammatory mediators (26). Thus, it is conceivable that HMGB1 secreted into the local milieu in the region of the gut epithelium, rather than HMGB1 in the general circulation, is the primary target for neutralizing anti-HMGB1 antibodies.
HMGB1 initiates cellular responses by interacting with at least 3 different cell-surface receptors: Toll-like receptor (TLR) 2, TLR 4, and RAGE (30,59,60,63,64). The relative importance of these various receptors for initiating HMGB1-mediated inflammation due to HS/R is not known. Recently, however, our laboratory carried out a series of studies wherein we compared the effects of HS/R on gut barrier function in wild-type and RAGE-deficient mice and in wild-type mice treated with a control protein or recombinant sRAGE, the ex-tracellular domain of RAGE (65). In these studies, RAGE-deficient mice and mice treated with sRAGE were protected from the late development of mucosal hyperpermeability and increased bacterial translocation induced by HS/R, findings that support the view that RAGE-dependent signaling is very important for the development of gut barrier dysfunction after hemorrhage. It is noteworthy, therefore, that we showed that HS/R induced ileal mucosal RAGE expression in mice, although treatment with anti-HMGB1 antibody failed to downregulate this response.
In summary, we showed for the first time in an in vivo model that treatment with a neutralizing anti-HMGB1 antibody can improve survival and ameliorate gut barrier dysfunction in a clinically relevant model of HS/R. Importantly, we used a posttreatment design (i.e., the therapeutic intervention was initiated at the time of resuscitation, rather than prior to traumatic insult). In the clinical setting, of course, only a posttreatment approach would be feasible. Our data suggest that anti-HMGB1 strategies warrant further evaluation for the prevention of organ system injury and mortality after trauma and hemorrhage.
ACKNOWLEDGMENTS
This work was supported by grants (GM53789 and GM37631) from the National Institutes of Health.
Footnotes
Online address: http://www.molmed.org
REFERENCES
- 1.Anderson RN, Smith BL. Deaths: Leading causes for 2002. National Vital Statistics Reports. 2005;53:1–90. [PubMed] [Google Scholar]
- 2.Sauaia A, et al. Epidemiology of trauma deaths: a reassessment. J Trauma. 1995;38:185–93. doi: 10.1097/00005373-199502000-00006. [DOI] [PubMed] [Google Scholar]
- 3.Acosta JA, et al. Lethal injuries and time to death in a level I trauma center. J Am Coll Surg. 1998; 186:528–33. doi: 10.1016/s1072-7515(98)00082-9. [DOI] [PubMed] [Google Scholar]
- 4.Sauaia A, Moore FA, Moore EE, Norris JM, Lezotte DC, Hamman RF. Multiple organ failure can be predicted as early as 12 hours after injury. J Trauma. 1998;45:301–3. doi: 10.1097/00005373-199808000-00014. [DOI] [PubMed] [Google Scholar]
- 5.Moore FA, Moore EE, Sauaia A. Blood transfusion: an independent risk factor for postinjury multiple organ failure. Arch Surg. 1997;132:620–4. [PubMed] [Google Scholar]
- 6.Tawadrous ZS, Delude RL, Fink MP. Resuscitation from hemorrhagic shock with Ringer’s ethyl pyruvate solution improves survival and ameliorates intestinal mucosal hyper-permeability in rats. Shock. 2002;17:473–7. doi: 10.1097/00024382-200206000-00006. [DOI] [PubMed] [Google Scholar]
- 7.Yang R, et al. Ethyl pyruvate modulates inflammatory gene expression in mice subjected to hemorrhagic shock. Am J Physiol Gastrointest Liver Physiol. 2002;283:G212–22. doi: 10.1152/ajpgi.00022.2002. [DOI] [PubMed] [Google Scholar]
- 8.Yang R, et al. Effect of hemorrhagic shock on gut barrier function and expression of stress-related genes in normal and gnotobiotic mice. Am J Physiol Regul Integr Comp Physiol. 2002;283:R1263–74. doi: 10.1152/ajpregu.00278.2002. [DOI] [PubMed] [Google Scholar]
- 9.Wattanasirichaigoon S, Menconi MJ, Fink MP. Lisofylline ameliorates intestinal and hepatic injury induced by hemorrhage and resuscitation in rats. Crit Care Med. 2000;28:1540–9. doi: 10.1097/00003246-200005000-00047. [DOI] [PubMed] [Google Scholar]
- 10.Wattanasirichaigoon S, Menconi MJ, Delude RL, Fink MP. Effect of mesenteric ischemia and reper-fusion or hemorrhagic shock on intestinal mucosal permeability and ATP content in rats. Shock. 1999;12:127–33. doi: 10.1097/00024382-199908000-00006. [DOI] [PubMed] [Google Scholar]
- 11.Yang R, et al. IL-6 is essential for development of gut barrier dysfunction after hemorrhagic shock and resuscitation in mice. Am J Physiol Gastrointest Liver Physiol. 2003;285:G621–9. doi: 10.1152/ajpgi.00177.2003. [DOI] [PubMed] [Google Scholar]
- 12.Russell DH, Barreto JC, Klemm K, Miller TA. Hemorrhagic shock increases gut macromolecu-lar permeability in the rat. Shock. 1995;4:50–5. doi: 10.1097/00024382-199507000-00008. [DOI] [PubMed] [Google Scholar]
- 13.Baker JW, Deitch EA, Li M, Berg RD, Specian RD. Hemorrhagic shock induces bacterial translocation from the gut. J Trauma. 1988;28:896–906. doi: 10.1097/00005373-198807000-00002. [DOI] [PubMed] [Google Scholar]
- 14.Ammori BJ, et al. Early increase in intestinal permeability in patients with severe acute pancreatitis: correlation with endotoxemia, organ failure, and mortality. J Gastrointest Surg. 1999;3:252–62. doi: 10.1016/s1091-255x(99)80067-5. [DOI] [PubMed] [Google Scholar]
- 15.Faries PL, Simon RJ, Martella AT, Lee MJ, Machiedo GW. Intestinal permeability correlates with severity of injury in trauma patients. J Trauma. 1998;44:1031–6. doi: 10.1097/00005373-199806000-00016. [DOI] [PubMed] [Google Scholar]
- 16.Oudemans-van Straaten HM, et al. Intestinal permeability, circulating endotoxin, and postoperative systemic responses in cardiac surgery patients. J Cardiothorac Vasc Anesth. 1996;10:187–94. doi: 10.1016/s1053-0770(96)80235-7. [DOI] [PubMed] [Google Scholar]
- 17.Doig CJ, et al. Increased intestinal permeability is associated with the development of multiple organ dysfunction syndrome in critically ill ICU patients. Am J Respir Crit Care Med. 1998;158:444–51. doi: 10.1164/ajrccm.158.2.9710092. [DOI] [PubMed] [Google Scholar]
- 18.Hua TC, Moochhala SM. Role of nitric oxide in hemorrhagic shock-induced bacterial translocation. J Surg Res. 2000;93:247–56. doi: 10.1006/jsre.2000.5991. [DOI] [PubMed] [Google Scholar]
- 19.Bustin M, Lehn DA, Landsman D. Structural features of the HMG chromosomal proteins and their genes. Biochim Biophys Acta. 1990; 1049:231–43. doi: 10.1016/0167-4781(90)90092-g. [DOI] [PubMed] [Google Scholar]
- 20.Wang H, et al. HMG-1 as a late mediator of endo-toxin lethality in mice. Science. 1999;285:248–51. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- 21.Yang H, et al. Reversing established sepsis with antagonists of endogenous HMGB1. Proc Natl Acad Sci U S A. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Andersson U, et al. High mobility group 1 protein (HMG-1) stimulates proinflammatory cy-tokine synthesis in human monocytes. J Exp Med. 2001;192:565–70. doi: 10.1084/jem.192.4.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rendon-Mitchell B, et al. IFN-gamma induces high mobility group box 1 protein release partly through a TNF-dependent mechanism. J Immunol. 2003;170:3890–7. doi: 10.4049/jimmunol.170.7.3890. [DOI] [PubMed] [Google Scholar]
- 24.Gardella S, et al. The nuclear protein HMGB1 is secreted by monocytes via a non-classical, vesicle-mediated secretory pathway. EMBO Rep. 2002;3:995–1001. doi: 10.1093/embo-reports/kvf198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Bonaldi T, et al. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it toward secretion. EMBO J. 2003;22:5551–60. doi: 10.1093/emboj/cdg516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liu S, et al. HMGB1 is secreted by immunostim-ulated enterocytes and contributes to cytomix-induced hyperpermeability of Caco-2 monolayers. Am J Physiol Cell Physiol. 2006;290:C990–9. doi: 10.1152/ajpcell.00308.2005. [DOI] [PubMed] [Google Scholar]
- 27.Scaffidi P, Misteli T, Bianchi ME. Release of chromatin protein HMGB1 by necrotic cells triggers inflammation. Nature. 2002;418:191–5. doi: 10.1038/nature00858. [DOI] [PubMed] [Google Scholar]
- 28.Ombrellino M, et al. Increased serum concentrations of high-mobility-group protein 1 in haemorrhagic shock. Lancet. 1999;354:1446–7. doi: 10.1016/S0140-6736(99)02658-6. [DOI] [PubMed] [Google Scholar]
- 29.Kim JY, et al. HMGB1 contributes to the development of acute lung injury after hemorrhage. Am J Physiol Lung Cell Mol Physiol. 2005;288:L958–65. doi: 10.1152/ajplung.00359.2004. [DOI] [PubMed] [Google Scholar]
- 30.Sappington PL, et al. HMGB1 B box increases the permeability of Caco-2 enterocytic monolayers and impairs intestinal barrier function in mice. Gastroenterology. 2002;123:790–802. doi: 10.1053/gast.2002.35391. [DOI] [PubMed] [Google Scholar]
- 31.Demetriades D, et al. Mortality prediction of head Abbreviated Injury Score and Glasgow Coma Scale: analysis of 7,764 head injuries. J Am Coll Surg. 2004;199:216–22. doi: 10.1016/j.jamcollsurg.2004.02.030. [DOI] [PubMed] [Google Scholar]
- 32.Song M, Kellum JA, Kaldas H, Fink MP. Evidence that glutathione depletion is a mechanism responsible for the anti-inflammatory effects of ethyl pyruvate in cultured LPS-stimulated RAW 264.7 cells. J Pharmacol Exp Ther. 2004;308:307–16. doi: 10.1124/jpet.103.056622. [DOI] [PubMed] [Google Scholar]
- 33.Siegel JH, et al. Oxygen debt criteria quantify the effectiveness of early partial resuscitation after hypovolemic hemorrhagic shock. J Trauma. 2003;54:862–80. doi: 10.1097/01.TA.0000066186.97206.39. [DOI] [PubMed] [Google Scholar]
- 34.Unno N, et al. Inhibition of inducible nitric oxide synthase ameliorates lipopolysaccharide-induced gut mucosal barrier dysfunction in rats. Gastroenterology. 1997;113:1246–57. doi: 10.1053/gast.1997.v113.pm9322519. [DOI] [PubMed] [Google Scholar]
- 35.Chavez A, Menconi MJ, Hodin RA, Fink MP. Cytokine-induced epithelial hyperpermeability: role of nitric oxide. Crit Care Med. 1999;27:2246–51. doi: 10.1097/00003246-199910000-00030. [DOI] [PubMed] [Google Scholar]
- 36.Jijon HB, et al. Inhibition of poly(ADP-ribose) polymerase attenuates inflammation in a model of chronic colitis. Am J Physiol Gastrointest Liver Physiol. 2000;279:G641–51. doi: 10.1152/ajpgi.2000.279.3.G641. [DOI] [PubMed] [Google Scholar]
- 37.Sorrells DL, et al. Inhibition of nitric oxide with aminoguanidine reduces bacterial translocation after endotoxin challenge in vivo. Arch Surg. 1996;131:1155–63. doi: 10.1001/archsurg.1996.01430230037007. [DOI] [PubMed] [Google Scholar]
- 38.Ma TY, et al. TNF-α-induced increase in intestinal epithelial tight junction permeability requires NT-κB activation. Am J Physiol Gastrointest Liver Physiol. 2004;286:G367–76. doi: 10.1152/ajpgi.00173.2003. [DOI] [PubMed] [Google Scholar]
- 39.Schmitz H, et al. Tumor necrosis factor-alpha (TNF-α) regulates the epithelial barrier in the human intestinal cell line HT-29/B6. J Cell Sci. 1999;112:137–46. doi: 10.1242/jcs.112.1.137. [DOI] [PubMed] [Google Scholar]
- 40.Ma TY, Boivin MA, Ye D, Pedram A, Said HM. Mechanism of TNF-α modulation of Caco-2 intestinal epithelial tight junction barrier: role of myosin light-chain kinase protein expression. Am J Physiol Gastrointest Liver Physiol. 2005; 288:G422–30. doi: 10.1152/ajpgi.00412.2004. [DOI] [PubMed] [Google Scholar]
- 41.Stevenson BR. Understanding tight junction clinical physiology at the molecular level. J Clin Invest. 1999;104:3–4. doi: 10.1172/JCI7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cereijido M, Shoshani L, Contreras RG. Molecular physiology and pathophysiology of tight junctions. I. Biogenesis of tight junctions and epithelial polarity. Am J Physiol Gastrointest Liver Physiol. 2000;279:G477–82. doi: 10.1152/ajpgi.2000.279.3.G477. [DOI] [PubMed] [Google Scholar]
- 43.Kislinger T, et al. N(epsilon)-(carboxymethyl)ly-sine adducts of proteins are ligands for RAGE that activate cell signaling pathways and modulate gene expression. J Biol Chem. 1999;274:31740–9. doi: 10.1074/jbc.274.44.31740. [DOI] [PubMed] [Google Scholar]
- 44.Yan S-D, et al. RAGE and amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature. 1996;382:685–91. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 45.Hofmann MA, et al. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 46.Hori O, et al. The receptor for advanced glycation end products (RAGE) is a cellular binding site for amphoterin: mediation of neurite outgrowth and co-expression of rage and amphoterin in the developing nervous system. J Biol Chem. 1995; 270:25752–61. doi: 10.1074/jbc.270.43.25752. [DOI] [PubMed] [Google Scholar]
- 47.Taguchi A, et al. Blockade of RAGE/amphoterin suppresses tumor growth and metastases. Nature. 2000;405:354–60. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 48.Kallijarvi J, Haltia M, Baumann MH. Amphoterin includes a sequence motif which is homologous to the Alzheimer’s beta-amyloid peptide (Abeta), forms amyloid fibrils in vitro, and binds avidly to Abeta. Biochemistry. 2001;40:10032–7. doi: 10.1021/bi002095n. [DOI] [PubMed] [Google Scholar]
- 49.Taniguchi N, et al. High mobility group box chromosomal protein 1 plays a role in the pathogenesis of rheumatoid arthritis as a novel cytokine. Arthritis Rheum. 2003;48:971–81. doi: 10.1002/art.10859. [DOI] [PubMed] [Google Scholar]
- 50.Fiuza C, et al. Inflammation-promoting activity of HMGB1 on human microvascular endothelial cells. Blood. 2003;101:2652–60. doi: 10.1182/blood-2002-05-1300. [DOI] [PubMed] [Google Scholar]
- 51.Hierholzer C, et al. Essential role of induced nitric oxide in the initiation of the inflammatory response after hemorrhagic shock. J Exp Med. 1998;187:917–28. doi: 10.1084/jem.187.6.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Deitch EA, et al. Hemorrhagic shock-induced bacterial translocation is reduced by xanthine oxi-dase inhibition or inactivation. Surgery. 1988;104:191–8. [PubMed] [Google Scholar]
- 53.Deitch EA, et al. Hemorrhagic shock-induced bacterial translocation: the role of neutrophils and hydroxyl radicals. J Trauma. 1990;30:942–52. doi: 10.1097/00005373-199008000-00002. [DOI] [PubMed] [Google Scholar]
- 54.Unno N, Menconi MJ, Smith M, Fink MP. Nitric oxide mediates interferon-gamma-induced hy-perpermeability in cultured human intestinal epithelial monolayers. Crit Care Med. 1995;23:1170–6. doi: 10.1097/00003246-199507000-00004. [DOI] [PubMed] [Google Scholar]
- 55.Adams RB, Planchon SM, Roche JK. IFN-γ modulation of epithelial barrier function: time course, reversibility, and site of cytokine binding. J Immunol. 1993;150:2356–63. [PubMed] [Google Scholar]
- 56.Luyer MD, et al. Pretreatment with high-fat enteral nutrition reduces endotoxin and tumor necrosis factor-alpha and preserves gut barrier function early after hemorrhagic shock. Shock. 2004;21:65–71. doi: 10.1097/01.shk.0000101671.49265.cf. [DOI] [PubMed] [Google Scholar]
- 57.Goldman G, et al. Tumour necrosis factor mediates bacterial translocation after haemorrhagic shock and endotoxaemia. Eur J Surg. 2001;167:299–304. doi: 10.1080/110241501300091543. [DOI] [PubMed] [Google Scholar]
- 58.Lotze MT, Tracey KJ. High-mobility group box 1 protein (HMGB1): nuclear weapon in the immune arsenal. Nature Rev Immunol. 2005;5:331–42. doi: 10.1038/nri1594. [DOI] [PubMed] [Google Scholar]
- 59.Park JS, et al. Involvement of Toll-like receptors 2 and 4 in cellular activation by high mobility group box 1 protein. J Biol Chem. 2004;279:7370–7. doi: 10.1074/jbc.M306793200. [DOI] [PubMed] [Google Scholar]
- 60.Kokkola R, et al. RAGE is the major receptor for the proinflammatory activity of HMGB1 in rodent macrophages. Scand J Immunol. 2005;61:1–9. doi: 10.1111/j.0300-9475.2005.01534.x. [DOI] [PubMed] [Google Scholar]
- 61.Messmer D, et al. High mobility group box protein 1: an endogenous signal for dendritic cell maturation and Th1 polarization. J Immunol. 2004;173:307–13. doi: 10.4049/jimmunol.173.1.307. [DOI] [PubMed] [Google Scholar]
- 62.Park JS, et al. Activation of gene expression in human neutrophils by high mobility group box 1 protein. Am J Physiol Cell Physiol. 2003;284:C870–9. doi: 10.1152/ajpcell.00322.2002. [DOI] [PubMed] [Google Scholar]
- 63.Rouhiainen A, et al. Regulation of monocyte migration by amphoterin (HMGB1) Blood. 2004;104:1174–82. doi: 10.1182/blood-2003-10-3536. [DOI] [PubMed] [Google Scholar]
- 64.Treutiger CJ, et al. High mobility group 1 B-box mediates activation of human endothelium. J Intern Med. 2003;254:375–85. doi: 10.1046/j.1365-2796.2003.01204.x. [DOI] [PubMed] [Google Scholar]
- 65.Raman KG et al. The role of RAGE in the pathogenesis of intestinal barrier dysfunction after hemorrhagic shock. Am. J. Physiol. Gastrointest. Liver Physiol. In press. [DOI] [PubMed]